

Химия твердого топлива, 2022, № 1, стр. 28-35
КАТАЛИТИЧЕСКИЙ ГИДРОКРЕКИНГ –C–O-МОСТИКОВЫХ СВЯЗЕЙ НА КАТАЛИЗАТОРЕ Mg2Si/γ-Al2O3
Жуйю Ванг 1, Бингкан Сунь 2, Цзилян Чжу 2, Син Фань 2, *
1 Институт низкоуглеродной энергетики, Китайский горно-технологический университет
221006 Сюйчжоу, Цзянсу, Китай
2 Головная лаборатория по переработке и эффективному использованию угля, Министерство образования, Китайский горно-технологический университет
221116 Сюйчжоу, Цзянсу, Китай
* E-mail: fanxing@cumt.edu.cn
Поступила в редакцию 15.01.2021
После доработки 22.09.2021
Принята к публикации 13.10.2021
- EDN: WXOSNU
- DOI: 10.31857/S0023117722010091
Аннотация
Методом золь-гель-синтеза получен носитель нано γ-Al2O3. Нанесением на носитель Mg2Si приготовлен катализатор Mg2Si/γ-Al2O3, который охарактеризован методами рентгеновской дифракции, адсорбции-десорбции N2, сканирующей электронной микроскопии и программированной десорбции CO2. Изучена каталитическая активность Mg2Si/γ-Al2O3 в реакции гидрокрекинга 1-метоксинафталина (1-MON) и дибензилового эфира (BE), моделирующих эфирные мостиковые связи в буром угле. Установлено равномерное распределение Mg2Si на поверхности и в порах γ-Al2O3 носителя. Катализатор Mg2Si/γ-Al2O3 имел высокую удельную поверхность, развитую мезопористость, проявлял сильные основные свойства и высокую каталитическую активность и селективность при гидрокрекинге 1-метоксинафталина (1-MON) и дибензилового эфира (BE) вследствие синергетического взаимодействия компонентов Mg2Si и γ-Al2O3. При начальном давлении водорода 3 МПа и времени реакции 60 мин величины конверсии 1-MON и BE достигали 100% при температурах, соответственно, 225 и 175°C. Селективность по нафталину и толуолу составляла почти 100%, продукты гидрирования присутствовали в следовых количествах. После 4-кратного цикла испытаний конверсия BE на катализаторе Mg2Si/γ-Al2O3 снижалась до 50%.
1. ВВЕДЕНИЕ
Прямое ожижение угля (DCL) – перспективный процесс для производства экологически безопасного жидкого топлива и химических веществ с высокой добавленной стоимостью [1, 2]. В Китае на бурый уголь приходится более 13% угольных ресурсов, он считается подходящим сырьем для процесса ожижения. В связи с уменьшением запасов ископаемых топлив и ростом спроса на энергию создание новых более эффективных процессов ожижения бурого угля привлекает большое внимание исследователей [3].
Катализаторы играют важную роль в DCL, они увеличивают скорость реакции гидрирования, изменяя путь расщепления молекулы водорода и снижая энергию активации реакций с его участием [4–6]. Широко изучены свойства катализаторов на основе Fe, Ni, Mo, Co, других металлов, а также кислотных катализаторов SnCl2 и ZnCl2. В основном исследования были направлены на увеличение дисперсности предшественников катализаторов [7–9], ингибирование агломерации активной фазы [10, 11] и установление синергетических эффектов полиметаллических/неметаллических катализаторов [12, 13]. Однако невысокая активность катализаторов, трудности их рецикла, а также высокая коррозионная активность ограничивают возможности их промышленного применения.
Органическая масса бурого угля содержит большое количество кислородсодержащих групп, значительная их часть представлена мостиковыми –C–O-связями между различными углеводородными фрагментами [14]. Эффективное расщепление этих мостиков является важным этапом превращения бурого угля в жидкие вещества. Было доказано, что щелочи, используемые в качестве катализатора в процессе DCL, проявляют высокую активность при расщеплении эфирных и сложноэфирных связей в макромолекулярной структуре бурого угля [15, 16], что в результате способствует увеличению выхода растворимых веществ [17–19]. В работе [20] бурый уголь Xilinhaote подвергали сверхкритическому метанолизу в присутствии щелочи в качестве катализатора, полученные растворимые продукты анализировали методом ГХ/МС. Результаты показали, что в условиях сверхкритического метанолиза происходит разрыв мостиковых эфирных связей, что приводит к увеличению растворимости в органическом растворителе. Авторы работы [21] изучали сверхкритический метанолиз бурого угля месторождения Шенгли в присутствии NaOH. Было показано, что величина конверсии и выход продуктов зависят от массового соотношения NaOH и угля. При температуре 300°C конверсия угля и выход продуктов достигали максимума при равном массовом соотношении NaOH и угля. Применение твердого CaO в качестве основного катализатора позволяло уменьшить давление водорода в реакторе и увеличить содержание растворимых веществ в продукте DCL [22, 23].
Твердые суперосновные катализаторы, к которым относят вещества с основностью (H) ≥ 26.0 проявляют высокую каталитическую активность в реакциях изомеризации, трансэтерификации, присоединения олефинов по Михаэлю, дегидрогенизации и конденсации спиртов [24]. По сравнению с жидкими твердые супероснования экологически безопасны и легко отделяются от продуктов реакции. Вследствие этого твердые гетерогенные основные катализаторы имеют хорошие перспективы применения. Силицид магния Mg2Si, являясь основанием Льюиса, может активировать молекулярный водород с образованием подвижного гидрид-иона Н–, способного расщеплять –C–O– мостиковые связи [25]. Было доказано [26], что бифункциональный катализатор Ni–Mg2Si/γ-Al2O3 проявляет высокую активность в гидрокрекинге и гидрировании экстракта и экстракционного остатка из бурого угля Yinggemajianfeng.
В настоящей работе методом золь-гель-синтеза получен наноноситель γ-Al2O3. Пропиткой носителя приготовлен катализатор Mg2Si/γ-Al2O3. Каталитические свойства приготовленного катализатора изучены в реакции гидрогенизационного расщепления –C–O-связей в дибензиловом эфире (BE) и в 1-метоксинафталине (1-MON), которые моделировали мостиковые связи в структуре органической массы бурого угля. Рассмотрены возможные маршруты реакции расщепления связей в 1-MON и BE.
2. ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
2.1. Материалы. Изопропилат алюминия был приобретен у компании Sinopharm Chemical Reagent Co., Ltd, (Шанхай, Китай), силицид магния – у Jinzhou Haixin Metal Materials Co., Ltd (Ляонин, Китай), модельные соединения 1-MON и BE – у Aladdin Industrial Inc. (Шанхай, Китай). Растворители толуол, изопропиловый спирт, четыреххлористый углерод и циклогексан, используемые в экспериментах, являются коммерчески приобретенными реагентами, перед применением очищены перегонкой с использованием роторного испарителя Büchi R-210.
2.2. Приготовление катализатора. Изопропилат алюминия массой 20.4 г растворяли в 200 мл толуола с использованием ультразвукового диспергатора, и полученный раствор обозначили как раствор I; 7.0 мл изопропилового спирта смешивали с 5.4 мл H2O, и раствор обозначили как раствор ΙΙ. К раствору II при перемешивании добавляли по каплям раствор Ι. Реакционную смесь перемешивали в течение 5 ч при 80°C, отфильтровывали, осадок на фильтре высушили в вакууме при 60°C в течение 12 ч, затем прокаливали в муфельной печи при 550°C в течение 3 ч, чтобы получить носитель γ-Al2O3.
Для приготовления катализатора Mg2Si/γ-Al2O3 в круглодонную колбу добавляли 5.0 г полученного γ-Al2O3, 0.5 г Mg2Si и 150 мл CCl4. Смесь встряхивали в ультразвуковом аппарате в течение 30 мин, затем кипятили с обратным холодильником при 90°C в течение 8 ч. После охлаждения до комнатной температуры, отфильтровывания осадка и сушки при 100°C в течение 4 ч получали катализатор Mg2Si/γ-Al2O3.
2.3. Характеристика катализатора. Дифрактограммы снимали на дифрактометре Bruker D8 Advance с излучением CuKα = 1.5406 Å при 30 кВ и 20 мА в диапазоне 2θ от 3° до 80°. Изотермы адсорбции-десорбции N2 получали с использованием объемного адсорбционного анализатора BELL Belsorp-Max при –196°C. Образцы предварительно дегазировали при 200°C в течение 5 ч. Общую удельную поверхность (ABET), удельную поверхность микропор (Vmicr), распределение мезопор (DBJH) и микропор по размерам (DHK) рассчитывали из изотерм с использованием методов BET, t-plot, BJH и HK соответственно.
Морфологию и химический состав катализатора исследовали с помощью сканирующего электронного микроскопа (SEM) (FEI, Quanta TM 250), оборудованного энергодисперсионным спектрометром (EDS) (Bruker, Quantax 400-10).
Основность катализатора определяли методом термопрограмированной десорбции CO2 на приборе TP-5080 с детектором по теплопроводности. Образец (навеска 50 мг) предварительно обрабатывали при 400°C в течение 0.5 ч. После охлаждения до 50°C подавали СО2 в течение 0.5 ч, после чего подавали азот в течение 1 ч для удаления физически адсорбированного СО2. Десорбцию СО2 осуществляли в температурном интервале 50–900°C в потоке азота (30 мл/мин).
2.4. Каталитическая реакция. В автоклав загружали 2.0 г BE (или 1.5 г 1-MON), 0.1 г Mg2Si/γ-Al2O3 и 20 мл циклогексана, после чего сосуд продували водородом и затем повышали давление H2 (до 1–4 МПа). Автоклав быстро нагревали до требуемой температуры и выдерживали при этой температуре в течение установленного периода времени. После окончания реакции и охлаждения до комнатной температуры реакционную смесь извлекали из реактора и фильтровали. Фильтрат анализировали на газовом хромато-масс-спектрометре Agilent 7890/5975 (ГХ/МС), оборудованном капиллярной колонкой (60 м × 0.25 мм × 0.25 мкм, фаза HP-5MS) и квадрупольным анализатором, работающим в режиме электронного удара (70 эВ). Капиллярную колонку нагревали от 60 до 300°C со скоростью 10°C/мин, затем выдерживали при 300°C в течение 6 мин.
3. РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
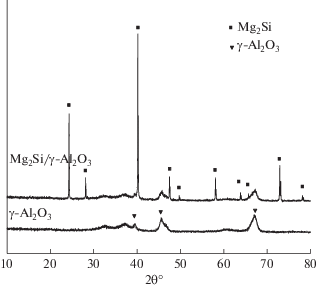
3.1. Рентгенофазовый анализ. На рис. 1 показаны дифрактограммы носителя γ-Al2O3 и катализатора Mg2Si/γ-Al2O3 в диапазоне 2θ от 10° до 80°. Пики при 2θ , равном 39.6°, 45.8°, 67.0°, относятся к кубической γ-Al2O3, а пики при 24.2°, 28.1°, 40.2°, 47.5°, 49.7°, 58.0°, 63.8°, 65.7°, 72.9°, 78.0° – к кристаллическим частицам Mg2Si. Дифракционные пики для Mg2Si узкие и интенсивные, что подтверждает эффективность нанесения на носитель γ-Al2O3.
3.2. Анализ текстурных характеристик. Изотермы адсорбции-десорбции N2 и распределение пор по размерам для носителя γ-Al2O3 и катализатора Mg2Si/γ-Al2O3 показаны на рис. 2, а, б. Оба образца, как γ-Al2O3 носитель, так и катализатор Mg2Si/γ-Al2O3, показывают типичную изотерму типа IV с петлей гистерезиса H1, которая, согласно классификации IUPAC, характерна для капиллярной конденсации в мезопорах. Это указывает на то, что носитель γ-Al2O3 и катализатор Mg2Si/γ-Al2O3 являются мезопористыми материалами. Основной объем порового пространства в носителе γ-Al2O3 и в катализаторе Mg2Si/γ-Al2O3 приходится на поры с размерами от 2.0 до 4.0 нм, средний диаметр пор в носителе γ-Al2O3 составляет около 2.5 нм – немного больше, чем в катализаторе Mg2Si/γ-Al2O3.
Рис. 2.
Изотермы адсорбции-десорбции азота (a) и распределение объема пор по размерам (б) в Mg2Si/γ-Al2O3 и носителе γ-Al2O3.
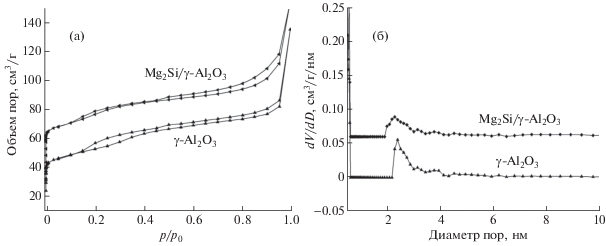
Текстурные характеристики, такие как удельная поверхность, объем пор, средний диаметр пор носителя γ-Al2O3 и катализатора Mg2Si/γ-Al2O3 приведены в табл. 1. Катализатор Mg2Si/γ-Al2O3, в сравнении с носителем, имеет несколько меньшую удельную поверхность, объем пор и диаметр пор, что обусловлено внедрением Mg2Si: удельная поверхность катализатора после нанесения Mg2Si уменьшалась от 350 до 315 м2/ г, объем пор от 0.34 до 0.20 см3/г, диаметр пор от 4.68 до 3.38 нм.
Таблица 1.
Параметры пористой структуры носителя γ-Al2O3 и катализатора Mg2Si/γ-Al2O3
| Образец | Удельная поверхность, м2/г | Объем пор, см3/г | Диаметр пор, нм |
|---|---|---|---|
| γ-Al2O3 | 350 | 0.34 | 4.68 |
| Mg2Si/γ-Al2O3 | 315 | 0.20 | 3.38 |
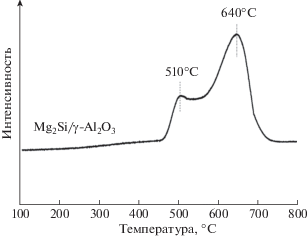
3.3. Термопрограммированная десорбция СО2для катализатора Mg2Si/γ-Al2O3. Основность катализатора Mg2Si/γ-Al2O3 изучена по данным термопрограмированной десорбции CO2, что показано на рис. 3. Катализатор Mg2Si/γ-Al2O3 имеет два десорбционных пика в диапазоне температур от 450 до 750°C, что указывает на присутствие сильных основных центров. Пики при 510 и 640°C можно отнести к десорбции СО2 с частиц Mg2Si прочно и слабо связанных с поверхностью γ-Al2O3 соответственно.
3.4. Результаты SEM и EDS. Микрофотографии, полученные на сканирующем электронном микроскопе, для носителя и катализатора Mg2Si/γ-Al2O3 показаны на рис. 4. По сравнению с носителем катализатор Mg2Si/γ-Al2O3 имеет шероховатую поверхность из-за нанесения силицида магния, который распределен на поверхности и в пустотах γ-Al2O3. Это согласуется с данными по распределению пор по размерам (рис. 2, б) и величине удельной поверхности и объема мезопор (табл. 1). В то время как индивидуальный носитель γ-Al2O3 характеризуется наличием мезопор с размерами от 2 до 4 нм, после нанесения Mg2Si наблюдается существенное уменьшение удельной поверхности, размера пор и объема пор.

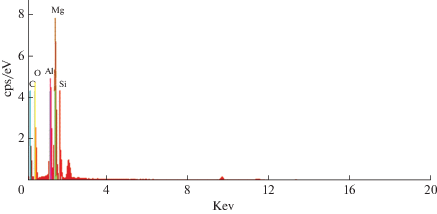
По данным энергодисперсионной спектроскопии (EDS), на катализаторе Mg2Si/γ-Al2O3 присутствуют углерод, кислород, алюминий, магний и кремний (рис. 5). Углерод можно отнести к проводящей углеродной пленке в процессе EDS, Al и O принадлежат носителю, а Mg и Si к нанесенному Mg2Si, примесей других элементов в катализаторе не обнаружено.
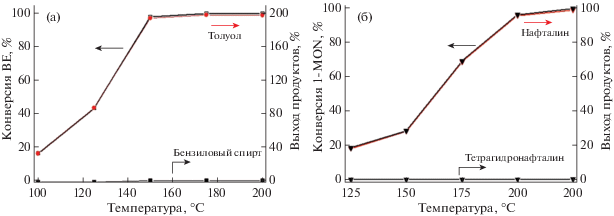
3.5. Каталитическая активность. Температура является наиболее важным фактором, определяющим активность катализатора. Было изучено влияние температуры реакции на величину конверсии BE и 1-MON на катализаторе Mg2Si/γ-Al2O3. Рисунок 6 показывает, что конверсия как BE, так и 1-MON увеличивается с повышением температуры. BE превращался почти полностью при достижении температуры 175°C. Толуол являлся основным продуктом, его выход составлял почти 200 мол. %, бензиловый спирт обнаруживался на уровне следов. Конверсия 1-MON достигала 100% при температуре 225°C, выходы нафталина и тетрагидронафталина составляли 99 и 1% соответственно. Таким образом, полученные результаты показывают, что катализатор Mg2Si/γ-Al2O3 проявляет высокую эффективность при гидрокрекинге 1-MON и BE, процесс протекает с селективным разрывом С–О-связей в обеих молекулах. Уникальные каталитические свойства обусловлены не только большой удельной поверхностью катализатора, но, возможно, и вследствие синергизма взаимодействия частиц Mg2Si с поверхностными центрами носителя γ-Al2O3.
Рис. 6.
Влияние температуры реакции гидрокрекинга BE (а) и 1-MON (б) на величину конверсии и выход продуктов на катализаторе Mg2Si/γ-Al2O3. Катализатор 100 мг, начальное давление водорода 3 МПа, 1 ч.
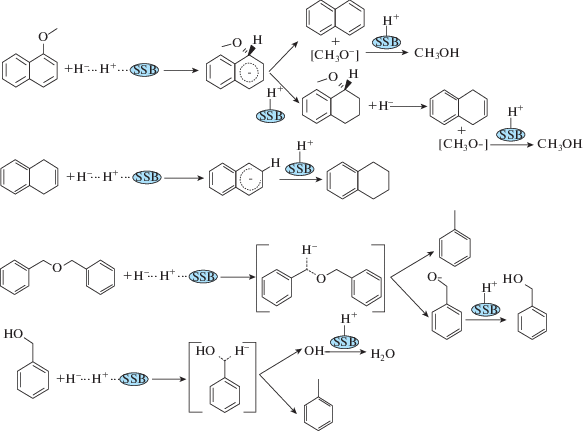
Как показано на рис. 7, в присутствии сильных основных центров Mg2Si молекулярный водород может подвергаться гетеролитической диссоциации с образованием активного подвижного гидрид-иона H– и неподвижного связанного с основным центром иона H+ [24]. При взаимодействии с молекулой 1-MON гидрид-ион H– атакует ипсо-положение нафталинового кольца, последующий разрыв связи C–O приводит к образованию нафталина и метанола. В случае BE гидрид-ион H– атакует мезо-положение бензильного атома углерода, при последующем расщеплении связи C–O образуются толуол и алкоголят-анион, который, отрывая Н+ от поверхностного центра катализатора, превращается в нейтральную молекулу бензилового спирта. Последний далее подвергается мезо-атаке гидрид-ионом Н– с расщеплением С–О-связи и образованием H2O и второй молекулы толуола.
Рис. 7.
Схема механизма гидрокрекинга двух соединений, моделирующих кислород-содержащие структурные мостики в буром угле. SSB - катализатор Mg2Si/γ-Al2O3.
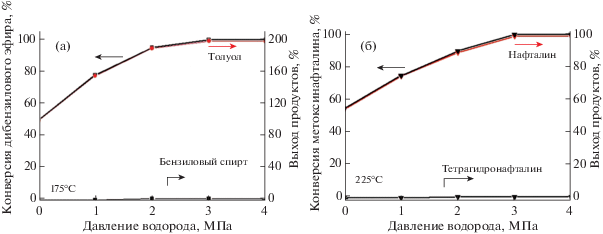
На рис. 8 видно, что конверсия BE и выход толуола значительно увеличиваются с повышением начального давления водорода и достигают максимума при 3 МПа. Выход бензилового спирта остается на низком уровне при изменении давления водорода от 0 до 4 МПа. Аналогичным образом влияло давление водорода на конверсию 1-MON и выход тетрагидронафталина. Эти результаты означают, что повышение давления водорода до 3 МПа значительно ускоряет гидрокрекинг BE до бензилового спирта, а 1-MON до тетрагидронафталина. Обе молекулы полностью превращаются при давлении водорода от 3 до 4 МПа.
Рис. 8.
Влияние начального давления водорода на показатели активности катализатора Mg2Si/γ-Al2O3 при гидрокрекинге BE (а) и 1-MON (б).
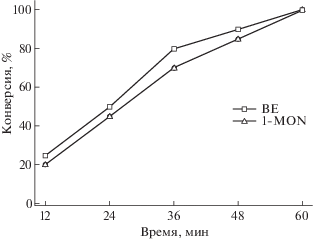
Было изучено также влияние времени реакции на превращение BE и 1-MON. Из рис. 9 следует, что величины конверсии BE и 1-MON монотонно увеличивались со временем реакции, через 1 ч достигалось 100%-ное превращение. По полученным результатам определены оптимальные условия реакции: начальное давление водорода 3 МПа, температура 175°C для BE и 225°C для 1-MON, продолжительность реакции 1 ч.
Суммируя результаты, отметим, что указанные условия гидрокрекинга 1-MON и BE на катализаторе Mg2Si/γ-Al2O3 мягче, чем опубликованные в работе [24]. В публикуемой работе использовался свежеприготовленный γ-Al2O3-носитель. Возможно синергетическое взаимодействие нанесенных частиц Mg2Si с активными центрами γ-Al2O3 носителя способствовало повышению каталитической активности катализатора Mg2Si/γ-Al2O3.
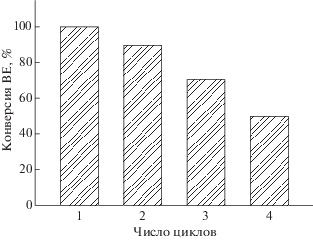
Были проведены опыты по рециклингу катализатора Mg2Si/γ-Al2O3, что показано на рис. 10. Конверсия BE уменьшалась с увеличением числа циклов. После четырехкратного каталитического рецикла активность катализатора снижалась до 50%, что, очевидно, связано с дезактивацией активных центров Mg2Si-катализатора.
ВЫВОДЫ
При гидрокрекинге на приготовленном катализаторе Mg2Si/γ-Al2O3 достигалось почти 100%-ное превращение BE до толуола (при 175°C) и 1-MON до нафталина (при 225°C), что свидетельствует об уникальной активности и селективности катализатора в отношении расщепления –CO-мостиковых связей в мягких условиях. Описан механизм гидрокрекинга на катализаторе Mg2Si/γ-Al2O3, согласно которому катализатор способствует образованию подвижного гидрид-иона Н- и его переносу в мезо-положение BE или ипсо-положение 1-MOM. Активность катализатора существенно снижалась после нескольких рециклов, что связано с дезактивацией активных центров Mg2Si. Катализатор Mg2Si/γ-Al2O3 представляет перспективное направление в создании новых эффективных катализаторов для процессов DCL.
Список литературы
Vasireddy S., Morreale B., Cugini A., Song C.S., and Spivey J.J. // Energy Environ. Sci. 2011. V. 4. P. 311.
Mochida I., Okuma O., and Yoon S.H. // Chem. Rev. 2013. V. 114. P. 1637.
Liu Z., Shi S., and Li Y. // Chem. Eng. Sci. 2010. V. 65. P. 12.
Luo H., Ling K., and Shen J. // Energ. Source Part A. 2014. V. 36. P. 949.
Priyanto U., Sakanishi K., Okuma O., and Mochida I. // Fuel Process. Technol. 2002. V. 79. P. 51.
Yue X.M., Wei X.Y., Sun B., Wang Y.H., Zong Z.M., and Liu Z.W. // Int. J. Min. Sci. Technol. 2012. V. 22. P. 251.
Kaneko T., Tazawa K., Okuyama N., Tamura M., and Shimasaki K. // Fuel. 2000. V. 79. P. 263.
Li Y.Z., Ma F.Y., Su X.T., Shi L.J., Pan B.B., Sun Z.Q., and Hou Y.L. // Ind. Eng. Chem. Res. 2014. V. 53. P. 6718.
Painter P.C., Cetiner R., Pulati N., Sobkowiak M., and Mathews J.P. // Energy Fuel. 2010. V. 24. P. 3086.
Zhu J.S., Yang J.L., Liu Z.Y., Dadyburjor D.B., Zhong B., and Li B.Q. // Fuel Process. Technol. 2001. V. 72. P. 199.
Lian Z., Yang J.L., Zhu J.S., Liu Z.Y., Li B.Q., Hu T.D., and Dong B.Z. // Fuel. 2002. V. 81. P. 951.
Li Y.Z., Ma F.Y., Su X.T., Sun C., Liu J.C., Sun Z.Q., and Hou Y.L. // Catal. Commun. 2012. V. 26. P. 231.
Trautmann M., Lang S., and Traa Y. // Fuel. 2015. V. 151. P. 102.
Zhou J., and Chen Y. // Energ. Source Part A. 2017. V. 39. P. 706.
Yu Y.R., Fan X., Zhao Y.P., Cao J.P., Kang S.G., and Wei X.Y. // Chin. J. Anal. Chem. 2017. V. 45. P. 1005.
Makabe M., Hirano Y., and Ouchi K. // Fuel. 1978. V. 57. P. 289.
Wang C.F., Fan X., Zhang F., Wang S.Z., Zhao Y.P., Zhao X.Y., Zhao W., Zhu T.G., Lu J.L., and Wei X.Y. // RSC Adv. 2017. V. 7. P. 20677.
Makabe M., and Ouchi K. // Fuel Process. Technol. 1981. V. 5. P. 129.
Chen J.W., Muchmore C.B., Lin T.C., and Tempelmeyer K.E. // Fuel Process. Technol. 1985. V. 11. P. 289.
Peng Y.L., Li Y., Zhou X., Xie R.L., Ma Y.M., Wei Y.B., and Wei X.Y. // Coal Engineering. 2009. V. 2. P. 88.
Lei Z.P., Liu M.X., Shui H.F., Wang Z.C., and Wei X.Y. // Fuel Process. Technol. 2010. V. 91. P. 783.
Lu H.Y., Wei X.Y., Sun B., Peng Y.L., Lv J., Xia T.C., Zong Z.M., Wang S.J., and Xu B. // J. Wuhan Univ. Technol. 2010. V. 33. P. 83.
Lei Z.P., Liu M.X., Gao L.J., Shui H.F., Wang Z.C., and Ren S.B. // Energy. 2011. V. 36. P. 3058.
Wei Y.D., Zhang S.G., Li G.S., Yin S.F., and Au C. // Chinese J. Catal. 2011. V. 32. P. 891.
Li W.T., Wei X.Y., Li X.K., Liu X.X., Li Z.K., and Zong Z.M. // Fuel. 2017. V. 200. P. 208.
Li X.K., Zong Z.M., Chen Y.F., Yang Z., Liu G.H., Liu F.J., Wei X.Y., Wang B.J., Ma F.Y., and Liu J.M. // Fuel. 2019. V. 249. P. 496.
Дополнительные материалы отсутствуют.
Инструменты
Химия твердого топлива