

Кинетика и катализ, 2019, T. 60, № 3, стр. 358-363
Кинетическое исследование стадии активации арилгалогенида в реакции прямого арилирования индола в условиях реального катализа
Е. В. Ларина 1, Е. В. Ярош 1, Н. А. Лагода 1, А. А. Курохтина 1, А. Ф. Шмидт 1, *
1 Иркутский государственный университет, химический факультет
664003 Иркутск, ул. К. Маркса, 1, Россия
* E-mail: aschmidt@chem.isu.ru
Поступила в редакцию 17.09.2018
После доработки 13.11.2018
Принята к публикации 14.01.2019
Аннотация
В работе представлены результаты кинетического исследования стадии активации арилиодидов и арилбромидов в условиях “безлигандного” катализа реакции прямого арилирования индола по С–Н-связи с использованием анализа интегральных кинетических зависимостей расходования конкурирующих арилгалогенидов. При проведении эксперимента в условиях конкуренции пары арилгалогенидов, имеющих различную реакционную способность, наблюдалось ускорение расходования менее активного субстрата, совпадающее по времени с практически полным израсходованием более активного. Полученные данные однозначно свидетельствуют о быстром характере стадии окислительного присоединения с участием не только реакционноспособных арилиодидов, но и неактивированных арилбромидов.
Реакция так называемого прямого арилирования (гетеро)ароматических соединений по С–Н-связи представляет собой интенсивно развивающуюся в последнее десятилетие область катализа. Наиболее привлекательными для этого процесса субстратами являются индол и его производные, поскольку продукты их арилирования представляют большой интерес для производства различных фармакологических, агрохимических и других современных функционализированных материалов [1]. Несмотря на значительное внимание исследователей к реакциям этого типа, заключающееся, в основном, в эмпирическом поиске каталитических систем для их осуществления, многие фундаментальные аспекты функционирования каталитического цикла образования арилированного продукта остаются невыясненными. В частности, в настоящий момент в литературе отсутствует единая точка зрения на природу реализующихся в каталитическом цикле элементарных стадий [2–5]. Тем не менее всеми исследователями признана необходимость активации молекулы арилгалогенида путем его окислительного присоединения к соединениям Pd(0) (схема 1 , А) с образованием ключевого интермедиата типа ArPdX (где Х – галоген), вступающего далее в превращения с участием молекулы гетероциклического субстрата (схема 1 , Б–Д). Следует отметить, что, несмотря на то, что протекание стадии окислительного присоединения в каталитическом цикле реакции прямого арилирования с использованием различных субстратов в настоящий момент не подвергается сомнению, экспериментальные исследования степени ее влияния на скорость образования продуктов реакции прямого арилирования практически отсутствуют.
Существуют единичные публикации, в которых приводятся лишь косвенные данные об относительной реакционной способности субстратов, оцениваемой не по скоростям реакций, а по выходам продуктов за достаточно продолжительный период времени [6, 7]. Кроме того, несмотря на строгие аргументы невозможности использования значений частных порядков по концентрациям веществ-участников реакции для формулирования корректных выводов о природе скорость-определяющей стадии из-за влияния сопряженных процессов формирования и дезактивации катализатора за пределами основного каталитического цикла [8], в ряде опубликованных работ значения частных порядков использовали для определения скорость-определяющих стадий реакций прямого арилирования [2, 4]. Таким образом, становится очевидной необходимость в более надежных методах проверки непосредственно в реальных каталитических условиях гипотез о быстром или медленном характере стадий каталитического цикла реакции прямого арилирования. Полученные этими методами результаты не должны быть подвержены влиянию нестационарности концентрации истинного катализатора, обусловленной протеканием процессов его формирования–дезактивации, являющихся характерными для всех реакций сочетания арилгалогенидов.

Схема 1 . Схема каталитического цикла реакции арилирования индола по С–Н-связи арилгалогенидами [5].
Нами был предложен и апробирован на примере нескольких реакций кросс-сочетания кинетический подход, базирующийся на методе конкурирующих реакций, однако использующий не традиционно оцениваемые в условиях конкурентного эксперимента относительные реакционные способности конкурирующих субстратов, а анализ интегральных кинетических зависимостей их расходования или накопления соответствующих продуктов [9–11]. В этом случае, с одной стороны, исследование проводится в условиях реального каталитического процесса (высокие соотношения субстрат/катализатор) и, с другой, при интерпретации получаемых экспериментальных данных нет необходимости в допущении экспериментально непроверяемого предположения о совпадении скорость- и селективность-определяющих стадий конкурентной реакции. Принятие такого допущения необходимо при сравнении величин относительных реакционных способностей в конкурентном эксперименте и в модельных стехиометрических реакциях, которое часто использовалось при обосновании гипотез скорость-определяющих стадий каталитических циклов родственных реакций кросс-сочетания.
Для установления характера стадий каталитического цикла с участием субстратов в реакции прямого арилирования можно проводить два типа конкурирующих экспериментов – с конкуренцией арилгалогенидов или конкуренцией индолов. Однако недавно нами было показано [5], что вхождение молекулы индола в каталитический цикл реакции прямого арилирования осуществляется через существенно обратимую стадию, вероятнее всего представляющую собой координацию молекулы индола к интермедиату ArPdX (схема 1 , Б) с последующим обратимым образованием σ-комплекса (схема 1 , В). При этом были получены надежные доказательства практически необратимого протекания стадии активации арилгалогенида [5]. Таким образом, с учетом надежно установленной обратимости стадии с участием молекулы индола предположение об ее скорость-определяющем характере может быть исключено из рассмотрения, а необратимый характер стадии с участием арилгалогенида требует экспериментальной проверки гипотез об ее быстром или медленном протекании.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Каталитические эксперименты
В стеклянный реактор, снабженный резиновой мембраной и магнитной мешалкой, помещали индол (2.5 ммоль), два конкурирующих арилгалогенида (по 1.25 ммоль каждого), PdCl2 (0.04 ммоль), NaOAc в качестве основания (1.625 ммоль), нафталин (0.5 ммоль) в качестве внутреннего стандарта и 5 мл смеси ДМФА–вода (4 : 1, по объему). Реакции проводили при 140°С при перемешивании. Каждый эксперимент повторяли трижды для проверки воспроизводимости. Скорости расходования субстратов оценивали по разности их концентраций в последовательно отбираемых пробах реакционной смеси.
Анализ состава реакционной смеси
Пробы реакционной смеси анализировали на газожидкостном хроматографе Хроматэк-Кристалл 5000 (“Хроматэк”, Россия, ДИП, колонка HP-5 длиной 15 м) и хромато-масс-спектрометре Shimadzu GCMS QP 2010 Ultra (“Shimadzu”, Япония) с ионизацией под действием электронного удара (энергия ионизации 70 эВ, колонка GsBP-5MS размером 0.25 мкм × 0.25 мм × 30 м, газ-носитель − гелий) с программированным нагревом от 100 до 250°С. Полученные масс-спектры сравнивали с библиотечными данными (Wiley, NIST, NIST05). Количественный состав проб вычисляли с использованием метода внутреннего стандарта (нафталин), калибровку проводили по аутентичным образцам.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Для того чтобы на качественном уровне продемонстрировать принцип используемого нами подхода (математическое обоснование которого дано в [9]), рассмотрим простейшую схему, описывающую реакции двух однотипных субстратов, конкурирующих за общий катализатор (схема 2 ).

Схема 2 . Схема реакции образования продуктов Р1 и Р2 из двух конкурирующих субстратов S1 и S2 и одного общего реагента R.
В том случае, если стадии А и Б (схема 2 ) являются быстрыми необратимыми или быстрыми обратимыми, когда равновесие смещено в сторону интермедиатов XS1 и XS2, бóльшая часть катализатора, распределенного между интермедиатами каталитических циклов, находится в виде XS1 и XS2 (catalyst resting state). Если же стадии А и Б (схема 2 ) являются медленными необратимыми или быстрыми обратимыми со сдвигом равновесия в сторону реагирующих субстратов и интермедиата Хобщ, то доминирующим в распределении катализатора является Хобщ. В первом случае, если один из конкурирующих субстратов (например, S1) имеет более высокую реакционную способность по сравнению со вторым субстратом (S2), соотношение их концентраций [S1]/[S2] будет уменьшаться в ходе реакции, что будет сопровождаться постепенным переходом катализатора из интермедиата XS1 в XS2 до полного израсходования субстрата S1. В работе [9] нами было показано, что в случае значительной разницы в реакционных способностях субстратов S1 и S2 переход катализатора из интермедиата XS1 в XS2 интенсивно протекает в момент практически полного израсходования S1, при этом скорость расходования субстрата S2 резко возрастает, что видно на кинетической кривой его расходования. Если же соотношение скоростей стадий каталитических циклов таково, что катализатор в основном находится в форме Хобщ, после израсходования более активного субстрата этот интермедиат продолжит преобладать в распределении катализатора. И в этом случае никаких видимых изменений на кривой расходования менее активного субстрата S2 наблюдаться не будет. Таким образом, эффект ускорения расходования менее активного субстрата является доказательством того, что лишь небольшая часть катализатора находится в виде общего интермедиата, реагирующего с конкурирующими субстратами в быстрых стадиях каталитических циклов. Данный вывод справедлив вне зависимости от того, являются эти стадии обратимыми или необратимыми. Отсутствие эффекта ускорения в свою очередь означает доминирование общего для конкурирующих каталитических циклов интермедиата. Эта ситуация может реализовываться в случае участия общего интермедиата в стадиях конкурирующих каталитических циклов, являющихся либо медленными, либо быстрыми обратимыми со сдвигом равновесия в сторону общего интермедиата и конкурирующих субстратов.
Описанный кинетический подход был применен нами для установления характера стадии взаимодействия катализатора с арилгалогенидом (активация арилгалогенида) в реакции прямого арилирования индолов (I). Как уже отмечалось, этой стадией, согласно общепринятой в настоящий момент точке зрения, является окислительное присоединение арилгалогенидов к Pd(0). Для успешного применения описанного выше подхода необходимо, чтобы конкурирующие субстраты существенно отличались по реакционной способности, а значения скоростей расходования этих субстратов позволяли бы проводить их достаточно точную количественную оценку в условиях кинетического эксперимента. Соответственно, пары конкурирующих арилиодидов и арилбромидов подбирали таким образом, чтобы они удовлетворяли этим требованиям.
В условиях конкуренции относительно реакционноспособных 4-иодацетофенона и иодбензола при больших конверсиях 4-иодацетофенона на кинетической кривой иодбензола начинало наблюдаться небольшое ускорение (рис. 1). Несмотря на то, что методика оценки мгновенной скорости расходования по разностям концентраций неизбежно сопровождается ошибками, связанными, прежде всего, с ошибками экспериментального определения концентраций, можно уверенно утверждать, что максимальная скорость расходования иодбензола пришлась на временной промежуток 45–75 мин, внутри которого происходило практически полное расходование более реакционноспособного 4-иодацетофенона (рис. 1). В соответствии с приведенными выше рассуждениями и с учетом установленного необратимого протекания стадии с участием арилгалогенидов [5], ускорение расходования менее активного иодбензола однозначно указывает на участие конкурирующих арилиодидов в быстрой стадии каталитического цикла. Аналогичные закономерности наблюдались и в условиях конкуренции другой пары арилиодидов – 4-иодацетофенона и 4-иодтолуола (рис. 2). Скорость расходования последнего проходила через максимум, совпадающий по времени с расходованием более активного 4-иодацетофенона, подтверждая вывод о быстром характере окислительного присоединения в реакции прямого арилирования индола арилиодидами.

Рис. 1.
Зависимости концентраций конкурирующих 4-иодацетофенона (▲) и иодбензола (◼), а также зависимость скорости расходования иодбензола (◻) от времени в реакции (I).

Рис. 2.
Зависимости концентраций конкурирующих 4-иодацетофенона (▲) и 4-иодтолуола (◼), а также зависимость скорости расходования 4-иодтолуола (◻) от времени в реакции (I).

На основании имеющихся в литературе данных об относительной реакционной способности арилгалогенидов в реакциях сочетания [12] можно ожидать падения скорости окислительного присоединения в случае использования в качестве субстратов менее реакционноспособных арилбромидов, а, следовательно, и возможной смены природы скорость-определяющей стадии. Скорости реакций с арилбромидами в аналогичных условиях действительно были существенно ниже, чем с арилиодидами. Тем не менее в реакции арилирования индола(I) с конкурирующими 4-бромацетофеноном и бромбензолом на кинетической кривой расходования последнего наблюдалось ускорение, совпадающее с моментом практически полного израсходования более реакционноспособного 4-бромацетофенона (рис. 3), а зависимость скорости расходования бромбензола от времени проходила через максимум, совпадающий по времени с моментом полного расходования более активного 4-бромацетофенона. Ускорение расходования менее активного субстрата в момент полного израсходования более активного зафиксировано и для другой пары конкурирующих арилбромидов – 4-бромацетофенона и 4-броманизола (рис. 4).
Рис. 3.
Зависимости концентраций конкурирующих 4-бромацетофенона (▲) и бромбензола (◼), а также зависимость скорости расходования бромбензола (◻) от времени в реакции (I).
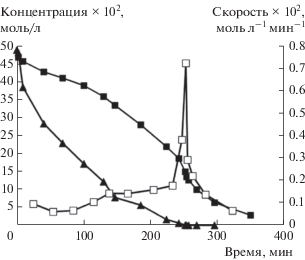
Рис. 4.
Зависимости концентраций конкурирующих 4-бромацетофенона (▲) и 4-броманизола (◼), а также зависимость скорости расходования 4-броманизола (◻) от времени в реакции (I).

Полученные результаты однозначно указывают на то, что, несмотря на снижение реакционной способности арилбромидов по сравнению с арилиодидами, арилбромиды продолжают участвовать не в медленной, а в быстрой стадии каталитического цикла. Таким образом, даже в случае использования менее реакционноспособных арилбромидов окислительное присоединение не является скорость-определяющей стадией каталитического цикла реакции прямого арилирования индола. Полученные данные указывают на возможность быстрой относительно других стадий каталитического цикла активации арилгалогенидов, включая неактивированные и дезактивированные арилбромиды, в “безлигандных” условиях реакции прямого арилирования. С учетом представленных в настоящей работе данных о быстром характере стадии с участием арилгалогенидов, а также ранее полученных данных о существенной обратимости стадии с участием молекулы индола [5] можно предположить, что скорость-определяющей является одна или совокупность нескольких мономолекулярных стадий, реализующихся в каталитическом цикле после последовательного вхождения в него арилгалогенида и индола (схема 1 , Г–Д).
Следует отметить, что экспериментально обоснованный в условиях реального катализа вывод о быстром протекании стадии активации арилбромидов (в том числе неактивированных) важен не только с фундаментальной точки зрения. Дело в том, что в литературе достаточно распространено мнение, что проблемы, связанные с низкой активностью неактивированных арилбромидов в реакциях сочетания, связаны с их неспособностью эффективно удерживать молекулярные комплексы Pd(0) в растворе в условиях отсутствия сильных органических лигандов [13]. Однако, как следует из полученных нами экспериментальных данных, основная часть палладия находится внутри каталитических циклов не в виде неустойчивых и склонных к агломерации интермедиатов Pd(0), а в виде интермедиатов Pd(II), образование которых происходит путем относительно быстрого окислительного присоединения арилгалогенида к Pd(0). В этом случае возможно некоторое снижение влияния дезактивации путем агломерации интермедиатов Pd(0) на функционирование каталитической системы и успешное применение “безлигандных” каталитических систем для конверсии малореакционноспособных арилбромидов в реакции прямого арилирования.
ЗАКЛЮЧЕНИЕ
С помощью анализа интегральных кинетических зависимостей расходования пары конкурирующих субстратов продемонстрирован быстрый характер стадии активации аригалогенидов (окислительного присоединения арилгалогенидов к нольвалентному палладию) в условиях “безлигандного” катализа реакции прямого арилирования индола не только в случае применения реакционноспособных арилиодидов, но и в случае неактивированных арилбромидов. С учетом полученных ранее и представленных в настоящей работе данных наиболее вероятной является гипотеза о скорость-определяющем характере одной (или совокупности нескольких) мономолекулярных стадий, реализующихся в каталитическом цикле после вхождения в него арилгалогенида и индола. Кроме того, полученные результаты показывают возможность успешного применения неактивированных арилбромидов в реакции прямого арилирования индола в условиях использования каталитических систем, не содержащих добавок сильных органических лигандов.
Список литературы
Lebrasseur N., Larrosa I. // Adv. Heterocycl. Chem. 2012. V. 105. P. 309.
Lane B.S., Brown M.A., Sames D. // J. Am. Chem. Soc. 2005. V. 127. P. 8050.
Joucla L., Batail N., Djakovitch L. // Adv. Synth. Catal. 2010. V. 352. P. 2929.
Tan Y., Barrios-Landeros F., Hartwig J.F. // J. Am. Chem. Soc. 2012. V. 134. P 3683.
Kurokhtina A.A., Larina E.V., Yarosh E.V., Lagoda N.A., Schmidt A.F. // Organometallics. 2018. V. 37. P. 2054.
Kim J., Hong S.H. // ACS Catal. 2017. V. 7. P. 3336.
Colletto C., Islam S., Julia-Hernandez F., Larrosa I. // J. Am. Chem. Soc. 2016. V. 138. P. 1677.
Rosner T., Le Bars J., Pfaltz A., Blackmond D.G. // J. Am. Chem. Soc. 2001. V. 123. P. 1848.
Шмидт А.Ф., Курохтина А.А., Смирнов В.В., Ларина Е.В., Чечиль Е.В. // Кинетика и катализ. 2012. Т. 53. № 2. С. 223.
Schmidt A.F., Kurokhtina A.A., Larina E.V. // Catal. Sci. Technol. 2014. V. 4. P. 3439.
Kurokhtina A.A., Larina E.V., Yarosh E.V., Schmidt A.F. // J. Mol. Catal. A. Chem. 2016. V. 425. P. 43.
Biffis A., Centomo P., Del Zotto A., Zecca M. // Chem. Rev. 2018. V. 118. P. 2249.
Kohler K., Kleist W., Prockl S.S. // Inorg. Chem. 2007. V. 46. P. 1876.
Дополнительные материалы отсутствуют.
Инструменты
Кинетика и катализ