

Химия твердого топлива, 2022, № 3, стр. 3-12
ИЗМЕНЕНИЕ СВОЙСТВ НАТИВНОГО НИЗКОМЕТАМОРФИЗОВАННОГО УГЛЯ ПРИ КОНТАКТЕ С ВОЗДУХОМ
С. А. Семенова 1, *, Ю. Ф. Патраков 1, **, А. В. Яркова 1, ***, С. Ю. Лырщиков 2, ****, Н. С. Захаров 2, *****
1 Институт угля Федерального исследовательского центра Угля и углехимии СО РАН
650065 Кемерово, Россия
2 Центр коллективного пользования Федерального исследовательского центра Угля и углехимии СО РАН
650000 Кемерово, Россия
* E-mail: semlight@mail.ru
** E-mail: yupat52@gmail.com
*** E-mail: nas.yarkova1998@yandex.ru
**** E-mail: serstud@mail.ru
***** E-mail: 2metil4@gmail.com
Поступила в редакцию 08.12.2021
После доработки 17.01.2022
Принята к публикации 26.01.2022
- EDN: GDEDCJ
- DOI: 10.31857/S0023117722030082
Аннотация
Представлены данные по анализу химического состава и физико-химических свойств нативного низкометаморфизованного угля в начальный момент его контактировании с воздушной средой. Для идентификации изменений органического вещества угля использованы методы ИК-спектроскопии диффузного отражения, ЭПР-, ЯМР-спектроскопии и газовой хроматографии, химический анализ кислородсодержащих групп, а также определение удельной поверхности и смачиваемости контактирующей поверхности. Динамика изменения количества парамагнитных центров и функциональных групп показывает, что наиболее интенсивные преобразования в поверхностном слое происходят в течение первых суток нахождения угля в воздушной атмосфере. Далее окисление при комнатной температуре протекает в периодическом режиме накопления и расходования радикалов и функциональных О-групп. По достижению 4 сут процесс низкотемпературного окисления с доступной внешней поверхности переходит в диффузионную область пористого пространства угля и постепенно замедляется.
При добыче угля уже на стадии обнажения поверхности пласта запускается процесс окисления, приводящий к изменению физико-химических свойств приграничной зоны, а именно химического состава поверхностных функциональных групп, смачиваемости, пористости, трещиноватости и т.д. Интенсивное развитие радикальных окислительных реакций без возможности отвода тепла может привести к самовозгоранию угля как на стадии добычи, так и при его транспортировке, хранении и первичной переработке. Наибольшая доля возгораний среди каменных углей относится к наименее метаморфизованным длиннопламенным углям, обладающим наиболее развитой поверхностью пор.
Проблемам самовозгорания, выяснения механизмов окисления различными формами кислорода, изменению состава и свойств углей при окислительном воздействии посвящены многочисленые работы советских, российских и зарубежных ученых, среди которых наиболее известны труды Б.Ф. Мефферта [1], Г.Л. Стадникова [2], В.С. Крыма [3], Б.В. Тронова [4], Н.М. Караваева [5], И.И. Аммосова и И.В. Еремина [6], В.С. Веселовского [7], А.И. Хрисанфовой [8], Т.А. Кухаренко [9], В.И. Саранчука [10], В.А. Проскурякова [11], И.В. Александрова и А.И. Камневой [12], Л.Ф. Бутузовой [13], М.Л. Улановского [14], Д.В. Мирошниченко [15]. Полученные знания обобщены в многочисленных обзорах методов оценки окисления и самовозгорания углей [14, 16–22].
Рассматривая множество предложенных теорий, в общем виде механизм окисления формулируется следующим образом. На первом этапе молекулы кислорода адсорбируются на активных участках поверхности угля с образованием уголь-кислородных комплексов, которые при дальнейшем окислении переходят в пероксидные и гидропероксидные соединения. Ввиду неустойчивости эти соединения распадаются с образованием радикалов, газообразных продуктов (Н2О, СО2, СО) и кислородсодержащих функциональных групп. Процесс окисления имеет радикально-цепной характер, количество кислородсодержащих групп (гидроксильных, карбоксильных, карбонильных) периодично изменяется [10, 13]. При определенных условиях (влияние температуры, влажности, фракционного состава, продолжительности контакта и др.) течение процесса может ускоряться и распространяться с поверхности в глубь частицы или слоя угля.
Отследить и оценить эти процессы в шахте или в условиях карьерной выработки весьма проблематично. Для ускорения медленных гетерогенных окислительных реакций в лабораторных условиях зачастую используют термическое стимулирование окисления угольного порошка при температурах 70–250оС [10, 13]. Вместе с тем с увеличением скорости процесса теряется информация о первичных превращениях в составе органической массы угля (ОМУ) при контакте с кислородом. На кинетических кривых отсутствует индукционный период [13]. Имеющаяся информация об изменении технологических свойств углей при окислении (зольность, влажность, теплота сгорания, спекаемость) [10, 15], заложенная в основу технологических регламентов по хранению на складах, учитывает уже глубокие преобразования в объеме угольного вещества. При этом первые изменения в поверхностном слое угольных частиц (или слоев) не оказывают ощутимого влияния на интегральные технологические характеристики угля.
Помимо этого, интерпретацию и изучение начальных стадий окисления осложняет отсутствие достоверных сведений о первичности исходной пробы, исключающей возможность предварительного контакта образцов с воздушной средой. Использование способов упаковки исследуемого материала в герметичные пакеты на месте отбора, опарафинивания, отделения в лаборатории контактировавшего слоя, измельчения, приготовления и хранения пробы с продувкой инертным газом не исключает вероятности контакта угля с воздухом на каких-либо этапах и впоследствии использования для кинетических исследований уже поверхностно-окисленного угля.
В ФИЦ УУХ СО РАН разработаны способы бесконтактного с воздухом отбора, доставки и разделки угольных проб, которые могут позволить в лабораторных условиях оценить изменения, которые происходят с ОМУ в первые моменты взаимодействия вскрытой активной угольной поверхности с кислородом.
Цель данной работы – оценить изменения в составе ОМУ при первичном контакте с воздушной средой, используя бесконтактные с воздухом методы отбора и разделки пробы.
МЕТОДИЧЕСКАЯ ЧАСТЬ
В качестве образца для исследования начальной стадии окисления использовался уголь марки Д (длиннопламенный) одного из перспективных месторождений республики Хакасия. Химико-технологическая характеристика угля приведена в табл. 1. Интерес к длиннопламенному углю данного месторождения республики Хакасия обусловлен его аномально высокой по сравнению с низкометаморфизованными каменными углями других бассейнов сорбционной способностью (SBET = 25–50 м2/г) (табл. 1), вследствие чего он отличается повышенной склонностью к самовозгоранию при отработке пластов, складировании и транспортировке.
Таблица 1.
Химико-технологические характеристики угля марки Д
| Петрографический анализ, % | Технический анализ, % | Элементный состав, % на daf | Удельная поверхность SBET, см2/г | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| показатель отраже-ния витринита Ro | витри-нит Vt | семивит-ринит Sv | инер-тинит I | влага W a | золь-ность Ad | выход летучих веществ Vdaf | C | H | O | N+ S | |
| 0.50 | 76 | 3 | 21 | 5.0 | 4.4 | 43.5 | 77.6 | 5.8 | 13.6 | 3.0 | 26.75 |
Пробу угля отбирали со свежеобнаженной поверхности угольного пласта в виде крупных кусков (150–200 мм). Образцы помещали в герметично закрывающийся, заполненный инертным газом и вакуумированный пластиковый контейнер. Таким образом обеспечивали минимальный контакт угля с атмосферным кислородом и исключали неконтролируемое окисление основной массы отобранной пробы. При доставке в лабораторию контейнер с углем через приемный шлюз помещали в заполненный инертным газом (азот особой чистоты ТУ 2114-003-05758954-2007) перчаточный бокс. Наличие кислорода в боксе и приемных шлюзах контролировали датчиком кислорода (газоанализатор ЭЛАН плюс, ЭКО-ИНТЕХ (Россия), точность измерения 0.005%). В герметичном перчаточном боксе в чистой инертной газовой среде проводили распаковку угля и все подготовительные операции для анализа нативного угля: откалывание внешней поверхности крупных кусков для удаления частично окисленного слоя угля, измельчение, рассев по классам крупности, взвешивание угольных проб, заполнение пробирок, ампул и кювет для последующих исследований, таблетирование угольного порошка и т.д. Внешнюю и избыточную влагу у нативного угля удаляли вакуумированием в инертной среде в приемном шлюзе перчаточного бокса.
Все методы анализа (если не указано дополнительно) выполнены с использованием мелкодисперсной фракции угля крупностью <0.1 мм.
Вскрытие ампул с пробами проводили в анализаторе спектрометра или непосредственно перед выполнением анализа. Предполагалось, что за короткий промежуток времени (не более 1 мин) пребывания угля на воздухе между вскрытием герметичной, заполненной азотом тары и началом анализа заполненные инертным газом поры и, соответственно, активные центры на поверхности угля еще находятся в неактивированном состоянии.
Кислородсодержащие (ОН+СООН)-группы анализировали рН-метрическим методом (рН-метр 150МИ, Россия) ионным обменом с гидроксидом натрия.
ИК-спектры регистрировали на Фурье-спектрометре Люмекс Инфралюм ФТ-08 с приставкой диффузного отражения PIKE Easydiff в области 400–4000 см–1. ИК-спектральный параметр Ко (показатель окисленности) определяли по отношению суммарной интенсивности (суммы интегральных оптических плотностей (D)) полос поглощения (п.п.) карбоксильных (1710 см–1) и фенольных + эфирных (1260 см–1) групп к сумме D СНх-алифатических (2920 см–1) и СН-ароматических (3040 см–1) групп:
Регистрацию спектров электронного парамагнитного резонанса (ЭПР) выполняли на ЭПР-спектрометре Bruker EMX micro 6/1 в прямоугольном резонаторе при комнатной температуре в атмосфере азота. Расчет числа парамагнитных центров (ПМЦ) проводили методом сравнения со стандартом (ионы Mn2+ в MgO). Основные настройки прибора при регистрации спектра: развертка магнитного поля 100 Гс; частота микроволнового излучения ≈9.85 ГГц; мощность СВЧ-генератора 1.85 мВт; частота модуляции 100 кГц; амплитуда модуляции 1 Гс; константа времени 40.96 мс; время преобразования 15 мс; время сканирования 60 с. Регистрацию и анализ спектров проводили с использованием пакета программ WinEPR. ЭПР-спектры были подвергнуты интегрированному разложению в программе Origin 8.
13С ЯМР-спектры высокого разрешения в твердом теле регистрировали на приборе Bruker Avance III 300 W на частоте 75 МГц с применением стандартной методики кросс-поляризации и вращением под магическим углом (CPMAS). Для получения количественных данных проводилось моделирование спектров с использованием программы Dmfit. На спектрах выделяли диапазоны, соответствующие резонансному поглощению следующих групп углеродных атомов, м.д.: (187-171) – атомы углерода карбоксильных групп и их производных (СОО–); (171-148) – атомы углерода ароматических систем, связанные с атомом кислорода (СarО); (148-129) – атомы углерода конденсированных ароматических систем (Сar); (129-93) атомы углерода ароматических систем с незамещенным атомом водорода (СНar); (93-67) – атомы углерода алифатических структур, связанные с атомом кислорода (С–О–С); (67-51) – атомы углерода метильных групп, связанные с атомом кислорода (О–СН3); (51-25) – атомы углерода метиленовых фрагментов (СН2); (25-0) – атомы углерода метильных групп (СН3). На основании анализа спектров определены значения нормализованных интегральных интенсивностей основных типов углеродных структур [23]. Степень ароматичности углей рассчитывали по формуле: fa = (СarО + Сar + СНar)/100.
Для определения угла смачивания поверхности угля использовали способ приготовления образца прессованием порошкообразной пробы под давлением 700 МПа в брикет цилиндрической формы диаметром 10 мм и высотой 5 мм. Брикет закрепляли на предметном стекле, горизонтально выравнивали и через капилляр подводили к исследуемой поверхности каплю жидкости. Равновесную форму и краевой угол сцепления капли с поверхностью угля регистрировали с помощью микроскопа, снабженного видеокамерой [24]. Для обеспечения воспроизводимости результатов использовали не менее 5 образцов угля и многократное закрепление капли. Относительная ошибка определения для разных способов подготовки поверхности составила 5–10%. В качестве образцов сравнения использовали брикеты из нативного угля, полученные в инертной атмосфере: непосредственно после извлечения из бокса и выдержанные в течение определенного времени в контакте с воздухом.
Анализ газообразных продуктов окисления угля определяли хроматографически с использованием газового хроматографа “Хроматэк – Газохром 2000”. Пробу газа (1 см3) с определенной периодичностью отбирали из герметичных сосудов (100 мл), заполненных воздухом и содержащих одинаковую навеску подготовленного в инертной среде нативного угля (5 г) крупностью 1–3 мм. Газовую пробу вводили в кран-дозатор анализатора для разделения смеси на хроматографических колонках. Обработка спектров осуществлялась с использованием программы Хроматэк-Аналитик.
Анализ образцов всеми перечисленными методами анализа осуществлялся в течение первых 7 сут нахождения угля в воздушной среде при комнатной температуре и влажности, близким к стандартным условиям.
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Среди множества современных физико-химических методов анализа состава и структуры углей метод ЭПР-спектроскопии характеризуется широкими возможностями изучения различных типов взаимодействия на начальном этапе, в том числе низкотемпературного окисления углей. Анализируемыми параметрами являются ширина и форма линии ЭПР-спектра, средняя интенсивность сигнала, величина фактора спектроскопического расщепления (g-фактора), а также расчетное количество парамагнитных центров ПМЦ.
ЭПР-спектр нативного длиннопламенного угля имеет гладкий профиль, слагающийся из суперпозиции сигналов нескольких типов радикалов. Гладкая форма линии профиля свидетельствует о наличии стабильных радикалов в структуре угля [25]. Спектр имеет форму Лоренца, характеризуется g-фактором, равным ~2.0031, шириной линии ∆Н = 3.97 Гс и количеством ПМЦ N = 4.40 ⋅ 1019 спин/г (табл. 2). Значение g-фактора связано с окружением радикала неспаренными электронами и может быть использовано в качестве определения типа радикальных структур. Исходный ЭПР-спектр угля интегрируется на две лоренцевы составляющие, позволяющие судить о существовании не менее двух типов ПМЦ – с g = 2.0050 (электроны локализованы на атоме кислорода) и с g = 2.0029 (электроны локализованы на атоме углерода) [13, 25, 26]. Количество ПМЦ второго типа превышает число ПМЦ первого типа на порядок (рис. 1), что указывает на доминирующий вклад стабильных углеводородных радикалов в химический состав поверхности нативного низкометаморфизованного угля марки Д.
Таблица 2.
Результаты химического и ЭПР-спектрального анализа угля марки Д
| Продолжительность контакта с О2, сут | Количество (ОН + + СООН)-групп, мг-экв/г | Количество ПМЦ N, спин/г | g-фактор | Интенсивность пика, отн. ед. | Ширина линии, Гс |
|---|---|---|---|---|---|
| 0 | 0.45 | 4.40 · 1019 | 2.0031 | 6.647 · 105 | 3.97 |
| 1 | 0.35 | 7.14 · 1019 | 2.0031 | 1.373 · 106 | 4.17 |
| 4 | 0.25 | 7.25 · 1019 | 2.0030 | 1.223 · 106 | 4.34 |
| 7 | 0.37 | 5.92 · 1019 | 2.0031 | 1.376 · 106 | 4.18 |
Рис. 1.
Изменение количества ПМЦ (а) и g-фактора (б) первого (электроны, локализованные на атомах кислорода) (1) и второго (электроны локализованы на атомах углерода) (2) типов по данным ЭПР-спектроскопии угля марки Д при его низкотемпературном окислении в воздушной среде.
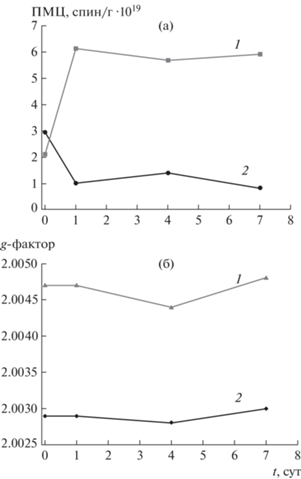
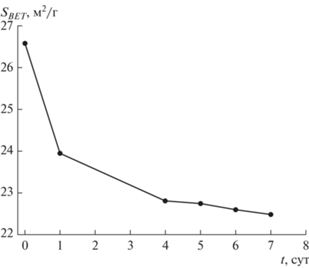
При попадании нативного угля в кислородсодержащую среду запускаются процессы сорбции кислорода угольной поверхностью и развитие радикально-цепных окислительных реакций. Это отражается в существенном снижении величины удельной поверхности, определяемой по сорбции азота (SBET) (рис. 2). По-видимому, этому способствуют блокировка физически адсорбированным кислородом адсорбционных центров на внутренних стенках пор, а также стерические препятствия, создаваемые газу-сорбату (азоту) продуктами хемосорбции – поверхностными кислородсодержащими группами. По данным ЭПР-спектроскопии, по истечении первых суток нахождения угля Д в воздушной среде наблюдается интенсивный рост числа ПМЦ, обусловленный более чем троекратным увеличением концентрации кислородсодержащих радикалов (g1) и двукратным превышением количества углеводородных радикалов (g2). Рост числа ПМЦ оказывает влияние на увеличение интенсивности и уширение линии ЭПР-спектра (рис. 1, табл. 2).
Рис. 2.
Изменение удельной поверхности (по азоту) угля марки Д при его низкотемпературном окислении в воздушной среде.
Увеличение количества ПМЦ, отвечающих СН- и СН2-радикалам (g2), на раннем этапе контактирования угля с воздушной средой, вероятно, связано с вовлечением углеводородных фрагментов ОМУ в реакции цепного окисления. По данным ИК-спектроскопии в первые сутки окисления фиксируется уменьшение относительного содержания СНn-алифатических (2920, 1380 см–1) и в меньшей степени СН-ароматических (3040, 820 см–1) групп (рис. 3). Стремительный рост кислородсодержащих радикалов (g1) может быть обусловлен активацией ОН- и С–О-групп, на что указывает снижение интенсивности поглощения фенольных (3400 см–1) и эфирных (1260 см–1) групп (рис. 3, табл. 2). Результатом окислительных преобразований на начальном этапе является накопление карбонильных (1650 см–1) и карбоксильных (1720 см–1) групп.
Рис. 3.
Изменение интенсивностей (I, отн. ед.) полос поглощения (см–1) углеводородных (а) и кислородсодержащих (б) фрагментов в ИК-спектрах угля марки Д при его окислении в воздушной среде.
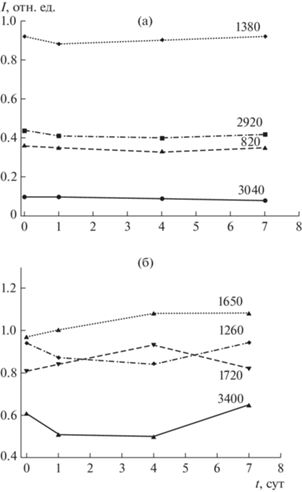
По достижении 4 сут скорость окисления угольной поверхности замедляется (рис. 1–3). Количество ПМЦ, соответствующих кислородсодержащим (g1) и углеводородным (g2) радикалам, изменяется незначительно (рис. 1, а), что может быть связано как с образованием новых радикалов алкильных и кислородных групп, так и с их вовлечением в реакции окисления. Изменение значений g-фактора указывает на то, что в процессе низкотемпературного окисления угля образовались новые радикалы, имеющие структуру, отличную от радикалов, первоначально существующих в угле. Снижение значений g-фактора с 2.0050 до 2.0047 (g1) и c 2.0029 до 2.0027 (g2) (рис. 1, б) может быть обусловлено уменьшением числа радикалов с локализацией электрона на атоме кислорода (например, феноксильных или алкилэфирных) и относительным увеличением количества стабильных углеводородных радикалов [25, 26]. Изменения химического состава поверхности угля на данном этапе связано, главным образом, с взаимопревращением периферийных функциональных групп (рис. 3).
По достижении 7 сут процесс стабилизируется (рис. 1–3). Концентрация ПМЦ, характеризующих электроны, локализованные на кислородных атомах (g1), держится около постоянного уровня; число ПМЦ, соответствующих углеводородным радикалам (g2), понижается (рис. 1, а). Это становится возможным благодаря одновременно образованию новых радикалов, их рекомбинации и гибели при формировании периферийных функциональных групп и газообразных продуктов, а также за счет перехода процесса с поверхности частиц в область пористого пространства угля (медленная диффузионная стадия). Снижение удельной поверхности (SBET), которое отмечалось на начальной стадии взаимодействия угля с кислородом воздуха, на этом этапе существенно замедляется (рис. 2).
Наиболее эффективным способом контроля развития процесса окисления является анализ изменений в составе газообразных продуктов взаимодействия угля с кислородом воздуха. Методом газовой хроматографии появление оксидов углерода в составе газовой среды регистрируется уже в первые сутки контакта угля с кислородом (табл. 3). По мере продолжительности окисления концентрация СО и СО2 возрастает, а наибольшая интенсивность их выделения соответствует 4 сут пребывания нативного угля на воздухе. Наряду с кислородсодержащими газами в составе продуктов окисления угля также отмечается повышение концентрации водорода. Выделение СО2, СО и Н2 наряду с Н2О связывают с разложением пероксидов – первичных продуктов окисления углей [13, 26]. Константа скорости процесса окисления, рассчитанная по методике [27], имеет максимальные значения (0.013 мг/г·ч) на начальном этапе и существенно снижается с повышением времени контакта нативного угля с воздухом. Это означает, что первоначальное поглощение кислорода происходит с высокой скоростью, которая со временем снижается из-за утрачивания доступных реакционноспособных центров.
Таблица 3.
Результаты газового анализа продуктов низкотемпературного окисления угля марки Д (фракция 1–3 мм)
| Продолжительность окисления, сут | Состав газовой фазы, отн. % | Константа скорости окисления, мг/г·ч | ||||
|---|---|---|---|---|---|---|
| N2 | O2 | CO | CO2 | H2 | ||
| 0 (воздух) | 78.9564 | 20.9456 | 0 | 0.0972 | 0.0008 | 0 |
| 1 | 79.7368 | 20.0783 | 0.0061 | 0.1779 | 0.0009 | 0.013 |
| 4 | 80.8738 | 18.7642 | 0.0188 | 0.3422 | 0.0010 | 0.008 |
| 7 | 80.9544 | 18.6571 | 0.0202 | 0.3671 | 0.0012 | 0.005 |
Несмотря на то что метод ядерного магнитного резонанса регистрирует эффекты в объеме угольного вещества, для оценки вклада поверхностных эффектов окисления результаты 13С ЯМР-спектроскопии оказались не менее информативны, чем данные ИКС- и ЭПР-спектроскопии (табл. 4). Однако при этом периодичность изменения структурных фрагментов ОМУ не совпадает по времени с данными ЭПР- и ИК-спектроскопии (рис. 2–5 ). Согласно результатам ЯМР 13С, основные изменения в ОМУ при низкотемпературном окислении связаны с уменьшением относительного содержания эфирных (ОСН3, С–О–С) групп. При этом суммарное количество алифатических СН3- и СН2-алкильных групп изменяется незначительно, что указывает на первичные преобразования главным образом кислородных атомов структурных единиц ОМУ. Ароматические структурные фрагменты также устойчивы к воздействию молекулярного кислорода при температуре окружающей среды, степень ароматичности угля в рамках продолжительности эксперимента остается практически неизменной. Количество фенольных групп убывает, а карбоксильных – возрастает. Таким образом, можно полагать, что в структуре низкометаморфизованного длиннопламенного угля активными центрами к взаимодействию с кислородом при температуре окружающей среды являются периферийные алкилэфирные и фенольные группы, что согласуется с данными ИК-спектроскопии и химического анализа.
Таблица 4.
Параметры фрагментарного состава угля марки Д по данным 13С ЯМР-спектрального анализа
| Продолжительность контакта с О2, сут | Распределение атомов углерода по структурным группам, отн. % | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| CH3 | CH2 | OCH3 | Cal–O | Car–H | Cаr–C | Cаr–O | COO- | fa | |
| 0 | 2.85 | 24.88 | 1.60 | 0.95 | 40.05 | 22.59 | 5.55 | 1.53 | 0.68 |
| 1 | 3.00 | 24.88 | 1.28 | 0.89 | 39.99 | 22.89 | 5.49 | 1.58 | 0.68 |
| 4 | 3.22 | 24.86 | 1.35 | 0.74 | 40,10 | 22.88 | 5.04 | 1.81 | 0.68 |
| 7 | 3.06 | 24.94 | 1.35 | 0.78 | 40.14 | 22.77 | 5.72 | 1.24 | 0.69 |
Рис. 4.
Изменение ИК-спектрального параметра Ко (степень окисленности) (а) и угла смачивания поверхности θ (б) угля марки Д при его низкотемпературном окислении в воздушной среде.
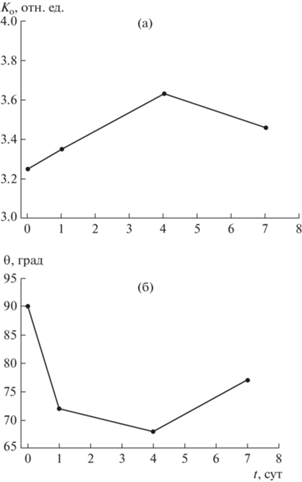
Изменение функционального состава О-групп в поверхностном слое угольных частиц оказывает влияние на смачиваемость угля – параметр, от которого зависит взаимодействие угля с жидкостями различного состава, в том числе ингибиторами процессов самовозгорания в шахте. Метод определения смачиваемости угольной поверхности оказался весьма чувствительным для фиксации и анализа результатов начального окисления. Краевой угол смачивания θ изменяется антибатно с ИК-спектральным показателем окисленности (Ко), определенным по отношению суммарных интенсивностей п.п. кислородсодержащих и углеводородных фрагментов ОМУ (рис. 4). С увеличением степени окисленности поверхностного слоя угля на начальном этапе его взаимодействия с воздушной средой смачиваемость возрастает (угол смачивания снижается). После стабилизации процесса и изменения функционального состава (перераспределение количества ОН- и СООН-групп (рис. 3)) по достижении 4 сут пребывания угля на воздухе угол смачивания вновь повышается, т.е. гидрофобность поверхности возрастает.
Установленные изменения состава структурных фрагментов угля в первые несколько суток его нахождения в воздушной среде не оказывают ощутимого влияния на изменение интегральных технологических характеристик угля – выхода летучих веществ, зольности, спекаемости, теплоты сгорания и т.д., и не могут быть идентифицированы стандартными методами определения окисленности углей (петрографический, щелочной), поскольку ограничены тонким поверхностным слоем.
С учетом сформированных представлений о молекулярной структуре низкометаморфизованных углей [16], по аналогии с [25] проиллюстрировать изменения, протекающие в составе ОМУ на начальной стадии низкотемпературного окисления, можно на примере схемы с участием исходных реакционноспособных стабильных радикалов и структурных фрагментов ОМУ рис. 5(схема).
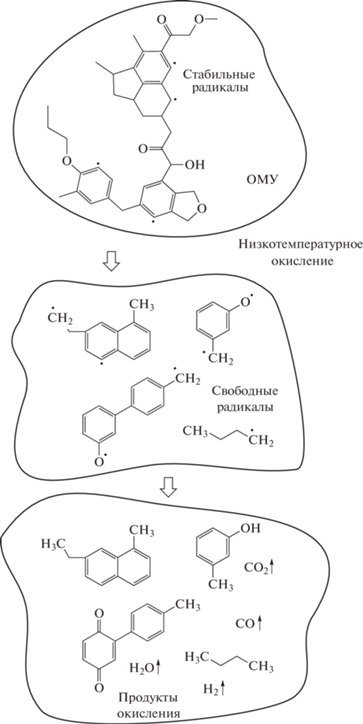
ЗАКЛЮЧЕНИЕ
Анализ изменения химического состава поверхности и продуктов низкотемпературного окисления нативного угля позволяет полагать, что инициирование окислительных радикальных реакций начинается уже в момент высвобождения (раскрытия) нативной поверхности с участием стабильных радикалов, присутствующих в структуре угля. Процессы окисления запускаются с образования свободных радикалов и их взаимодействия с сорбированным на поверхности угля кислородом. Ввиду гетерофазности течения процесса, ограниченного толщиной диффузионного слоя, продолжительного развития цепи, по-видимому, не происходит. Свободные радикалы на обнаженной поверхности угля рекомбинируют с образованием газообразных продуктов, стабильных радикалов и макромолекулярных кислородсодержащих фрагментов (функциональных групп). В случае длиннопламенного угля доминирующая роль реакций с образованием свободных радикалов утрачивает свое значение спустя 24 ч пребывания угля в воздушной среде. Далее следуют реакции, связанные с взаимопревращением периферийных функциональных групп. По достижении 7 сут процесс окисления замедляется и для его дальнейшего развития требуется воздействие внешних физико-химических факторов, способствующих повторной активации стабильных радикалов (увлажнение, измельчение, температура, УФ-излучение и т.д.).
Для низкометаморфизованного длиннопламенного угля наиболее активными к взаимодействию с сорбированными на поверхности молекулами кислорода, вероятно, являются кислородсодержащие радикалы алкилэфирных и фенольных групп и алкильные радикалы.
Угли с повышенной пористостью характеризуются высокой скоростью развития окислительных процессов на начальной стадии низкотемпературного окисления вследствие свободной диффузии окислителя в глубь пористого пространства.
Список литературы
Мефферт Б.В. О выветривании минерального угля. Тр. Геологического комитета. Новая серия. Вып. 60. Санкт-Петербург: Тип. М.М. Стасюлевича, 1910. 89 с.
Стадников Г.Л. Происхождение углей и нефти. М.: Госхимтехиздат, 1933. 222 с.
Крым В.С. Химия твердого топлива. Ч. 1. Ископаемые угли. Харьков: ДНТВУ ОНТИ НКТП, Гос. научно-техн. изд-во Украины, 1934. 286 с.
Тронов Б.В. Окисление углей кислородом воздуха / Матер. для участников Всес. совещ. по подземным пожарам. М.: Полиграфкнига, 1941. 28 с.
Караваев Н.М. Химия твердого топлива. Сб. 1. М.: Изд-во иностр. лит., 1951. 408 с.
Аммосов И.И., Еремин И.В. Трещиноватость углей. М.: Изд-во АН СССР, 1960. 110 с.
Веселовский В.С. Химическая природа горючих ископаемых. М.: Изд-во АН СССР, 1955. 424 с.
Хрисанфова А.И., Шубников А.К., Захаров А.Н., Гусев Р.П. Ингибиторы для борьбы с окислением и самовозгоранием ископаемых углей. М.: Изд-во АН СССР, 1959. 137 с.
Кухаренко Т.А. Окисленные в пластах бурые и каменные угли. М.: Недра, 1972. 216 с.
Саранчук В.И. Окисление и самовозгорание угля. Киев: Наук. думка, 1982. 166 с.
Проскуряков В.А., Чистяков А.Н. // ХТТ. 1972. № 2. С. 82.
Камнева А.И., Александров И.В. // ХТТ. 1977. № 4. С. 105.
Кучер Р.В., Компанец В.А., Бутузова Л.Ф. Структура ископаемых углей и их способность к окислению. Киев: Наук. думка, 1980. 168 с.
Улановский М.Л. // Кокс и химия. 2012. № 7. С. 5.
Мирошниченко Д.В., Кафтан Ю.С. // Кокс и химия. 2017. № 5. С. 2. https://doi.org/10.3103/S1068364X17050052
Krevelen Dirk W. van. Coal: typology, physics, chemistry, constitution. Amsterdam, New York: Elsevier, 1993. 979 p.
Wang H., Dlugogorski B.Z., Kennedy E.M. // Prog. Energy Combust. Sci. 2003. V. 29. P. 487.
Taraba B., Pavelek Z. // Fuel. 2014. V. 125. P. 101. https://doi.org/10.1016/j.fuel.2014.02.024
Onifade M., Genc B. // Int. J. Mining Sci. Technol. 2020. V. 30. P. 303. https://doi.org/10.1016/j.ijmst.2020.03.001
Кузнецов П.Н., Малолетнев А.С., Исмагилов З.Р. // Химия в интересах устойчивого развития, 2016. Т. 24. С. 335. https://doi.org/10.15372/KhUR20160308
Эпштейн С.А., Монгуш М.А., Нестерова В.Г. // ГИАБ. 2008. № 12. С. 211.
Семенова С.А., Патраков Ю.Ф., Майоров А.Е. // Кокс и химия. 2020. № 5. С. 12. https://doi.org/10.3103/S1068364X20050063
Rausa R., Calemma V., Ghelli S., Girardi E. // Fuel. 1989. V. 68. N. 9. P. 1168. https://doi.org/10.1016/0016-2361(89)90190-7
Патраков Ю.Ф., Семенова С.А., Харлампенкова Ю.А., Созинов С.А. // Кокс и химия. 2019. № 12. С. 2. https://doi.org/10.3103/S1068364X19120081
Cai Ji., Yang Sh., Zheng W., Song W., Gupta R. // Fuel. 2021. V. 292. 120256. https://doi.org/10.1016/j.fuel.2021.120256
Green U., Aizenshtat Z., Ruthstein Sh., Cohen H. // Phys. Chem. Chem. Phys. 2012. V. 14. P. 13046. https://doi.org/10.1039/c2cp41696d
Федеральные нормы и правила в области промышленной безопасности “Инструкция по определению инкубационного периода самовозгорания угля”. Сер. 05. Вып. 38. М.: ЗАО НТЦ ПБ, 2013. 22 с.
Дополнительные материалы отсутствуют.
Инструменты
Химия твердого топлива