

Кинетика и катализ, 2019, T. 60, № 3, стр. 394-402
Влияние природы модификатора на каталитические свойства и термическую стабильность катализаторов разложения аммиака Ru–Cs(Ba)/сибунит
В. А. Борисов 1, *, К. Н. Иост 1, Д. А. Петрунин 2, В. Л. Темерев 1, И. В. Муромцев 1, А. Б. Арбузов 1, М. В. Тренихин 1, Т. И. Гуляева 1, Н. С. Смирнова 3, Д. А. Шляпин 1, П. Г. Цырульников 1
1 ФГБУН Институт проблем переработки углеводородов СО РАН
644040 Омск, ул. Нефтезаводская, 54, Россия
2 ФГБОУ ВО Омский государственный технический университет
644050 Омск, просп. Мира, 11, Россия
3 ФГБУН Институт общей и неорганической химии им. Н.С. Курнакова РАН
119991 Москва, Ленинский просп., 31, Россия
* E-mail: borisovtiger86@mail.ru
Поступила в редакцию 21.09.2018
После доработки 17.12.2018
Принята к публикации 28.12.2018
Аннотация
На основе углеродного композита Сибунита были получены катализаторы Ru–Cs(Ba)/Сибунит c мольным соотношением Cs(Ba) : Ru равным 0.5, 1.5 и 2.5. Сопоставлены каталитическая активность полученных промотированных катализаторов в реакции разложения аммиака и их термическая устойчивость к метанированию. В реакции разложении аммиака удельная активность (Wуд) барийсодержащих катализаторов оказалась в 2 раза ниже, чем удельная активность Cs-содержащих образцов при одинаковом мольном соотношении М : Ru. Так, Wуд при 500°С для 0.5Cs–Ru/Sib составила 29.4 ммоль H2 ${\text{г }}_{{{\text{к а т }}}}^{{ - {\text{1}}}}$ мин–1, тогда как для 0.5Ba–Ru/Sib – 15.4 ммоль H2 ${\text{г }}_{{{\text{к а т }}}}^{{ - {\text{1}}}}$ мин–1. Показано, что введение бария в интервале мольных соотношений Ba : Ru = 0.5–2.5 существенно увеличивает термическую стабильность образцов, определяемую как отношение массы водорода, полученного на катализаторе, к массе углерода, подвергшейся метанированию. Для непромотированного катализатора Ru/Sib на 1 г потерь углерода приходится 52 г H2, для образца 0.5Ba–Ru/Sib – 370 г H2, для 1.5Ba–Ru/Sib – 200 г H2, а для 2.5Ba–Ru/Sib – 150 г H2.
ВВЕДЕНИЕ
Реакция разложения аммиака 2NH3 ⇌ N2 + 3H2 имеет большое практическое значение для очистки промышленных выбросов, создания защитных (восстановительных) атмосфер [1], а также может служить источником чистого водорода для топливных элементов [2]. Поскольку эта реакция обратима, то с позиции термодинамики благоприятно проводить ее при как можно более низких давлениях и высоких температурах [3]. Однако в настоящее время актуален поиск катализаторов, активных при низких температурах, что важно с точки зрения перспективы использования аммиака для получения чистого водорода непосредственно на борту электромобилей [4]. Наиболее активная каталитическая система в реакции разложения аммиака – рутений, нанесенный на различные углеродные носители (нанотрубки, графен, графит, активированный уголь) [5, 6]. Для повышения активности в каталитическую систему Ru/C вводят промоторы (модификаторы) – соединения щелочных и щелочноземельных металлов. В работе [7] было обнаружено, что промотирующий эффект для рутениевых катализаторов разложения аммиака на углеродных нанотрубках увеличивается с падением электроотрицательности соответствующего элемента и возрастает в ряду Ca < La < Ba < Ce < Li < Na < K. Это означает, что в пределах одной группы элементов (K, Na и Li; Ba и Ca) активность промотора в разложении NН3 тем меньше, чем выше его электроотрицательность. Сравнение рутениевых катализаторов на активированном угле [8] также показало, что катализатор Cs–Ru/C в реакции разложении аммиака в 1.6 раза активнее, чем K–Ru/C, и в 4 раза – чем Ba–Ru/C. Применение других носителей подтверждает вышеуказанную зависимость: Cs–Ru/MgAl2O4 активнее, чем Ba–Ru/MgAl2O4 [9]. При использовании в качестве носителей катализаторов амидов щелочноземельных металлов активность также возрастает в ряду Ru–Mg(NH2)2 < Ru–Ca(NH2)2 < < Ru–Ba(NH2)2 [10].
Катализаторы Ru/C имеют существенный недостаток, связанный с тенденцией углеродных носителей вступать в водородсодержащей среде в побочную реакцию метанирования, которую катализирует рутений [11]. Подобное взаимодействие, особенно заметное при высоких температурах и давлениях водорода, приводит к деградации пористой структуры носителя, спеканию частиц рутения и дезактивации катализатора [12]. В общем случае присутствие промоторов повышает устойчивость носителя к метанированию, что связано с частичной блокировкой поверхности непосредственно по границам частиц рутения и углеродного носителя и уменьшением количества активированного водорода [13]. Ранее было показано [14], что графитизация углеродного носителя при высоких температурах также способствует снижению его способности вступать в реакцию метанирования.
Целью настоящего исследования являлось изучение влияния модификатора на каталитическую активность катализаторов Ru–Cs(Ba)/Сибунит (в дальнейшем Ru–Cs(Ba)/Sib) в разложении аммиака и их термическую стабильность в водородсодержащей среде.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Методика подготовки носителя
В качестве углеродного носителя применяли графитоподобный мезопористый углерод – углеродный композит Сибунит [15–18]. Удельная поверхность носителя составляла 320 м2/г. После процедуры фракционирования носитель отмывали от угольной пыли и сушили при 120°С на воздухе в течение 3 ч. Для приготовления катализаторов использовали фракцию 0.4–0.8 мм.
Методика синтеза катализатора
Предшественником активного компонента служил аммиачный комплекс рутения (II) [Ru(NH3)nClm]Clр (n = 5–6; m = 0–1; p = 1–2), синтезированный по методике [17]. Нанесение раствора аммиачного комплекса на Сибунит проводили методом пропитки по влагоемкости. Затем образцы сушили при 120°С на воздухе в течение 3 ч. Нанесенный предшественник рутения восстанавливали при 450°С (предварительно нагревая образец от комнатной до заданной температуры со скоростью 10°С/мин) в токе Н2 (60 мл/мин) в течение 4 ч. Содержание рутения во всех образцах было равно 4 мас. %. Полученные образцы Ru/Sib пропитывали водным раствором предшественника промотора (CsNO3, Ва(СН3СОО)2) и сушили при 120°С на воздухе в течение 3 ч. Мольное соотношение M : Ru составляло 0.5, 1.5 и 2.5 (соответствующее значение указано в маркировке образца). Для разложения предшественника промотора высушенные образцы подвергали дальнейшей термообработке при 350°С (5°С/мин) 2 ч в аргоне (60 мл/мин) и 2 ч в водороде (60 мл/мин). Затем образец охлаждали в токе аргона до комнатной температуры.
Просвечивающая электронная микроскопия
Для электронно-микроскопического исследования образцов применяли метод просвечивающей электронной микроскопии (ПЭМ) с использованием просвечивающего электронного микроскопа JEM-2100 (“JEOL”, Япония, ускоряющее напряжение – 200 кВ и разрешение решетки – 0.145 нм) с энергодисперсионным рентгеновским спектрометром INCA-250 (“Oxford Instruments”, Великобритания). Суспензии образцов в спирте подвергали ультразвуковому диспергированию на приборе УЗДН-2Т (“Электрон”, СССР) с последующим закреплением на перфорированной углеродной пленке, нанесенной на медную сетку. Калибровку линейных размеров при измерениях диаметра частиц проводили относительно кристаллической решетки частиц золота. Определение параметров кристаллических плоскостей в отдельных наночастицах и компьютерную обработку ЭМ снимков выполняли с помощью программного пакета Digital Micrograph “Gatan”. Химический элементный анализ полученных образцов осуществляли методом рентгеновской энергодисперсионной спектроскопии (ЭДС).
КР-спектроскопия
Спектры комбинационного рассеяния света (КРС) записывали на дисперсионном спектрометре DXR Smart Raman (“Thermo Fisher Scientific”, США), снабженном приставкой для регистрации обратно рассеянного света, в диапазоне 50–3500 см–1. Длина волны возбуждающего лазерного излучения составляла 633 нм. Мощность лазера была 5 мВт, время выдержки – 60 с, число накопления спектров – 15. Перед регистрацией спектров КРС образцы прессовали на ручном прессе в специальном магнитном держателе. Из полученных спектров КРС вычитали спектр КРС воздуха. Степень упорядоченности углеродных материалов оценивали по известным методикам [19, 20].
Рентгенофазовый анализ
Рентгенофазовый анализ (РФА) проводили на порошковом рентгеновском дифрактометре D8 Advance (“Bruker”, Германия) в монохроматизированном CuKα-излучении (длина волны 0.15418 нм). Использовали следующие режимы измерения: шаг сканирования — 0.050, время накопления сигнала – 2 с/точка, напряжение и ток накала – 40 кВ и 40 мA соответственно, диапазон сканирования 2θ = 10°–80°. Полученные дифрактограммы расшифровывали с помощью с базы данных по порошковой дифракции ICDD PDF-2.
Определение удельной поверхности
Параметры пористой структуры исходных носителей находили по изотермам адсорбции–десорбции азота при –195.7°С на приборе Sorptomatic-1900 (“Carlo Erba”, Италия). Расчет удельной поверхности (Sуд) выполняли по методу БЭТ в интервале равновесных относительных значений паров азота P/P0 = 0.05–0.33 по изотерме адсорбции. Погрешность измерения величин удельной поверхности (Δ) стандартных образцов на вышеуказанном приборе равна ±2.8 отн. %.
Определение дисперсности частиц рутения
Дисперсность рутения в полученных катализаторах определяли методом импульсной хемосорбции СО на анализаторе AutoChem II 2920 (“Micromeritics”, США). Хемосорбцию СО проводили после предварительного восстановления образцов смесью 10% Н2/Ar при 350°С в течение 60 мин с последующей продувкой гелием при этой же температуре в течение 30 мин и охлаждением образца до комнатной температуры. Смесь 10 об. % СО/Не импульсами через равные промежутки времени подавали в поток инертного газа-носителя (гелия). Объем калиброванной петли составлял 1 мл. Значения дисперсности и размера частиц рассчитывали, принимая линейную хемосорбцию СО на рутении (стехиометрический коэффициент СО : Ru = 1 : 1) [21, 22].
Испытания в реакции разложения аммиака
Активность катализаторов в реакции разложения аммиака изучали в проточном реакторе с неподвижным слоем при атмосферном давлении в интервале температур 420–500°С. Масса загружаемого катализатора составляла 0.1 г, расход аммиака – 34 000 мл NH3 ${\text{г }}_{{{\text{к а т }}}}^{{ - {\text{1}}}}$ ч–1, время контакта – 0.21 с. Газовую смесь на выходе из реактора анализировали с использованием хроматографа ЦВЕТ-500М (“Цвет”, Россия) с детектором по теплопроводности, газ-носитель – водород. Колонку длиной 1.5 м наполняли сорбентом Haeyesep C, позволяющим разделить NH3 и N2. Условия хроматографирования: скорость газа-носителя (водорода) – 60 мл/мин, давление – 1 атм, напряжение моста – 4 В, температура колонки – 70°С. На основании полученных данных рассчитывали конверсию аммиака и удельную каталитическую активность (Wуд) (ммоль H2 ${\text{г }}_{{{\text{к а т }}}}^{{ - {\text{1}}}}$ мин–1).
Исследование термической стабильности катализаторов в водородсодержащей атмосфере
Устойчивость катализаторов к метанированию определяли методом измерения термической стабильности в восстановительной среде [12]. Эксперимент проводили в изотермическом режиме. Установка для испытаний включала в себя систему подачи газов (гелия и водорода) и кварцевый U-образный реактор диаметром 10 мм, помещаемый в трубчатую печь. Аналитический блок, состоящий из газового хроматографа с пламенно-ионизационным детектором и поликапиллярной колонки [23], позволял осуществлять экспресс-анализ выходящей газовой смеси каждые 15 с.
При проведении эксперимента образец массой 100 мг загружали в реактор, подавали гелий со скоростью 60 мл/мин при нагреве со скоростью 10°С/мин и запускали хроматографический анализ выходящей газовой смеси. Когда температура достигала 500°С, производили замену гелия на водород и выдерживали образец при заданных условиях в течение 20 ч. На основании анализа образующегося метана в выходящей смеси рассчитывали общее количество потерь углерода (мас. %).
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Физико-химические исследования были проведены для катализаторов с мольным соотношением М : Ru = 2.5, так как для этих образцов имеется бóльшая вероятность обнаружить формирование совместной фазы рутения и модификатора в случае ее образования. В ранее проведенных исследованиях [24] было показано, что мольное соотношение рутений/модификатор влияет только на количество совместных фаз, но не их природу.
КР-спектроскопия
Спектры комбинационного рассеяния углеродного носителя Сибунита, а также катализаторов на его основе характеризуются наличием интенсивной линии при 1596 см–1, соответствующей полносимметричным продольным валентным колебаниям связей С=С (G-полоса) и линии при 1330 см–1 (D-полоса) (табл. 1). Отношение интенсивностей соответствующих линий ID/IG > 1, что свидетельствует о разупорядоченной структуре используемого углеродного носителя. Также в спектре наблюдаются очень слабые полосы с колебательными частотами 1167, 2654 (2D-полоса) и 2924 см–1 [19, 25], характерные для графита. Отношение интенсивностей I2D/IG = 0.055 указывает на то, что расстояние между графеновыми слоями отличается от эталонного графита, что свойственно носителю с наличием турбостратной структуры [26].
Таблица 1.
Интенсивности D- и G-полос Сибунита и катализаторов на его основе
| Время, мин | Интенсивность | ID/IG | |
|---|---|---|---|
| D-полоса | G-полоса | ||
| Sib | 34.7 | 23.2 | 1.49 |
| 2.5Ba–Ru/Sib | 17.0 | 11.1 | 1.54 |
| 2.5Cs–Ru/Sib | 23.1 | 15.0 | 1.54 |
Как видно из табл. 1, отношения ID/IG для исследуемых образцов носителя и рутениевых модифицированных катализаторов на его основе существенно не отличаются, т.е. введение рутения и модификаторов и проведение процедур термообработок и восстановления практически не влияет на структуру носителя.
Фазовый состав образцов
Дифрактограммы для образцов Sib, Ru/Sib, 2.5Cs–Ru/Sib и 2.5Ba–Ru/Sib представлены на рис. 1.
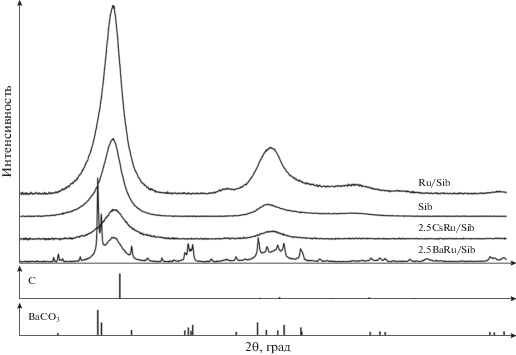
Согласно результатам рентгенофазового анализа, исходный Сибунит отличается увеличенным межплоскостным расстоянием базального рефлекса (d002) – 0.349 нм, для эталонного графита этот показатель составляет 0.335 нм. Такие же значения d002 характерны для углерода в образцах Ru/Sib, 2.5Cs–Ru/Sib и 2.5Ba–Ru/Sib, из чего следует, что в процессе приготовления катализатора углеродный носитель не претерпевает изменений.
Для образца Ru/Sib был обнаружен очень слабый пик при 2θ = 44.01°, относящийся к рутению. Для промотированных образцов 2.5Cs–Ru/Sib и 2.5Ba–Ru/Sib пики, соответствующие рутению, не найдены, что может быть обусловлено снижением интенсивности сигнала вследствие диспергирования рутения в промотированных образцах, а также эффектом падением интенсивности дифракционных пиков в присутствии тяжелых металлов. Рефлексы на дифрактограмме образца 2.5Ba–Ru/Sib говорят о присутствии бария в образце в форме BaCO3, которую можно рассматривать как результат разложения используемого нами предшественника (ацетата бария), протекающего при температуре 450–470°C с образованием ацетона и BaCO3 [27]. Средние размеры областей когерентного рассеяния (ОКР) для BaCO3 составили 26 нм, для Ru размер ОКР определить не удалось.
Удельная поверхность носителя и дисперсность активного компонента по данным хемосорбции СО
Результаты определения удельной поверхности образцов представлены в табл. 2. Наибольшее значение удельной поверхности (309 м2/г) наблюдается для непромотированного образца Ru/Sib. Введение промотора сильно уменьшает Sуд: удельная поверхность образца, промотированного цезием, падает в 2 раза, а для образца 2.5Ba–Ru/Sib наблюдается снижение этого показателя на 36%. Значительное сокращение удельной поверхности объясняется частичной блокировкой пор носителя промотором, как уже было показано ранее [24]. Меньшее снижение Sуд для образца 2.5Ba–Ru/Sib по сравнению с 2.5Cs–Ru/Sib может быть следствием присутствия в катализаторе значительной части промотора в виде крупных частиц карбоната бария [12]. Идентифицировать частицы металлического цезия или его соединения примененными методами не удалось. В одном из ранних исследований [28] с помощью метода РФЭС было показано, что цезий может находится на поверхности в форме субоксида, например Cs11O3. Формы нахождения Cs в образце, изученном в настоящей работе, неизвестны, но, возможно, он интеркалирован в решетку углерода [27], однако подтверждение или отклонение этого предположения требует специальных дополнительных исследований.
Таблица 2.
Удельная площадь поверхности катализаторов и дисперсность активного компонента
| Катализатор | Sуд, м2/г | dRu, нм | DCO, % |
|---|---|---|---|
| Ru/Sib | 309 | 9.8 | 13.9 |
| 2.5Cs–Ru/Sib | 166 | 8.7 | 15.7 |
| 2.5Ba–Ru/Sib | 205 | 45 | 3 |
Размеры частиц рутения, рассчитанные из величин хемосорбции СО для непромотированного образца, составили 9.8 нм, а для промотированных цезием и барием катализаторов – 8.7 и 45 нм соответственно. Возможно, повышение среднего размера частиц для промотированного барием катализатора связано с частичной блокировкой поверхности носителя соединениями бария, что приводит к недоступности активного компонента для CO. Для лучшего понимания сути этого явления были проведены электронно-микроскопические исследования.
Электронно-микроскопическое исследование образцов
Из анализа результатов ПЭМ (рис. 2) следует, что носитель в образцах катализаторов представлен частицами сферической формы с толщиной оболочки от 5 до 20 нм. Также проведен анализ формы, размеров и распределения активного компонента (Ru) и промоторов (Сs и Ba) в объеме углеродного носителя.
Рис. 2.
ПЭМ-изображения частиц катализаторов разложения аммиака: а, в – 2.5Cs–Ru/Sib; б, г – 2.5Ba–Ru/Sib.

Согласно результатам ПЭМ и ЭДА во всех образцах катализаторов частицы рутения с размерами от 3 до 13 нм равномерно распределены по носителю. Рутений находится в металлическом состоянии. Фазы, соответствующие соединениям цезия, в образце 2.5Cs–Ru/Sib определить не удалось, однако данные ЭДА свидетельствуют о равномерном распределении этого модификатора, предположительно в оксидной форме [28]. Места локализации соединений цезия в виде отдельных частиц не выявлены. Как показали наши предыдущие исследования [29], а также работы других авторов [30], Cs-содержащие частицы не всегда удается обнаружить с помощью метода ПЭМ. Для образца 2.5Cs–Ru/Sib на поверхности отдельных частиц рутения наблюдается присутствие соединения цезия, возможно, Cs11O3, как было показано в работе [28].
В образце 2.5Ba–Ru/Sib методом ПЭМ были определены рутений и карбонат бария. Рутений присутствует преимущественно в виде округлых металлических частиц. Частиц рутения, покрытых соединениями бария, не найдено. Крупные (10–100 нм) частицы неправильной формы соответствуют карбонату бария (рис. 2а), о чем свидетельствуют значения межплоскостных расстояний. Эти результаты хорошо согласуются с данными РФА. Размеры частиц рутения, определенные методами ПЭМ и хемосорбции СО, для образца 2.5Ba–Ru/Sib отличаются в 4–5 раз. Вероятно, частицы BaCO3 закупоривают крупные поры, в результате чего доля частиц, доступных для СО значительно уменьшается.
Каталитическая активность образцов в реакции разложения аммиака
Результаты исследования каталитической активности образцов Ru–Ba(Cs)/Сибунит в разложении аммиака представлены в табл. 3.
Таблица 3.
Результаты каталитических испытаний
| Катализатор | Температура, °С | Wуд, ммоль H2${\text{г }}_{{{\text{к а т }}}}^{{ - 1}}$ мин–1 | Конверсия, % | *Ea, кДж/моль | Источник |
|---|---|---|---|---|---|
| Ru/Sib 0.5Cs–Ru/Sib 1.5Cs–Ru/Sib 2.5Cs–Ru/Sib 0.5Ba–Ru/Sib 1.5Ba–Ru/Sib 2.5Ba–Ru/Sib |
500 | 8.71 29.4 12.7 7.06 15.4 5.23 3.90 |
22.9 77.2 33.4 18.6 40.4 13.8 10.2 |
65.1 31.7 48.9 71.1 57.2 59.5 56.4 |
Настоящая работа |
| Ru/CNT Ru/AC Ru/K-CNT |
500 | 27.7 17.6 47.9 |
16.5 10.5 28.6 |
64.9 81.3 59.0 |
[31] |
| Ru/Graphene | 450 | 28.7 | 85.8 | 47.9 | [32] |
Как видно из полученных данных (табл. 3), 0.5Cs–Ru/Sib не уступает по удельной активности катализатору Ru/Grapfene и на 35% менее активен, чем образец Ru/K-CNT. Введение небольшого количества промоторов увеличивает Wуд по сравнению с непромотированным образцом: для 0.5Cs–Ru/Sib – более чем в 3 раза, а для 0.5Ba–Ru/Sib – почти в 2 раза. Объяснением промотирующего эффекта цезия является уменьшение работы выхода электронов с поверхности частиц рутения, покрытого слоем ионного промотора [33]. Однако при увеличении мольного соотношения M : Ru наблюдается резкое падение Wуд вплоть до того, что активность образцов с мольным соотношением 2.5 оказывается ниже (для 2.5Cs–Ru/Sib – 7.06 ммоль Н2 ${\text{г }}_{{{\text{к а т }}}}^{{ - {\text{1}}}}$ мин–1, а для 2.5Ba–Ru/Sib – 3.90 ммоль Н2${\text{г }}_{{{\text{к а т }}}}^{{ - {\text{1}}}}$ мин–1), чем у непромотированного образца (8.71 ммоль Н2${\text{г }}_{{{\text{к а т }}}}^{{ - {\text{1}}}}$ мин–1). Rarog-Pilecka W. et al. [34] показали, что в условиях синтеза катализатора Cs–Ru/C нитрат цезия превращается в субстехиометрический оксид CsхOу (x/y = = 2.7–3.6), покрывающий поверхность катализатора и, возможно, препятствующий адсорбции аммиака. Присутствуя в избытке, цезий, по-видимому, частично блокирует доступ к активным центрам рутения, что снижает скорость образования водорода [24].
Энергия активации (Еа) разложения аммиака на непромотированном катализаторе Ru/Sib составляет 65.1 кДж/моль. Добавление цезия в соотношении Cs : Ru, равном 0.5 и 1.5, снижает энергию активации до величин, меньших, чем на непромотированном катализаторе – 31.7 и 48.9 кДж/моль соответственно. Однако при отношении Cs : Ru = 2.5 значение Еа становится выше, чем у непромотированного катализатора – 71.1 кДж/моль. Косвенно это свидетельствует, что при большом избытке цезия (Cs : Ru = 2.5) поверхность рутения может покрываться пленкой промотора или его соединений с рутением. Для катализаторов Ba–Ru/Sib зависимости энергии активации от соотношения Ba : Ru выявлено не было. Для всех образцов она составляет примерно 57–58 кДж/моль, и с учетом возможной погрешности определения Ea на уровне 10% можно считать ее значения одинаковыми. Такие несущественные изменения величин Ea, вероятно, связаны с тем, что промотирующее действие соединений бария реализуется за счет хорошей электропроводности носителя, но прямого взаимодействия промотора с рутением нет. Уменьшение активности происходит за счет блокировки пор Сибунита BaCO3 и снижением доступности рутения для аммиака.
Энергия активации непромотированного катализатора близка к значению этого показателя, найденному в [35] – 54–72 кДж/моль. В работах других авторов [30, 36] для эффективных катализаторов также наблюдалось падение Ea промотированных образцов: в присутствие соединений калия и лития более чем в полтора раза (с 85.8 до 51.3 и 53.2 кДж/моль соответственно) снижалась Ea для Ru/MgO; при введении Cs до соотношения Cs : Ru = 2.8 энергия активации уменьшалась с 97 кДж/моль в отсутствие цезия до 60 кДж/моль в его присутствии.
Термическая стабильность
Ранее нами для каталитических систем Ru/Сибунит, Cs–Ru/Сибунит [37] было показано, что образование метана начинается при 350°С и с ростом температуры его количество возрастает. Добавление цезия сдвигает температуру начала образования метана на 30°С – до 380°С. Равномерное распределение соединений цезия по катализатору препятствует взаимодействию активированного на рутении водорода с поверхностью углеродного носителя. Вместе с тем, высокодисперсные соединения цезия затрудняют поверхностную миграцию частиц рутения, замедляя, таким образом, спекание активного компонента [13]. На термическую стабильность непромотированных образцов положительно влияет предварительная обработка Сибунита при 1900°С [38]: потери носителя в пересчете на углерод снижаются в 5 раз (с 0.5 до 0.1 мас. % С).
По сравнению с цезийсодержащими образцами катализаторы, модифицированные барием, в 2–4 раза более устойчивы в водородсодержащей среде. Поэтому в настоящей работе исследована термическая стабильность образцов Ba–Ru/Синибут (рис. 3).
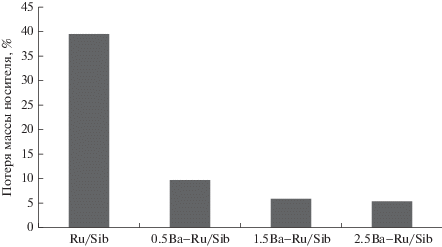
Добавление даже небольшого количества бария приводит к уменьшению потерь углерода в 4 раза. Дальнейшее допирование барием снижает степень метанирования не столь значительно: потери углерода для 1.5Ba–Ru/Sib составляют 6%, а для 2.5Ba–Ru/Sib – 5%, что в сочетании с падением активности катализаторов в разложении аммиака делает нецелесообразным дальнейшее увеличение количества промотора. Для подтверждения этого мы рассчитали отношение массы водорода, полученного на катализаторе, к массе углерода, подвергшейся метанированию. Для непромотированного катализатора на 1 г потерь углерода приходится 52 г H2, для образца 0.5Ba–Ru/Sib – 370 г H2, для 1.5Ba–Ru/Sib – 200 г H2, а для 2.5Ba–Ru/Sib – 150 г H2. Полученные данные свидетельствуют о том, что по критерию “термическая стабильность–каталитическая активность” наилучший результат получен для образца 0.5Ba–Ru/Sib.
ЗАКЛЮЧЕНИЕ
Данные РФА и КР-спектроскопии согласованно указывают на низкую упорядоченность Сибунита и неизменность его структурных характеристик в процессе приготовления катализаторов. Согласно результатам РФА и ПЭМ в образце 2.5Ba–Ru/Sib присутствует хорошо окристаллизованные частицы BaCO3 с размерами 10–100 нм. Размеры частиц рутения, определенные методами ПЭМ и хемосорбции СО, для образца 2.5Ba–Ru/Sib отличаются в 4–5 раз, что может свидетельствовать о блокировке пор Сибунита частицами BaCO3. В результате закупорки пор BaCO3 при увеличении отношения Ba : Ru падает удельная активность катализаторов в разложении аммиака, но в тоже время повышается их термическая стабильность. Так, потеря массы углеродного носителя за счет метанирования снижается с 39.5 мас. % для образца Ru/Sib до 5.3 мас. % для 2.5Ba–Ru/Sib.
В разложении аммиака удельная активность Ba–Ru-образцов оказалась в 2 раза ниже активности Cs–Ru-катализаторов при одинаковом мольном соотношении М : Ru. Так, Wуд при 500°С для 0.5Cs–Ru/Sib составила 29.4 ммоль H2 ${\text{г }}_{{{\text{к а т }}}}^{{ - 1}}$ мин–1, для 0.5Ba–Ru/Sib – 15.4 ммоль H2 ${\text{г }}_{{{\text{к а т }}}}^{{ - 1}}$ мин–1. По своей активности образец 0.5Cs–Ru/Sib не уступает катализаторам на углеродных нанотрубках и графене.
БЛАГОДАРНОСТИ
Физико-химические исследования выполнены с использованием приборной базы Омского регионального ЦКП СО РАН (Омск).
ФИНАНСИРОВАНИЕ
Работа выполнена в рамках государственного задания ИППУ СО РАН в соответствии с Программой фундаментальных научных исследований государственных академий наук на 2013–2020 годы по направлению V. 46, проект V.46.2.5 (номер госрегистрации в системе ЕГИСУ НИОКТР AAAA-A17-117021450096-8). Исследования проводили на приборной базе Омского регионального ЦКП СО РАН (Омск).
Список литературы
Li G., Kanezashi M., Tsuru T. // Catalysts. 2017. V. 7. P. 23.
Моисеев М.М., Будейкина Е.В., Моисеева И.Д. // Изв. вузов. Химия и хим. технология. 2009. Т. 52. № 6. С. 48.
Ефремов В.Н., Стрекалов Ю.В., Кашинская А.В., Голосман Е.З. // Катализ в промышленности. 2015. Т. 15. № 6. С. 14.
García-García F.R., Yi Hua Ma, Rodríguez-Ramos I., Guerrero-Ruiz A. // Catal. Commun. 2008. V. 9. № 3. P. 482.
Mukherjee S., Devaguptapu S.V., Sviripa A., Lund C.R.F., Wu G. // Appl. Catal. B: Environ. 2018. V. 226. P. 162.
Pat. 4163775 USA, 1979.
Wang S.J., Yin S.F., Li L., Xu B.Q., Ng C.F., Au C.T. // Appl. Catal. B: Environ. 2004. V. 52. P. 287.
Raróg-Pilecka W., Szmigiel D., Kowalczyk Z., Jodzis S., Zielinski J. // J. Catal. 2003. V. 218. P. 465.
Szmigiel D., Rarog-Pilecka W., Miskiewicz E., Kaszkur Z., Kowalczyk Z. / Appl. Catal. A: General. 2004. V. 264. P. 59.
Yu P., Guo J., L Liu., Wang P., Chang F., Wang H., Ju X., Chen P. // J. Phys. Chem. C. 2016. V. 120. P. 2822.
Goethel P. J., Yang R.T. // J. Catal. 1988. V. 111. P. 220.
Иост К.Н., Темерев В.Л., Смирнова Н.С., Шляпин Д.А., Борисов В.А., Муромцев И.В., Тренихин М.В., Киреева Т.В., Шилова А.В., Цырульников П.Г. // Журн. прикладной химии. 2017. Т. 90. №. 6. С. 731.
Rossetti I., Pernicone N., Forni L. // Appl. Catal. A: General. 2001. V. 208. P. 271.
Цырульников П.Г., Иост К.Н., Шитова Н.Б., Темерев В.Л. // Катализ в промышленности. 2016. Т. 16. № 4. С. 20.
Dobrynkin N.M., Tsyrulnikov P.G., Noskov A.S., Shitova N.B., Polukhina I.A., Savelieva G.G., Duplyakin V.K., Likholobov V.A. // Stud. Surf. Sci. Catal. 1988 V. 118. P. 213.
Ynusov S.M., Likholobov V.A., Shur V.B. // Appl. Catal. A: General. 1997. V. 158. P. L35
Smirnova N.S., Borisov V.A., Iost K.N., Temerev V.L., Surovikin Ju.V., Guljaeva T.I., Arbuzov A.B., Cyrul’nikov P.G. // Proc. Eng. 2015. V. 113. P. 84.
Smirnova N.S., Iost K.N., Temerev V.L., Gulyaeva T.I., Trenikhin M.V., Muromtsev I.V., Khramov E.V., Zubavichus Y.V., Shlyapin D.A., Tsyrul’nikov P.G. // Mol. Catal. 2017. V. 433. P. 235.
Букалов С.С., Михалицын Л.А., Зубавичус Я.В., Лейтес Л.А., Новиков Ю.Н. // Рос. хим. журн. 2006. Т. 50. № 1. С. 83.
Ferrari A.C. // Solid State Commun. 2007. V. 143. P. 47.
Maroto-Valiente A., Cerro-Alarcón M., Guerrero-Ruiz A., Rodríguez-Ramos I. // Appl. Catal. A: General. 2005. V. 283 P. 23.
Bergeret G., Gallezot P. Particle size and dispersion measurements, Handbook of Heterogeneous Catalysis, Wiley-VCH Verlag GmbH & Co, 2008. P. 738.
Nikolaeva O.A., Patrushev Y.V., Sidelnikov V.N. // J. Chromatography. A. 2017. V. 1488. P. 126.
Шитова Н.Б., Добрынкин Н.М., Носков А.С. Просвирин И.П., Бухтияров В.И., Кочубей Д.И., Цырульников П.Г., Шляпин Д.А. // Кинетика и катализ. 2004. Т. 45. С. 414.
Li X.K., Ji W.J., Zhao J., Wang S.J., Au C.T. // J. Catal. 2005. V. 236. P. 181.
Плаксин Г.В., Бакланова О.Н., Лавренов А.В., Лихолобов В.А. // Химия твердого топлива. 2014. № 6. С. 26.
Rossetti I., Mangiarini F., Forni L. // Appl. Catal. A. General. 2007. V. 323. P. 219.
Ларичев Ю.В., Просвирин И.П., Бухтияров В.И., Шляпин Д.А., Шитова Н.Б., Цырульников П.Г. // Кинетика и катализ. 2005. Т. 46. № 4. С. 635.
Борисов В.А., Иост К.Н., Темерев В.Л., Леонтьева Н.Н, Муромцев И.В., Арбузов А.Б., Тренихин М.В., Савельева Г.Г., Смирнова Н.С., Шляпин Д.А. // Кинетика и катализ. 2018. Т. 59. № 2. С. 161.
Hill A.K., Torrente-Murciano L. // Intern. J. Hydrogen Energy. 2014. V. 39. P. 7646.
Yin S.F., Zhang Q.H., Xu B.Q., Zhu W.X., Ng C.F., Au C.T. // J. Catal. 2004. V. 224. P. 384
Li G., Nagasawa H., Kanezashi M., Yoshioko T., Tsuru T. // J. Mater. Chem. A. 2014. V. 2. P. 9185.
Larichev Y. V., Moroz B.L., Zaikovskii V.I., Yunusov S.M., Kalyuzhnaya E.S., Shur V.B., Bukhtiyarov V.I., // J. Phys. Chem. C. 2007. V. 111. P. 9427.
Rarog-Pilecka W., Miskiewicz E., Jodzis S., Petryk J., Lomot D., Kaszkur Z., Karpinski Z., Kowalczyk Z. // J. Catal. 2006. V. 239. P. 313
Huang D.C., Jiang C.H., Liu F.J., Cheng Y.C., Chen Y.C., Hsueh K.L. // Int. J. Hydrogen Energy. 2013. V. 38. P. 3233.
Guo J., Chen Zh., Wu A., Chang F., Wang P., Hu D., Wu G., Xiong Zh., Yuab P., Chen P. // Chem. Commun. 2015. V. 51. P. 15161.
Иост К.Н., Темерев В.Л., Смирнова Н.С., Шляпин Д.А., Суровикин Ю.В., Тренихин М.В., Леонтьева Н.Н., Шитова Н.Б., Цырульников П.Г. // Химия в интересах устойчивого развития. 2015. Т. 23. № 6. С. 691.
Iost K.N., Borisov V.A., Temerev V.L., Surovikin Y.V., Pavluchenko P.E., Trenikhin M.V., Lupanova A.A., Arbuzov A.B., Shlyapin D.A., Tsyrulnikov P.G., Vedyagin A.A. // Surf. Interf. 2018. V. 12. P. 95.
Дополнительные материалы отсутствуют.
Инструменты
Кинетика и катализ