

Химическая физика, 2023, T. 42, № 2, стр. 14-27
Люминесценция как метод исследования механизмов фотохимических реакций
В. Ф. Разумов *
Федеральный исследовательский центр проблем химической физики и медицинской химии Российской академии наук
Черноголовка, Россия
* E-mail: razumov@icp.ac.ru
Поступила в редакцию 21.08.2022
После доработки 16.09.2022
Принята к публикации 20.09.2022
- EDN: IXMXCZ
- DOI: 10.31857/S0207401X23020139
Аннотация
В данной обзорной статье сделан краткий экскурс в современное состояние теории люминесценции и на примере собственных работ автора, выполненных совместно с коллегами начиная с 70-х годов прошлого столетия и вплоть до настоящего времени, показано, насколько эффективно применение люминесцентного анализа к изучению механизмов фотохимических превращений.
ВВЕДЕНИЕ
Явление люминесценции известно очень давно. Люминесценцией интересовался еще Аристотель, а понять ее природу пытались такие известные ученые, как Бойль, Ньютон, Ломоносов, Эйлер, Брюстер, Стокс, Беккерель и многие другие. Однако создание физической теории люминесценции стало возможным только с появлением квантовой механики. В развитии учения о люминесценции выдающуюся роль сыграли работы С.И. Вавилова и его школы, в которых были сформулированы основные законы, управляющие процессами излучения световой энергии в люминофорах.
Люминесценция является предметом как фундаментальных, так и прикладных исследований. С одной стороны, она играет большую роль с точки зрения практического применения, а с другой стороны, благодаря исследованиям люминесценции были не только открыты новые физические явления, но и сам люминесцентный метод стал мощным инструментом изучения структуры и свойств вещества.
Еще в 1864 году на заседании Королевского общества Дж. Стокс предложил использовать люминесценцию как метод химического анализа для идентификации различных органических веществ, но люминесцентный анализ в современном понимании возник только лишь в середине прошлого века. Основным достоинством люминесцентного анализа является его исключительно высокая чувствительность. Даже на обычном спектрофлуориметре легко регистрируются концентрации вещества ∼10–5–10–7 M, а предел обнаружения как минимум еще на 2–3 порядка меньше.
Однако эти оценки относятся к так называемому качественному химическому анализу. В случае многокомпонентных систем и необходимости количественного определения их состава методом люминесцентного анализа дело обстоит гораздо сложнее. Как правило, спектры люминесценции сложных органических молекул имеют вид бесструктурных широких полос, и поэтому считается, что это сильно ограничивает селективность и избирательность даже качественного люминесцентного анализа. Тем не менее за последние десятилетия в результате многочисленных исследований отечественных и зарубежных ученых достигнут значительный прогресс как в направлении развития и совершенствования люминесцентного анализа, так и в области исследования люминесценции в целом.
1. ЗАКОНЫ ЛЮМИНЕСЦЕНЦИИ
Люминесценцией, согласно определению С.И. Вавилова, называют свечение, превышающее по интенсивности собственное тепловое излучение вещества в данном спектральном диапазоне при данной температуре, которое возникает в результате внешнего воздействия на люминофор и характеризуется эффектом “послесвечения”, т.е. люминесценция после внешнего воздействия возникает с временем задержки, значительно превосходящим характерный период колебаний световой волны. Характерное время люминесценции составляет величину, большую по крайней мере 10–12–10–10 с.
Необходимым условием возникновения люминесценции является наличие в структуре энергетических уровней запрещенной зоны. По этой причине металлы не обладают люминесценцией. Физическая природа люминесценции по своей сути состоит в излучательных переходах электронов из электронно-возбужденного состояния в основное электронное состояние вещества.
Спектром люминесценции называют зависимость интенсивности люминесцентного излучения от длины волны испускаемого света. В силу эффекта послесвечения спектр люминесценции индивидуального вещества не зависит от способа его возбуждения. Далее мы будем говорить только о фотолюминесценции, т.е. люминесценции, возникающей в результате поглощения возбуждающего света.
Спектры молекулярной люминесценции определяются переходами с поглощением или излучением света между различными электронно-колебательными уровнями молекулы. Вероятности этих переходов определяются принципом Франка–Кондона, суть которого состоит в том, что за время элементарного акта поглощения или излучения кванта света относительное расположение ядер и их кинетическая энергия не изменяются и переход с большей вероятностью происходит на тот электронно-колебательный уровень, который имеет то же значение межъядерного расстояния. Впервые этот принцип был сформулирован в 1925 году Дж. Франком на основе полуклассических представлений, а Э. Кондон в 1926 году дал его квантово-механическую трактовку [1].
Первым законом люминесценции, сформулированным еще в середине XIX века, было правило Стокса–Ломмеля, согласно которому спектр фотолюминесценции сдвинут относительно спектра поглощения в сторону длинных волн. Разница длин волн максимумов спектров поглощения и флуоресценции называется стоксовым сдвигом. Данное явление связано с потерей некоторой части поглощенной световой энергии на тепловое движение. Однако теперь мы знаем, что возможен и обратный процесс, когда излучаемый квант света имеет бóльшую энергию, чем поглощенный. Это явление называется антистоксовой люминесценцией, при которой тепловая энергия переходит в световую. Таким образом, энергетический КПД фотолюминесценции может превышать 100%, но как показал Л.Д. Ландау, эффективность антистоксовой люминесценции определяется вторым законом термодинамики [2], который накладывает определенные ограничения на его значения:
В результате большого цикла исследований, проведенных в 20–40-х годах прошлого века С.И. Вавиловым, был сформулирован основной закон люминесценции, устанавливающий зависимость квантового выхода фотолюминесценции от длины волны возбуждающего света [4]. Согласно закону Вавилова, квантовый выход люминесценции постоянен при изменении в широких пределах длины волны возбуждающего света в стоксовой области и падает, если длина волны возбуждающего света лежит в антистоксовой области спектральной полосы поглощения. В соответствии с постоянством квантового выхода энергетический выход люминесценции растет с увеличением длины волны возбуждающего света и падает в антистоксовой области.
Закон Вавилова связан с независимостью спектра люминесценции от длины волны возбуждающего света и обусловлен быстрой по сравнению с временем жизни электронного возбуждения колебательной релаксацией на каждом электронном уровне. Падение квантового и энергетического выходов при возбуждении люминесценции в антистоксовой области обусловлено уменьшением вероятности электронного перехода на возбужденный уровень, т.е. в антистоксовой области возрастает доля “неактивного” поглощения света. Однако константы скорости флуоресценции и безызлучательного перехода в основное состояние не могут зависеть от того, каким образом молекула оказалась в возбужденном состоянии. Поэтому квантовый выход люминесценции не должен зависеть от длины волны возбуждающего света и в антистоксовой области, если его рассчитывать по отношению к “активному” поглощению.
Физический смысл закона Вавилова состоит в том, что за время жизни люминесценции многоатомная молекула, находящаяся в возбужденном состоянии, приходит в термодинамическое равновесие с окружением и “забывает” о предыстории, связанной с возбуждением, потому что характерное значение константы скорости излучения по крайней мере на 2–3 порядка меньше константы скорости колебательной релаксации. Другими словами, для люминофора поглощение и излучение света являются независимыми между собой процессами. Тем не менее оказалось, что существуют связи между спектрами поглощения и излучения люминофора. Об одной из них – правиле Стокса–Ломмеля мы уже говорили. А в 1931 году В.Л. Лёвшин сформулировал еще одно правило, устанавливающее связь спектров поглощения и люминесценции, согласно которому электронно-колебательные спектры поглощения и люминесценции молекул, представленные в зависимости от волнового числа v, зеркально симметричны относительно частоты электронного перехода [5]. Объяснение зеркальности спектров было дано Д.И. Блохинцевым в 1939 году, из которого следует, что правило Лёвшина выполняется в том случае, когда колебательные частоты молекулы в основном и возбужденном электронных состояниях одинаковы, а прямые и обратные электронные переходы равновероятны [6].
В 1950 году американский ученый Михаэль Каша обобщил описанные выше закономерности люминесценции в виде простого тезиса (правило Каша), согласно которому для органических молекул в конденсированной фазе излучение фотона (люминесценция) всегда происходит с низшего возбужденного уровня данной мультиплетности. Поэтому независимость в целом спектров и квантового выхода люминесценции от длины волны возбуждающего света можно рассматривать как следствие правила Каша.
2. СООТНОШЕНИЕ КЕННАРДА–СТЕПАНОВА–ВАН РОСБРУКА–ШОКЛИ
В 1957 году Б.И. Степанов [7], рассматривая излучательные переходы между равновесно заселенными колебательными подуровнями основного и возбужденного электронных состояний в многоатомных молекулах, спектры люминесценции которых не зависят от длины волны возбуждающего света, получил следующее соотношение между спектрами поглощения, ε(λ), и люминесценции, f(λ):
где λ – длина волны света, h – постоянная Планка, c – скорость света, kB – постоянная Больцмана, T – температура. Это соотношение по форме аналогично формуле Кирхгофа, определяющей универсальную связь между излучательной, A(λ), и поглощательной, B(λ), способностями любого материального тела:
Отличие формулы (2) от формулы (1) состоит лишь в том, что в (1) вместо знака равенства стоит знак пропорциональности, и при этом формула Планка P(λ, T) – функция спектрального распределения излучения черного тела представлена в приближении Вина, что вполне естественно, потому что при обычных температурах энергия квантов люминесценции намного больше kBT. На первый взгляд, соотношение (1) представляется достаточно неожиданным. Почему для люминесценции, которая по определению является неравновесным излучением, выполняется соотношение, справедливое для равновесного теплового излучения? Здесь следует также вспомнить, что в своей основополагающей работе [8] Кирхгоф особо отметил, что полученное им соотношение (2) “…справедливо только лишь в том случае, когда поток световой энергии, поглощаемый телом, преобразуется в тепло. Таким образом, этот универсальный закон может оказаться недействительным в некоторых частных случаях, например, когда тело испускает свет не в результате теплового движения материи, а в результате какого-то внешнего нетеплового возмущения”. Среди таких частных случаев Кирхгоф выделил различные типы люминесценции.
Тем не менее вывод формулы (1), представленный Степановым в работе [7], достаточно очевиден. Скорость люминесценции и скорость поглощения света на заданной длине волны определяются суммированием скоростей всех элементарных переходов между соответствующими подуровнями возбужденного, S1, и основного, S0, электронных состояний, приводящих к излучению или поглощению кванта света с энергией hc/λ, как это показано на рис. 1. Следовательно,
где ρ(E*) = N*g*(E*)exp(–E*/kBT) и ρ(E) = Ng(E) × × exp(–E*/kBT) – функции больцмановского распределения заселенности колебательных подуровней нижнего возбужденного, S1, и основного, S0, электронных состояний; g*(E*) и g(E) – статистические веса соответствующих уровней энергии, A($E_{{{\lambda }}}^{*},$ λ) – коэффициент Эйнштейна, определяющий вероятность в единицу времени спонтанного перехода с колебательного подуровня E* возбужденного электронного состояния S1 на колебательный подуровень E основного электронного состояния S0 с излучением кванта света с длиной волны λ; B(Eλ, λ) – коэффициент Эйнштейна, определяющий вероятность в единицу времени вынужденного поглощения кванта света с частотой λ с переходом с колебательного подуровня энергии E основного электронного состояния S0 на колебательный подуровень энергии E* возбужденного электронного состояния S1. Отношение коэффициентов Эйнштейна определяется известной формулой:
(4)
$\frac{{A(E_{{{\lambda }}}^{*},{{\lambda }})}}{{B({{E}_{{{\lambda }}}},{{\lambda }})}} = \frac{{g({{E}_{{{\lambda }}}})}}{{g{\text{*}}(E_{{{\lambda }}}^{*})}}\frac{{8{{\pi }}}}{{{{{{\lambda }}}^{4}}}}.$Рис. 1.
К выводу формулы Степанова: схема переходов с поглощением и излучением кванта света заданной энергии.

Для заданной длины волны λ значения энергий E и E* связаны между собой уравнением (рис. 1)
(5)
$E_{{{\lambda }}}^{*} + {{E}_{0}} = {{E}_{{{\lambda }}}} + {{hc} \mathord{\left/ {\vphantom {{hc} \lambda }} \right. \kern-0em} \lambda }.$Проведя в интеграле в числителе формулы (3) замену переменных, согласно (5), и, приняв во внимание (4), приходим к уравнению (1).
Соотношение Степанова в свое время вызвало большой интерес. Появились многочисленные исследования по его экспериментальной проверке, которые показали, что оно выполняется для большинства красителей [9, 10]. Оказалось также, что немного ранее, в 1954 году, Вилли Ван Росбрук и Уильям Шокли получили похожее соотношение для полупроводников [11], которое также было экспериментально подтверждено [12–15].
В работе [16] было указано, что почти идентичный результат был получен еще в 1918 году в работе Эрла Кеннарда [17] из более общих термодинамических соображений. Тогда этот результат не был воспринят научным сообществом и о нем забыли. Сейчас формулу (1) называют соотношением Кеннарда–Степанова.
В работе [17] Е. Кеннард, исходя из того, что термодинамическое равновесие должно устанавливаться для любых материальных тел, а следовательно, и для люминофоров тоже, предложил следующую более общую формулировку закона Кирхгофа: “Поглощение любого тела при заданной температуре пропорционально сумме термического и люминесцентного излучения тела для каждой длины волны света”. Таким образом, формулу (2) Кеннард переписал в следующем виде:
где h(λ) – термическое излучение, а f(λ) – фотолюминесценция, которая определяется интегральным соотношением(7)
$f({{\lambda }}) = \int {F({{\lambda }}{\kern 1pt} {\text{'}},{{\lambda }})P({{\lambda }}{\kern 1pt} {\text{'}},T)d{{\lambda }}{\kern 1pt} {\text{'}}} ,$Как далее предположил Кеннард, в силу независимости процессов термического и люминесцентного излучения поглощательную способность тела можно также представить в виде суммы термического и “люминесцентного” поглощения:
Согласно принципу детального равновесия скорость поглощения света, отвечающего за люминесценцию, должна быть равна скорости излучения света в виде люминесценции, точно так же как скорость “теплового” поглощения должна быть равна скорости теплового излучения. Однако если для “тепловой” составляющей принцип детального равновесия приводит к равенству скоростей излучения и поглощения на каждой отдельно взятой длине волны света:
то для фотолюминесценции длина волны излучаемого света в общем случае не равна длине волны поглощаемого света. Поэтому для фотолюминесценции принцип детального равновесия приводит к следующему уравнению:(10)
$P({{\lambda }}{\kern 1pt} {\text{'}},T)F({{\lambda }}{\kern 1pt} {\text{'}},{{\lambda }}) = P({{\lambda }},T)F({{\lambda }},{{\lambda }}{\kern 1pt} {\text{'}}).$Если для люминофора выполняется закон Вавилова о независимости спектра люминесценции от длины волны возбуждающего света, то из уравнения детального баланса (10) для люминесценции следует соотношение Степанова, так как в этом случае характеристическую функцию люминофора можно представить в виде
и, подставляя выражение (11) в уравнение (10), после небольшого преобразования получим
Это равенство может выполняться для всех длин волн λ и λ' только в том случае, если обе части равенства не зависят от длины волны. Отсюда в приближении Вина следует соотношение (1).
3. УНИВЕРСАЛЬНЫЙ ЗАКОН ФОТОЛЮМИНЕСЦЕНЦИИ
Несмотря на фундаментальный характер соотношения (10), оно до самого недавнего времени не привлекало к себе вообще никакого внимания исследователей: во всяком случае, публикаций на эту тему не было. Кроме того, это соотношение не является априорно очевидным и требует более строгого доказательства, потому что если прямой процесс при λ < λ' является стоксовой люминесценцией, то обратный процесс будет неизбежно антистоксовой люминесценцией, и в равновесии их скорости должны быть равны. В 1926 году Кеннард предпринял попытку проверить соотношение (10) на модельной системе, используя подход Эйнштейна при выводе формулы Планка [18]. Однако очевидно, что если соотношение (10) действительно является общим для любого люминофора, то должно существовать и его строгое теоретическое доказательство, в котором не используются никакие модельные соображения. В нашей работе [19] впервые был представлен такой вывод и проведена экспериментальная проверка выполнения принципа детального равновесия для люминесценции на примере раствора коллоидных квантовых точек фосфида индия с оболочкой сульфида цинка (InP/ZnS), для которых не выполняется закон Вавилова. Оказалось, что точное выражение для принципа микроскопической обратимости для люминофора, находящегося в термодинамическом равновесии с тепловым излучением, имеет следующий вид:
(12)
$\begin{gathered} \frac{1}{{{{\lambda }}_{1}^{4}}}[1 + n({{{{\lambda }}}_{1}},T)]n({{{{\lambda }}}_{1}},T)F\left( {{{{{\lambda }}}_{1}},{{{{\lambda }}}_{2}},t} \right) = \\ = \frac{1}{{{{\lambda }}_{2}^{4}}}[1 + n({{{{\lambda }}}_{2}},T)]n({{{{\lambda }}}_{2}},T)F\left( {{{{{\lambda }}}_{2}},{{{{\lambda }}}_{1}},t} \right), \\ \end{gathered} $Строго говоря, формула (12) выражает не принцип детального равновесия, а равенство вероятностей двух процессов: индуцированного флуктуацией теплового фона на длине волны λ1 излучения люминофора на длине волны λ2 и индуцированного флуктуацией теплового фона на длине волны λ2 излучения люминофора на длине волны λ1. Это следует из того, что в соответствии со статистикой Бозе–Эйнштейна дисперсия флуктуации теплового излучения на длине волны λ пропорциональна [1 + n(λ, T)]n(λ, T).
При n(λ, T) $ \ll $ 1 формулу (12) можно переписать в виде
(13)
$\frac{{{\text{PL}}({{{{\lambda }}}_{0}},{{\lambda }},t)}}{{{\text{PLE}}({{\lambda }},{{{{\lambda }}}_{0}},t)}} \approx \frac{{P({{\lambda }},T)}}{{P({{{{\lambda }}}_{0}},T)}} \approx \frac{{{{\lambda }}_{0}^{4}}}{{{{{{\lambda }}}^{4}}}}\exp \left[ {\frac{{hc}}{{{{k}_{B}}T}}\left( {\frac{1}{{{{{{\lambda }}}_{0}}}} - \frac{1}{{{\lambda }}}} \right)} \right],$Таким образом, уравнение (13) есть математическое выражение нового универсального закона фотолюминесценции, в соответствии с которым для любого люминесцирующего тела отношение спектра люминесценции PL(λ0, λ, t) в момент времени t при возбуждении на заданной длине волны λ0 к спектру возбуждения люминесценции PLE(λ, λ0, t) при наблюдении в тот же момент времени t на той же длине волны λ0 является универсальной функцией, определяемой спектром излучения абсолютно черного тела, P(λ, T) [20].
Теперь рассмотрим уравнение (12) при фиксированных значениях λ1 и λ2 только как функцию времени и представим его в следующем виде:
(14)
$\frac{{F\left( {{{{{\lambda }}}_{1}},{{{{\lambda }}}_{2}},t} \right)}}{{F\left( {{{{{\lambda }}}_{2}},{{{{\lambda }}}_{1}},t} \right)}} = \frac{{P\left( {{{{{\lambda }}}_{2}},T} \right)}}{{P\left( {{{{{\lambda }}}_{1}},T} \right)}}{\text{.}}$В работе [21] мы провели экспериментальную проверку этого теоретического вывода для наноразмерных кластеров коллоидных квантовых точек InP/ZnS, люминесцентные свойства которых кроме всего прочего определяются безызлучательным ферстеровским переносом энергии внутри кластера, состоящего из 103–104 отдельных наночастиц. Кинетика затухания люминесценции в этом случае является неэкспоненциальной и зависит от длины волны возбуждения, но при этом инвариантность кинетики спада люминесценции при перестановке длин волн возбуждения и наблюдения выполнялась достаточно точно.
Одним из практических применений уравнения (13) может быть дистанционное определение температуры с помощью регистрации фотолюминесценции. Действительно, если выбрать некоторую длину волны λ0 и измерить спектр фотолюминесценции при возбуждении на этой длине волны, PL0(λ) ≡ EEM(λ0, λ), и спектр возбуждения фотолюминесценции при наблюдении на этой же длине волны, PLE0(λ) ≡ EEM(λ, λ0), то можно построить следующую зависимость от обратной длины волны:
По тангенсу угла наклона графика зависимости от обратной длины волны логарифма отношения спектра люминесценции ${\text{P}}{{{\text{L}}}_{0}}\left( {{\lambda }} \right) \equiv {\text{PL}}({{\lambda }_{0}},~\lambda )$ к спектру возбуждения люминесценции ${\text{PL}}{{{\text{E}}}_{0}}\left( {{\lambda }} \right) \equiv $ $ \equiv {\text{PLE}}(\lambda ,~{{\lambda }_{0}},~t),$ умноженного на ${{{\text{(}}{{{\lambda }} \mathord{\left/ {\vphantom {{{\lambda }} {{{{{\lambda }}}_{0}}}}} \right. \kern-0em} {{{{{\lambda }}}_{0}}}})}^{4}},$ вычисляется температура T.
4. СПЕКТРАЛЬНЫЙ ЛЮМИНЕСЦЕНТНЫХ АНАЛИЗ МНОГОКОМПОНЕНТНЫХ СИСТЕМ
В идеале спектральный анализ многокомпонентной системы должен давать спектры отдельных компонент и их вклады в спектр смеси. Одним из наиболее распространенных видов спектрального анализа является анализ с использованием оптических спектров поглощения или спектров люминесценции многокомпонентной системы. Сложность решения задачи анализа зависит от наличия априорной информации относительно спектров поглощения или люминесценции отдельных компонент, при этом люминесцентные данные и данные спектров поглощения рассматриваются независимо друг от друга. И в том, и в другом случае, если не известны спектры отдельных компонент, то для решения задачи анализа необходимо иметь набор независимых экспериментальных данных с разным соотношением этих компонент. Этот набор данных может быть результатом определенного внешнего воздействия на исследуемую многокомпонентную систему. Примером такого воздействия может быть, например, фотохимическое превращение одних компонент системы в другие при облучении светом. В наших работах [22–24] путем применения метода сингулярного разложения к набору экспериментальных спектров трехкомпонентной смеси продуктов реакций фотоизомеризации и фотоциклизации обнаружен одноквантовый процесс образования транс-1,2-ди(2-нафтил)этилена (ДНЭ) при фотолизе 4а,4b-дигидробензфенантрена. Необычным эффектом в этом экспериментальном исследовании было то, что процесс цис-транс-фотоизомеризации происходил при поглощении кванта света с энергией, существенно меньшей, чем барьер реакции цис-транс-перехода.
Существенным шагом в люминесцентном анализе многокомпонентных систем был переход к совместному анализу спектров люминесценции и поглощения путем рассмотрения матрицы “возбуждение–люминесценция”. Впервые на важность матрицы “возбуждение–люминесценция” для люминесцентного анализа многокомпонентных систем было обращено внимание в работе [25]. Действительно, измерение матрицы “возбуждение–люминесценция” для смеси всего лишь одного состава сразу позволяет определить число независимых компонент. Дальнейшее развитие этого подхода показало плодотворность применения матрицы “возбуждение–люминесценция” к анализу многокомпонентных систем [26–29].
Универсальное соотношение (13) открывает новые возможности для спектрально-люминесцентного анализа многокомпонентных систем матрицы “возбуждение–люминесценция”, которую для смеси простых фотолюминофоров, подчиняющихся закону Вавилова, можно представить в виде
5. КОНФОРМАЦИОННАЯ ИЗОМЕРИЯ ДИАРИЛЭТИЛЕНОВ И НАРУШЕНИЕ ЗАКОНА ВАВИЛОВА
Как уже было сказано выше, для большинства известных люминофоров при обычных условиях спектр люминесценции и ее квантовый выход не зависят от длины волны возбуждения, а кинетика распада возбужденного состояния является моноэкспоненциальной с характерным временем, также не зависящим от длины волны возбуждения. Однако, как будет подробно изложено далее, для транс-изомеров диарилэтиленов (ДАЭ) закон Вавилова не выполняется; при этом наблюдается неэкспоненциальная кинетика затухания люминесценции и существенная зависимость спектров и квантового выхода люминесценции от длины волны возбуждающего света. Как оказалось, это обусловлено наличием конформационной изомерии в транс-ДАЭ.
На рис. 2 показаны три конформера транс-1,2-ди(2-нафтил)этилена, между которыми устанавливается термическое равновесие в основном состоянии за счет вращения арильных фрагментов молекулы вокруг одинарных связей. Барьер вращения составляет всего несколько сотых долей эВ. Однако в электронно-возбужденном состоянии величина барьера вращения арильного фрагмента вокруг одинарной связи значительно повышается за счет ослабления центральной этиленовой связи и перераспределения электронной плотности на соседние одинарные связи. Поэтому переход между различными конформациями молекулы люминофора в возбужденном состоянии по времени происходит гораздо медленнее распада этого состояния, и с точки зрения процесса люминесценции каждая конформация может фактически рассматриваться как независимое индивидуальное соединение. А поскольку различные конформации молекул обладают различными спектральными характеристиками, то в этом случае для всей системы будет наблюдаться зависимость характеристик люминесценции от длины волны возбуждения.
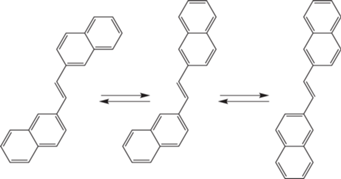
Впервые на аномальные люминесцентные свойства ДНЭ, обусловленные конформационной изомерией, было обращено внимание в наших работах [31, 32], а потом в 1980-х годах эффект был многократно подтвержден (см. обзоры [33, 34] и статью [35]), а в работе [36] даже удалось с помощью определенным образом модифицированного метода главных компонент получить спектры люминесценции отдельных конформеров. В 2002 году вышла работа [37], в которой тем же методом наряду со спектрами люминесценции отдельных конформеров были получены также их спектры возбуждения, а в 2018 году в нашей работе [38] спектры поглощения и люминесценции отдельных конформеров ДНЭ были установлены путем анализа матрицы “возбуждение–люминсценция”.
В конечном счете исследования по конформационной изомерии позволили найти очень элегантный подход к изучению необычных механизмов фотоизомеризации арильных производных этилена, о чем речь пойдет в следующем разделе.
6. МЕХАНИЗМ “HULA-TWIST” ЦИС-ТРАНС-ФОТОИЗОМЕРИЗАЦИИ ДИАРИЛЭТИЛЕНОВ
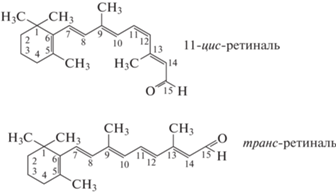
Цис-транс-фотоизомеризация является одной из основных реакций в фотохимии, которая выполняет роль “спускового крючка” для ряда важных биохимических процессов. Одним из таких наиболее известных процессов является фоторецепция. При поглощении света в зрительном белке родопсине его хромофорная группа (11-цис-ретиналь) изомеризуется, переходя в транс-форму (рис. 3), и запускает цепь биохимических реакций, которые сопровождаются изменением проницаемости мембран в палочках и колбочках сетчатки глаза и создают потенциал действия, приводящий в конечном итоге к возникновению соответствующего нервного импульса.
Всегда считалось, что механизм цис-транс-фотоизомеризации представляет собой вращение соответствующего фрагмента молекулы вокруг этиленовой связи: так называемый механизм OBF (one bond flip). При фотовозбуждении С=С-связь значительно ослабляется и барьер вращения, составляющий в основном состоянии величину ∼102 кДж/моль, становится сравнимым с тепловой энергией: 2–3 кДж/моль. Однако, как показали времяразрешенные измерения, цис-транс-переход в ретинале происходит в субпикосекундном интервале времен. В работе [39] приводятся данные, полученные при возбуждении ретиналя импульсом света с длиной волны 500 нм и длительностью 35 фс, свидетельствующие о том, что характерное время цис-транс-превращения хромофора ретиналя в родопсине составляет величину порядка 200 фс. Фактически это самая быстрая известная в настоящее время фотохимическая реакция. Возникает естественный вопрос: как за такое короткое время может произойти вращение такого большого фрагмента молекулы в стесненных условиях белковой молекулы родопсина? В 1976 году Варшел постулировал механизм “велосипедной педали” [40], в котором две соседних одинарных C–H-связи, находящихся между двумя чередующимися двойными связями, вращаются согласованно (рис. 4). Конечным результатом является одновременная изомеризация двух связей, при этом лишь небольшая часть хромофора подвергается внеплоскостному вращению. Очевидно, что для этого требуется меньший объем, чем просто для вращения вокруг этиленовой связи, но все равно не ясно, как это может произойти за 0.1 пс.
Рис. 4.
Два альтернативных механизма цис-транс-фотоизмеризации 11-цис-ретиналя; BP – bicycle-pedal, HT – hula-twist.

В 1985 году был предложен еще один оригинальный механизм фотоизомеризации, получивший название “hula-twist” (HT), в котором соседняя пара одинарных и двойных связей вращается одновременно [41, 42]. Этот механизм предполагает, что только одна группа СН этиленовой связи вращается с выходом из плоскости молекулы, а заместители реорганизуются, оставаясь в плоскости (рис. 4). Прямым экспериментальным доказательством принципиальной возможности НТ-механизма фотоизомеризации оказалась наша работа, опубликованная двумя годами раньше [35], в которой обсуждался процесс фотохимического получения неравновесной концентрации конформеров диарилэтиленов в стеклообразных растворах при 77 K. Суть работы сводилась к следующему. Как было установлено ранее, в стеклообразных растворах фотоизомеризация диарилэтиленов является необратимой и протекает только в сторону образования транс-формы. Это объяснялось тем, что в силу стерической напряженности цис-изомера и его существенно непланарной структуры цис-форма занимает в стеклообразной матрице больший объем, чем транс-изомер (рис. 5). Таким образом, непланарная молекула цис-изомера как бы запасает некоторый свободный объем для изомеризации, которая в конечном счете происходит с уменьшением молекулярного объема. В то же время обратный процесс, т.е. транс-цис-фотоизомеризация, должен идти с увеличением молекулярного объема, и поэтому в твердой стеклообразной матрице не происходит.
Рис. 5.
Структура цис-изомера (слева) и транс-изомера (справа) 1,2-ди(1-нафтил)этилена в кристаллической решетке по данным рентгеноструктурного анализа [43].

В нашей работе [35] было показано, что спектры поглощения и люминесценции транс-формы, полученной в результате цис-транс-фотоизомеризации в стеклообразных растворах, существенно отличаются от спектров транс-изомеров, полученных путем охлаждения жидкого раствора транс-изомеров до 77 К. Двумя годами позже это же исследование было проведено в работе [44].
Мы объяснили это явление тем, что в процессе цис-транс-фотоизомеризации в стеклообразных растворах образуется другой (неравновесный) состав конформеров транс-формы. Действительно, если после цис-транс-фотоизомеризации в стеклообразном растворе постепенно повышать температуру последнего, то спектры изменяются и в конечном счете система приходит к равновесному конформационному составу (рис. 6).
Рис. 6.
Спектры поглощения 1-фенил-(2-нафтил)этилена. а – Кинетика в процессе облучения при 77 К в твердом стеклообразном растворе цис-изомера (красные кривые); конечный спектр, соответствующий транс-форме, показан жирной синей кривой. б – Спектр поглощения конечного продукта (транс-изомера), полученного в процессе облучения (сплошная синяя линия), изменение спектра поглощения при нагревании до 200 К (штриховая красная линия) и в результате последующего охлаждения до 77 К (сплошная красная линия) [45].

Прочитав нашу статью [35], профессор Роберт Лью (Robert Liu), предложивший новый механизм HT цис-транс-фотоизомеризации, понял, что детальный анализ неравновесного состава конформеров может быть прямым экспериментальным подтверждением того или иного механизма цис-транс-фотоизомеризации в стеклообразных растворах. Это следует из того, что конформационная структура транс-изомера, получающаяся в результате цис-транс-фотоизомеризации, зависит от того, по какому механизму, OBF или HT, происходит переход из цис-изомера в транс-изомер. Это наглядно продемонстрировано на примере фенил-(2-нафтил)этилена (рис. 7).
Рис. 7.
Схема цис-транс-фотоизомеризации ДАЭ: если изомеризация идет по механизму HT, то получается транс-изомер в конформации 1, а если по механизму OBF – то транс- изомер в конформации 2.

Последовавший за этим большой цикл работ убедительно показал, что механизм НТ действительно реализуется для различных производных этилена и дает новую основу для понимания механизма фоторецепции, а также и ряда других биохимических процессов [45–50].
7. ФОТОИЗОМЕРИЗАЦИЯ В ЭТАНОЛЬНЫХ СТЕКЛАХ В ИНТЕРВАЛЕ ТЕМПЕРАТУР 4.2–100 К
Отметим еще один немаловажный аспект исследований цис-транс-фотоизомеризации в стеклообразных растворах, состоящий в том, что эту реакцию можно использовать как тест на структурную неоднородность стеклообразного состояния вещества. Описанные выше исследования проводились в этанольных стеклах при 77 К, что примерно соответствует температуре стеклования этих оптически прозрачных матриц. А что же произойдет, если температура твердого раствора будет существенно ниже точки стеклования? В работе [51] мы провели исследования цис-транс-фотоизомеризации в этанольных стеклах в интервале температур 4.2–100 К.
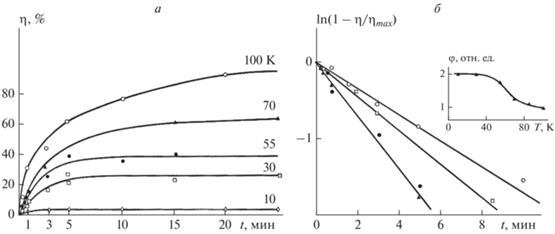
Оказалось, что с понижением температуры уменьшается максимальный выход транс-изомеров ηmax (рис. 8а). Это говорит о том, что при снижении температуры уменьшается доля молекул, способных к фотоизомеризации, которая определяется наличием в стекле микрогетерогенных областей с достаточно высокой молекулярной подвижностью, в то время как квантовый выход фотоизомеризации в расчете на долю изомеризующихся молекул даже увеличивается с понижением температуры (рис. 8б). Заметим, что при 4.2 К наблюдается заметная глубина превращения с ηmax = 3%. Сам факт наблюдения в твердой фазе при столь низких температурах фотоизомеризации свидетельствует о сохранении в стеклообразных растворах “жидкоподобных” областей, так что уменьшение ηmax при понижении температуры раствора обусловлено уменьшением их объемной доли.
8. ЦИС-ТРАНС-ФОТОИЗОМЕРИЗАЦИЯ В КРИСТАЛЛИЧЕСКОМ СОСТОЯНИИ
В 1978 году в работе [52] нами было показано, что в кристаллическом и аморфном состояниях диарилэтиленов, так же как и в стеклообразных растворах, имеет место реакция цис-транс-фотоизомеризации. При этом обратная реакция не идет, а в аморфном состоянии наблюдается и фотоинициированный фазовый переход – кристаллизация переохлажденного аморфного состояния диарилэтилена [53–58].
Механизм цис-транс-фотоизомеризации в монокристаллах диарилэтиленов был установлен нами в результате проведения комплексного кристаллохимического исследования и люминесцентного анализа [43]. Оказалось, что в монокристаллах размером 2–3 мкм цис-транс-фотоизомеризация идет без разрушения кристаллической решетки вещества.
Можно предположить, что, по-видимому, реакция происходит непосредственно в узлах кристаллической решетки, но при этом, как показали рентгеноструктурные исследования, конформационная структура цис- и транс-изомеров соответствует механизму OBF (рис. 9). Довольно очевидно, что для вращения нафтильного фрагмента явно не хватает свободного пространства в достаточно плотно упакованной кристаллической решетке.
Рис. 9.
Кристаллическая структура монокристаллов цис-изомера (слева) и транс-изомера (справа) 1,2-ди(1-нафтил)этилена.

Предложенный нами механизм реакции включает две стадии (рис. 10): первая стадия – это поглощение кванта света цис-изомером и образование нестабильного циклобутанового димера в электронно-возбужденном состоянии, а вторая – вращение нафтильного фрагмента молекулы в возбужденном димерном комплексе с образованием транс-изомера и затем дезактивация возбужденного состояния димера с образованием транс-изомера в основном электронном состоянии.

9. ЛЮМИНЕСЦЕНЦИЯ И КИНЕТИКА ФОТОИЗОМЕРИЗАЦИИ В КРИСТАЛЛИЧЕСКОМ СОСТОЯНИИ
Достаточно необычным представляется кинетический аспект протекания цис-транс-фотоизомеризации в кристаллическом и аморфном состояниях, а именно в изменении во времени люминесценции и концентрации транс-изомера (рис. 11). Он состоит в том, что кинетические зависимости изменения люминесценции и концентрации транс-изомера в твердой фазе существенно разные. В нашей работе [59] было показано, что при облучении светом в кристалле происходит не только цис-транс-фотоизомеризация, но и миграция электронного возбуждения по узлам кристаллической решетки.
Рис. 11.
Кинетика изменения фотолюминесценции (сплошная кривая) и концентрации транс-изомера в процессе облучения цис-изомера в твердом состоянии.

Решение этой достаточно сложной кинетической задачи приводит к следующей зависимости интенсивности люминесценции F(η) от глубины превращения η – относительной доли молекул транс-изомера в твердой фазе:
где J – скорость поглощения света кристаллом в расчете на одну молекулу, γ – квантовый выход цис-транс-фотоизомеризации отдельно взятой молекулы цис-изомера, α и β – вероятности внутримолекулярной дезактивации возбужденного состояния находящихся в кристаллической решетке молекул цис- и транс-изомеров соответственно. Величины (1 – α – γ) и (1 – β) определяют вероятности передачи возбуждения на соседний узел кристаллической решетки от молекул цис- и транс-изомеров соответственно. Таким образом, цис-транс-фотоизомеризация в кристалле происходит не только в результате поглощения кванта света непосредственно молекулой цис-изомера, но и в результате переноса энергии электронного возбуждения с соседних молекул цис- или транс-изомеров. Именно этим обстоятельством определяется необычное изменении во времени люминесценции и концентрации транс-изомера.ЗАКЛЮЧЕНИЕ
Подводя итог данной обзорной работы, следует отметить, что люминесцентный анализ является достаточно простым, удобным и эффективным методом для изучения кинетики химических и особенно фотохимических превращений в конденсированной фазе. Люминесцентный метод позволяет регистрировать кинетику химических реакций в масштабе реального времени, а также с достаточно высоким временны́м разрешением, предельное значение которого определяется характерным временем электронных возбужденных состояний, т.е. вплоть до 10–8–10–9 с. Последнего вопроса мы практически не касались в данном обзоре, потому что это вообще отдельная и достаточно обширная тема, хотя время жизни люминесценции является очень важной характеристикой с точки зрения химического анализа.
Применение современных математических методов обработки спектральных данных, включая совместную обработку спектров поглощения и люминесценции на основе метода матрицы “возбуждение–люминесценция”, дало существенное продвижение и развитие новых направлений люминесцентного анализа за последние несколько десятилетий.
Существенно новые перспективы в развитии люминесцентного анализа появились буквально в последние несколько лет в связи с открытием универсального соотношения между спектрами люминесценции и возбуждения люминесценции, что позволяет симметризовать матрицу “возбуждения–люминесценция” и тем самым реализовать чрезвычайно эффективные методы разложения матрицы “возбуждения–люминесценция” многокомпонентных систем на ее составляющие, т.е. фактически найден эффективный способ решения основной задачи люминесцентного спектрального анализа. Однако в этом направлении предстоит еще много поработать, сейчас у нас есть пока только общее понимание того, куда двигаться дальше, а разработка новых алгоритмов и экспериментальная проверка их эффективности требуют немалых усилий.
В заключение я хотел бы выразить глубокую благодарность все тем, с кем мне посчастливилось вместе работать на протяжении этих 50 лет и, конечно, в первую очередь – моему научному руководителю академику Михаилу Владимировичу Алфимову, а также сотрудникам моей лаборатории, которые в разное время работали и внесли неоценимый вклад в те исследования, о которых было упомянуто в этом обзоре, в том числе Бричкину Сергею Борисовичу, Гадомской Анне Владимировне, Иванченко Антонине Григорьевне, Кварацхелии Татьяне Георгиевне, Казакову Сергею Петровичу, Мартьяновой Елене Геннадьевне, Попову Леониду Сергеевичу, Рачинскому Александру Геннадьевичу, Спирину Максиму Геннадьевичу, Товстуну Сергею Александровичу, Шевченко Геннадию Александровичу, а также моим коллегам из других лабораторий: Алдошину Сергею Михайловичу, Галашину Анатолию Евгеньевичу, Кузьмину Владимиру Александровичу, Рябенко Александру Григорьевичу, Филиппову Павлу Геннадьевичу и многим другим, с которыми мне так или иначе довелось общаться по работе за эти годы.
Автор благодарит С.А. Товстуна за помощь в оформлении обзора и полезную дискуссию при обсуждении его содержания.
Работа выполнена по теме госзадания № АААА-А19-119070790003-7.
Список литературы
Кондон Э.Ю. // Успехи физ. наук. 1948. Т. 35. С. 35.
Landau L. // J. Phys. (Moscow). 1946. V. 10. P. 503.
Чукова Ю.П. // Письма в ЖЭТФ. 1969. Т. 10. С. 458.
Степанов Б.И. // Успехи физ. наук. 1956. Т. 58. С. 3.
Левшин В.Л. // ЖФХ. 1931. Т. 2. С. 641.
Блохинцев Д.И. // ЖЭТФ. 1939. Т. 9. С. 459.
Степанов Б.И. // Докл. Акад. наук СССР. 1957. Т. 112. С. 839.
Kirchhoff G. // Annalen der Physik. 1860. V. 185. P. 275.
Ketskeméty I., Dombi J., Horvai R. // Acta Phys. Acad. Sci. Hungaricae. 1960. V. 12. P. 263.
Левшин В.Л. // ЖПС. 1967. Т. 7. № 4. С. 466.
van Roosbroeck W., Shockley W. // Phys. Rev. 1954. V. 94. P. 1558.
Ihara T., Maruyama S., Yoshita M. et al. // Phys. Rev. B. 2009. V. 80. 033307.
Temkin H., Keramidas V.G., Pollack M.A., Wagner W.R. // J. Appl. Phys. 1981. V. 52. P. 1574.
Muth J.F., Lee J.H., Shmagin I.K. et al. // Appl. Phys. Lett. 1997. V. 71. P. 2572.
Ullrich B., Munshi S.R., Brown G.J. // Semicond. Sci. Technol. 2007. V. 22. P. 1174.
Мазуренко Ю.Т., Непорент Б.С. // Оптика и спектроскопия. 1962. Т. 12. № 5. С. 571.
Kennard E.H. // Phys. Rev. 1918. V. 11. P. 29.
Kennard E.H. // Phys. Rev. 1926. V. 28. P. 672.
Tovstun S.A., Razumov V.F., Spirin M.G., Martyanova E.G., Brichkin S.B. // J. Lumin. 2017. V. 190. P. 436.
Разумов В.Ф. // Изв. РАН. Сер. физ. 2020. Т. 84. № 5. С. 654.
Разумов В.Ф., Товстун С.А., Кузьмин В.А. // Письма в ЖЭТФ. 2019. Т. 110. С. 307.
Разумов В.Ф., Казаков С.П. // Изв. АН. Сер. хим. 1998. С. 2366.
Разумов В.Ф., Казаков С.П., Рябенко А.А. // Оптика и спектроскопия. 1999. Т. 86. № 4. С. 604.
Казаков С.П., Разумов В.Ф., Алфимов М.В. // Химия высоких энергий. 2004. Т. 38. С. 283.
G. Weber // Nature. 1961. V. 190. P. 27.
Warner I.M., Christian G.D., Davidson E.R., Callis J.B. // Anal. Chem. 1977. V. 49. P. 564.
JiJi R.D., Andersson G.G., Booksh K.S. // J. Chemometrics. 2000. V. 14. P. 171.
Andersen C.M., Bro R. // J. Chemometrics. 2003. V. 17. P. 200.
Маслов Н.А., Папаева Е.О. // Письма в ЖТФ. 2016. № 14. С. 7.
Lee D.D., Seung H.S. // Nature. 1999. V. 401. P. 788.
Sheck Yu.B., Kovalenko N.P., Alfimov M.V. // J. Lumin. 1977. V. 15. P. 157.
Разумов В.Ф., Алфимов М.В., Шевченко Г.А., Коваленко Н.П. // Докл. АН СССР. 1978. Т. 238. № 4. С. 885.
Mazzucato U., Momicchioli F. // Chem. Rev. 1991. V. 91. P. 1679.
Разумов В.Ф., Алфимов М.В. // Журн. науч. и прикл. фотографии. 2003. Т. 48. № 6. С. 28.
Alfimov M.V., Razumov V.F., Rachinsky A.G., Listvan V.N., Scheck Yu.B. // Chem. Phys. Lett. 1983. V. 101. P. 593.
Sun Ya.-P., Sears D.F., Saltiel J. et al. // J. Amer. Chem. Soc. 1988. V. 110. P. 6974.
Bartocci G., Spalletti A. // J. Phys. Chem. A. 2002. V. 106. P. 7068.
Гадомская А.В., Разумов В.Ф., Товстун С.А. // Химия высоких энергий. 2018. Т. 52. С. 267.
Wang Q., Schoenlein R.W., Peteanu L.A., Mathies R.A., Shank C.V. // Science. 1994. V. 266. P. 422.
Warchel A. // Nature. 1976. V. 260. P. 679.
Liu. R.S.H., Asato A.E. // Proc. Natl. Acad. Sci. USA. 1985. V. 82. P. 259.
Liu R.S.H., Brown D.T. // Acc. Chem. Res. 1986. V. 19. P. 42.
Aldoshin S.M., Alfimov M.V., Atovmyan L.O. et al. // Mol. Cryst. Liq. Cryst. 1984. V. 108. P. 1.
Castel N., Fisher E. // J. Mol. Struct. 1985. V. 127. P. 159.
Yang L.-Yi., Liu R.S.H. // Photochem. Photobiol. 2007. V. 83. P. 1436.
Müller A.M., Lochbrunner S., Schmid W.E., Fuß W. // Angew. Chem. Intern. Ed. 1998. V. 37. P. 505.
Liu R.S.H., Hammond G.S. // Proc. Natl. Acad. Sci. USA. 2000. V. 97. P. 11153.
Liu R.S.H. // Acc. Chem. Res. 2001. V. 34. P. 555.
Krishnamoorthy G., Asato A.E., Liu R.S.H. // Chem. Commun. 2003. P. 2170.
Imamoto Ya., Kuroda T., Kataoka M. et al. // Angewandte Chemie, International Edition. 2003. V. 42. P. 3630.
Филлипов П.Г., Разумов В.Ф., Рачинский А.Г., Алфимов М.В. // Докл. АН СССР. 1987. Т. 295. С. 434.
Alfimov M.V., Razumov V.F. // Mol. Cryst. Liq. Cryst. 1978. V. 49. P. 95.
Алфимов М.В., Разумов В.Ф. // Химия высоких энергий. 1979. Т. 13. С. 561.
Кварацхелия Т.Г., Разумов В.Ф., Попов Л.С., Алфимов М.В. // Докл. АН СССР. 1981. Т. 259. С. 1389.
Alfimov M.V., Razumov V.F. // J. Photographic Sci. 1983. V. 31. P. 217.
Алфимов М.В., Аптекарь И.Л., Галашин А.Е. и др. // Докл. АН СССР. 1984. Т. 276. № 2. С. 360.
Алфимов М.В., Галашин А.Е., Кварацхелия Т.Г., Разумов В.Ф. // Химия высоких энергий. 1986. Т. 20. С. 378.
Разумов В.Ф., Кварацхелия Т.Г., Алфимов М.В. // ЖФХ. 1987. Т. 61. С. 1821.
Разумов В.Ф., Иванченко А.Г., Рачинский А.Г., Алфимов М.В. // Докл. АН СССР. 1987. Т. 293. С. 666.
Дополнительные материалы отсутствуют.
Инструменты
Химическая физика