

Кинетика и катализ, 2019, T. 60, № 4, стр. 499-508
Влияние модифицирования ланатаном на состояние и структурные особенности ванадия в нанесенных V2O5/SiO2-катализаторах
Т. С. Харламова a, *, Х. Х. Уразов a, О. В. Водянкина a
a ФГБОУ ВО Томский государственный университет
634050 Томск, просп. Ленина, 36, Россия
* E-mail: kharlamova83@gmail.ru
Поступила в редакцию 15.01.2019
После доработки 16.03.2019
Принята к публикации 20.03.2019
Аннотация
С применением методов электронной спектроскопии диффузного отражения, спектроскопии комбинационного рассеяния и температурно-программированного восстановления водородом изучено формирование поверхностных фаз в нанесенных V2O5/SiO2-катализаторах, модифицированных лантаном. Показано, что порядок нанесения активного компонента оказывает существенное влияние на фазовый состав и каталитические свойства образцов в реакции окислительного дегидрирования пропана.
ВВЕДЕНИЕ
Нанесенные ванадийоксидные системы привлекают большое внимание исследователей как катализаторы различных окислительно-восстановительных реакций, в том числе окислительного дегидрирования (ОДГ) легких углеводородов [1, 2], окисления метанола в формальдегид [3–5], формальдегида в муравьиную кислоту [6], этанола в ацетальдегид и уксусную кислоту [6–8], селективного восстановления NOx аммиаком [9–11]. Каталитические свойства таких систем определяются присутствием на поверхности двухмерных или трехмерных ванадийоксидных фаз, образование которых зависит от количества наносимого компонента, природы носителя, а также природы предшественника ванадия, используемого для приготовления образцов [1, 3, 12–15].
Образование на поверхности двухмерных VOx-фаз, чаще всего представляющих наибольший интерес для катализа, реализуется при нанесении активного компонента в пределах монослоя [1, 2, 14, 15]. Для большинства носителей монослойное покрытие поверхности VOx-фазами соответствует плотности покрытия 8–9 атомов V/нм2. Исключение составляет оксид кремния, для которого ввиду низкой реакционной способности поверхности характерно формирование фазы трехмерного V2O5 уже при плотности покрытия поверхности ~3 атома V/нм2.
Носитель влияет как на максимальную плотность покрытия поверхности двумерными VOx-фазами, так и на их реакционную способность [1–3, 8, 14, 15]. Для большинства окислительных реакций на нанесенных ванадиевых катализаторах наблюдается уменьшение активности в ряду носителей CeO2 > ZrO2 > TiO2 > Al2O3 > SiO2 [1–3, 14, 15]. Указанная зависимость реакционной способности нанесенных ванадиевых катализаторов от природы носителя отражает важную роль взаимодействия активного компонента с носителем и прочности образующейся мостиковой связи V–O–носитель в механизме и кинетике протекающих на них окислительных реакций. Полагают, что катионы оксидного носителя выступают в качестве лигандов во второй координационной сфере ванадия, изменяя окислительно-восстановительный потенциал поверхностных VOx-фаз [1, 2].
Для реакций, протекающих по последовательному механизму с образованием более реакционноспособного целевого продукта в качестве промежуточного, таких как ОДГ пропана, влияние носителя на окислительно-восстановительный потенциал поверхностных ванадийоксидных фаз сказывается не только на их реакционной способности в отношении активации молекулы исходного вещества, но и на протекании последующих реакций глубокого окисления целевого продукта [1, 2]. В этой связи наиболее подходящиe носители для ванадиевых катализаторов ОДГ пропана – Al2O3 и SiO2, для которых наблюдаются относительно высокие селективности при конверсии пропана до 10% [1, 16].
Аморфный оксид кремния является одним из широко используемых недорогих инертных носителей, обладающий высокой удельной площадью поверхности (Sуд) и развитой пористой структурой, регулируемой в широком диапазоне. Однако на его поверхности вероятность образования кристаллического V2O5, характеризующегося низкой селективностью в ОДГ легких алканов, выше, чем для Al2O3, TiO2, Nb2O5 или ZrO2, что приводит к падению селективности по отношению к целевому продукту при относительно малых количествах нанесенного ванадия. Это требует развития подходов к формированию активной и селективной поверхности V2O5/SiO2-катализаторов с высоким содержанием ванадия.
Grant и соав. показали [17, 18], что нанесение ванадия на оксид кремния, модифицированный натрием, позволяет повысить плотность покрытия поверхности носителя двумерными VOx-фазами без образования кристаллического оксида ванадия вплоть до 9 атомов V/нм2. При этом отмечалось, что добавка натрия не приводит к существенным изменениям каталитической активности и селективности катализаторов при низких молярных соотношениях натрия к ванадию (Na : V < 0.25). Это связано с влиянием добавки только на формирование двумерных ванадийоксидных фаз на поверхности оксида кремния и отсутствием воздействия малых количеств натрия на электронную структуру VOx [17]. Увеличение количества натрия в образце приводит к образованию фазы кристаллического метаванадата натрия, характеризующегося, как и V2O5, низкой селективностью [17, 18]. В то же время изменения, происходящие в первой и второй координационных сферах ванадия в поверхностных ванадийоксидных фазах в результате образования связей V–O–модификатор, могут влиять на их окислительно-восстановительный потенциал и каталитические свойства [19–22]. В этой связи интересно исследование добавок, оказывающих влияние как на формирование двумерных ванадийоксидных фаз на поверхности оксида кремния, так и на их окислительно-восстановительный потенциал.
Оксиды редкоземельных элементов, в частности оксиды церия и лантана, находят широкое применение в качестве модификаторов и промоторов различных катализаторов, в том числе никельоксидных катализаторов окислительного дегидрирования легких углеводородов [23–29]. При этом большую роль в улучшении свойств модифицированных образцов играет взаимодействие модификатора с носителем и/или активным компонентом. В тоже время использование оксидов церия и лантана в качестве модификаторов нанесенных ванадийоксидных катализаторов окислительного дегидрирования алканов остается малоизученным.
В представленной работе исследовано влияние модифицирования оксидом лантана нанесенных V2O5/SiO2-катализаторов при малых поверхностных плотностях наносимого ванадия на формирование в них поверхностных двумерных ванадийоксидных фаз, их структурные особенности, окислительно-восстановительные и каталитические свойства в реакции ОДГ пропана.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Приготовление образцов
Серия нанесенных V2O5/SiO2-катализаторов, модифицированных оксидом лантана, была приготовлена методом последовательной пропитки носителя по влагоемкости водными растворами предшественников. В качестве предшественников были взяты VOSO4 (“РЕАХИМ”, 99.0%) и La(NO3)3 ⋅ 6H2O (“ВЕКТОН”, 99.0%). В качестве носителя использовали силикагель (Sуд = 190 м2/г, Vпор = 0.61 см3/г) марки КСКГ (Россия), подвергнутый гидротермальной обработке для увеличения размера пор. Гидротермальную обработку проводили в водном растворе аммиака с последующим прокаливанием при 900°С [30]. Образцы после каждой пропитки прокаливали на воздухе при 500°С в течение 2 ч. Промежуточные однокомпонентные V2O5/SiO2 и La2O3/SiO2 применяли в качестве катализаторов сравнения. Номинальное массовое содержание оксида ванадия в образцах составляло 0.059 г V2O5 на 1 г SiO2, что соответствует плотности покрытия поверхности носителя ванадием 0.25 монослоя или 2 атомам V/нм2 носителя. Мольное отношение V : La = 1 : 1 во всех двухкомпонентных образцах. Использование совместной пропитки для приготовления катализаторов было затруднено вследствие нестабильности смешанного раствора предшественников (наблюдалось образование осадка и выделение газа).
Исследование физико-химических свойств образцов
Физико-химические свойства синтезированных образцов были исследованы комплексом методов: низкотемпературная адсорбция азота, рентгенофазовый анализ (РФА), УФ-видимая электронная спектроскопия диффузного отражения (ЭСДО), спектроскопия комбинационного рассеяния (КР), инфракрасная спектроскопия (ИК) и температурно-программируемое восстановление водородом (ТПВ-Н2).
Фазовый состав образцов исследовали методом РФА на дифрактометре XRD6000 (“Shimadzu”, Япония) с монохроматическим CuKα-излучением (λ = 1.54056 Å) в диапазоне углов 2θ = = 10°–60°. Полученные рентгенограммы анализировали с помощью базы данных PDF4.
Спектры ЭСДО получены на спектрометре Evolution 600 (“ThermoScientific”, США). В качестве стандарта использован MgO. Для регистрации спектров дегидратированных образцов их выдерживали в специальных реакторах с клапаном для обеспечения герметичности при 200°C в течение 2 ч на воздухе, а затем в течение 1.5 мин в вакууме (10–2 Торр). Спектры отражения исследуемых образцов переводили в спектры поглощения с помощью функции Кубелки–Мунка:
где R – коэффициент отражения вещества.КР-спектры синтезированных образцов снимали на конфокальным микроскопе комбинационного рассеяния Raman (“Renishaw”, Великобритания) с длиной волны 532 нм и анализировали с применением программы OMNIC (“Thermo Nicolet Corporation”, США).
Окислительно-восстановительные характеристики полученных образцов изучали методом ТПВ-Н2 на хемосорбционном анализаторе Chemisorb 2750 (“Micromeritics”, США) с детектором по теплопроводности. Все образцы перед началом опытов предварительно подвергали окислительной обработке в потоке воздуха (20 мл/мин) со скоростью нагрева 10 град/мин до 500°С. Все ТПВ-Н2-эксперименты проводили в температурном диапазоне от 25 до 800°С. Поток газа подавали со скоростью 20 мл/мин, скорость нагрева составляла 10 град/мин. Образующуюся в ходе реакции восстановления воду вымораживали до детектора в специальной ловушке, охлаждаемой смесью жидкого азота с изопропиловым спиртом при температуре –86°С.
Исследование каталитических свойств образцов
Каталитические свойства полученных образцов в реакции окислительного дегидрирования пропана исследовали в проточном реакторе со стационарным слоем катализатора в диапазоне температур от 500 до 600°С и атмосферном давлении. Анализ продуктов до и после реактора осуществляли в online режиме на хроматографе Кристалл 5000.2 (“Хроматэк”, Россия). Реактор представлял собой кварцевую трубку с внутренним диаметром 10 мм с коаксиально расположенным карманом для термопары с внешним диаметром 4 мм. В реактор помещали 0.20–0.25 г образца (фракция 0.25–0.5 мм, удельный объем 0.5–0.6 г/см3), разбавленного 1 см3 кварцевой крошкой (фракция 0.5–1.0 мм). Перед проведением эксперимента образцы подвергали окислительной предварительной обработке в потоке воздуха при 500°С в течение 30 мин. В качестве реакционной смеси использовали газовую смесь 5 об. % С3Н8 и 5 об. % О2 в азоте. Температуру в реакторе контролировали термопарой, размещенной в центре слоя катализатора.
Конверсию пропана Х(C3H8) и селективность по пропилену S(C3H6) рассчитывали по формулам:
Константы скорости определяли по уравнению первого порядка для реактора идеального вытеснения:
где t – время контакта, с; С 0 – начальная концентрация превращаемого вещества, об. %; С – конечная концентрация превращаемого вещества, об. %; $\text{v}$ – объемная скорость реакционной смеси, см3/с; V – объем катализатора, см3; x – степень превращения, доли. При расчете константы скорости окисления пропилена k2 полагали, что его начальная концентрация определяется конверсией пропана, а конечная – селективностью реакции ОДГ по пропилену.РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Состав и текстурные характеристики образцов
В табл. 1 представлены условные обозначения, состав и текстурные характеристики синтезированных образцов. Содержание наносимых компонентов в исследуемых объектах изменяется в интервалах 5.1–5.8 мас. % для V2O5 и 10.1–10.7 мас. % для La2O3. Уменьшение содержания оксида ванадия в La2O3/V2O5–SiO2 по сравнению с V2O5/SiO2 и оксида лантана в V2O5/La2O3–SiO2 по сравнению с La2O3/SiO2 связано с изменением доли соответствующего компонента в результате введения второго компонента. Различия в содержании оксидов лантана и ванадия в двухкомпонентных катализаторах связаны с экспериментальными погрешностями входе приготовления образцов.
Таблица 1.
Обозначение, состав и текстурные характеристики образцов
| Образец | Состав, мас. % | Sуд, м2/г | Vпор, см3/г | Dпор, нм | |
|---|---|---|---|---|---|
| V2O5 | La2O3 | ||||
| V2O5/SiO2 | 5.8 | – | 164 | 0.55 | 10.0 |
| La2O3/V2O5–SiO2 | 5.2 | 10.2 | 151 | 0.50 | 10.0 |
| La2O3/SiO2 | – | 10.7 | 168 | 0.54 | 9.7 |
| V2O5/La2O3–SiO2 | 5.6 | 10.1 | 163 | 0.49 | 9.3 |
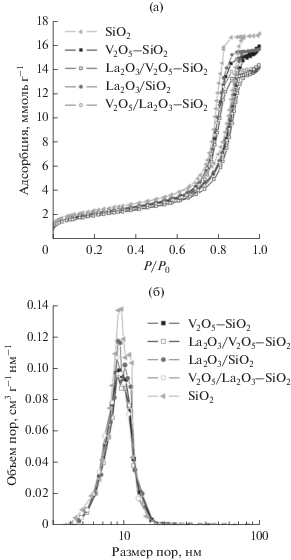
На рис. 1 приведены изотермы адсорбции–десорбции азота и распределение пор по размерам для синтезированных образцов и исходного носителя. Изотермы адсорбции–десорбции азота приготовленных катализаторов аналогичны изотерме исходного носителя с учетом изменения массы образцов в результате последовательного нанесения оксидов ланатана и ванадия (рис. 1а). Для всех образцов на изотерме адсорбции–десорбции наблюдается петля капиллярно-конденсационного гистерезиса в области относительных давлений 0.7–0.95 с перегибом при ~0.86, что говорит о наличии в них мезопор. Согласно данным о распределении пор по размерам пористая структура образцов и исходного носителя представлена порами с размером в интервале 4–20 нм. Наблюдаемые изменения в распределении пор по размерам говорят о равномерном заполнении пор носителя при последовательном нанесении компонентов (рис. 1б). Таким образом, представленные результаты свидетельствуют, что текстурные характеристики приготовленных катализаторов определяются текстурными характеристиками носителя. Все образцы имеют высокую удельную поверхность 151–168 м2/г и общий объем пор 0.49–0.55 см3/г при среднем диаметре пор ~10 нм (табл. 1).
Рис. 1.
Изотермы адсорбции–десорбции азота (а) и распределение пор по размерам (б) приготовленных образцов и исходного носителя.
На рис. 2 представлены данные РФА. Рентгенограмма образца V2O5/SiO2 аналогична рентгенограмме исходного SiO2 (на рис. 2 не приведена) и содержит широкое гало в интервале 2θ = 15°–35°, обусловленное наличием в образце аморфного оксида кремния. Рефлексов кристаллической фазы V2O5 обнаружено не было, что говорит о присутствии в образце ванадия в виде поверхностных ванадийоксидных фаз или высокодисперсного оксида ванадия, недоступного для наблюдения методом РФА. Модифицирование образца V2O5/SiO2 оксидом лантана приводит к появлению на рентгенограмме La2O3/V2O5–SiO2 широких рефлексов, отнесенных к фазе LaVO4 с моноклинной структурой (соответствующая штрих-диаграмма приведена на рис. 2). Уширение рефлексов фазы LaVO4 свидетельствует о ее высокой дисперсности и/или дефектности.
Рис. 2.
Рентгенограммы образцов V2O5/SiO2 (1), La2O3/V2O5–SiO2 (2), La2O3/SiO2 (3), V2O5/La2O3–SiO2 (4) и штрих-диаграммы ожидаемых кристаллических фаз V2O5, La2O3, LaVO4 и La2SiO5.
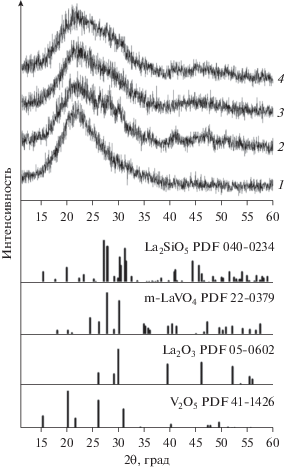
Рентгенограмма образца La2O3/SiO2 характеризуется присутствием широких гало: интенсивного в интервале 2θ = 15°–35° и слабоинтенсивного в интервале 2θ = 36°–55°. Рефлексы кристаллических фаз модификатора La2O3 или продуктов его взаимодействия с носителем – силикатов лантана – на рентгенограмме не идентифицированы. Однако наблюдаемые на рентгенограмме широкие пики не могут быть объяснены только аморфной фазой оксида кремния и связаны с образованием на поверхности носителя аморфного силиката лантана [31, 32]. Последующее нанесение оксида ванадия не приводит к существенным изменениям на рентгенограмме образца V2O5/La2O3–SiO2, что свидетельствует о присутствии ванадия в образце в виде высокодисперсных поверхностных оксидных фаз.
Исследование образцов методом КР-спектроскопии
На рис. 3 приведены КР-спектры гидратированных образцов в интервале 123–1050 см–1. В спектре носителя (не представлен) в указанном интервале никаких полос не наблюдали. В спектре образца V2O5/SiO2 имеются относительно узкие полосы при 130 и 178 см–1 и широкие полосы в интервалах 250–380, 410–740 и 750–980 см–1. Полосы при 130 и 178 см–1 обусловлены колебаниями решетки, а полосы в интервалах 250–380 и 450–740 см–1 относятся к колебаниям V–O–V-связей в гидратированных VOx-структурах на поверхности оксида кремния [33]. Полоса при 750–980 см–1 соответствует колебаниям связей V–O–Si в поверхностных силикатах ванадия [33].
Рис. 3.
КР-спектры гидратированных образцов: 1 – La2O3/SiO2; 2 – V2O5/La2O3–SiO2; 3 – V2O5/SiO2; 4 – La2O3/V2O5–SiO2.
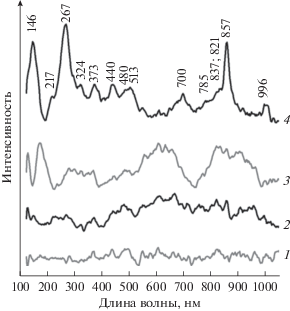
Модифицирование образца V2O5/SiO2 оксидом лантана приводит к существенному изменению вида спектра. В спектре La2O3/V2O5–SiO2 присутствуют полосы при 857, 837, 821, ~785, 440, 373, 324 и 217 см–1, относящиеся к ортованадату лантана LaVO4 [34, 35], а также полосы при 146, 480, 513, 706, 996 см–1, характерные для V2O5 [36, 37]. Уширение полос свидетельствует о высокой дисперсности и дефектности этих фаз. Учитывая данные РФА, можно заключить, что бóльшая часть ванадия находится в образце в виде ортованадата лантана, а оксид ванадия является примесной фазой.
В спектре образца La2O3/SiO2 видны только колебания сигнала на уровне шума. Полос, обусловленных колебаниями связей La–O, не наблюдается, что исключает присутствие фазы оксида лантана. В спектре V2O5/La2O3–SiO2 полос, характерных для кристаллических фаз LaVO4 и V2O5, не обнаружено [34–37]. На фоне шума различимы широкие полосы в интервале 400–980 см–1, которые были отнесены к колебаниям V–O–V-связей в гидратированных поверхностных VOx-структурах на поверхности оксида кремния и связей V–O–Si в поверхностных силикатах ванадия [33]. Однако интенсивность соответствующих полос, особенно в области 750–980 см–1, ниже, чем в спектре образца V2O5/SiO2, что может быть связано с наличием в образце лантана. Присутствие лантана на поверхности носителя может препятствовать образованию связей V–O–Si, а также способствовать формированию LaVOx-структур на поверхности оксида кремния.
Исследование образцов методом ЭСДО
На рис. 4 представлены электронные спектры гидратированных и дегидратированных образцов в диапазоне 200–900 нм. В спектре La2O3/SiO2 наблюдается слабая полоса в интервале 200–280 нм, которая не вносит существенный вклад в спектры ванадийсодержащих образцов. В спектрах всех ванадийсодержащих образцов в интервале 200–600 нм обнаружены полосы переноса заряда (ППЗ) металл–лиганд V(V) [38, 39]. Малоинтенсивные полосы в диапазоне 700–900 нм в спектрах V2O5/SiO2 и La2O3/V2O5–SiO2 обусловлены d-d-переходами в ванадии(IV), присутствующем в образцах в виде примеси в результате частичного восстановления V(V). В спектре V2O5/La2O3–SiO2 полос, обусловленных d-d-переходами в V(IV), не наблюдается, что говорит о стабилизации в нем ванадия(V). Вид ППЗ V(V) в спектрах образцов, зависящий от локального окружения ванадия, различен и определяется способом приготовления катализаторов и условиями получения спектров.
Рис. 4.
Спектры ЭСДО гидратированных (а) и дегидратированных (б) образцов, а также образцов V2O5/SiO2 (в) и V2O5/La2O3–SiO2 (г) после дегидратации (черный спектр) и последующего взаимодействия с влажным воздухом (серые спектры). На частях (б) и (г) рисунка стрелкой показано увеличение интенсивности полосы при 400 нм со временем при взаимодействии с влажным воздухом.
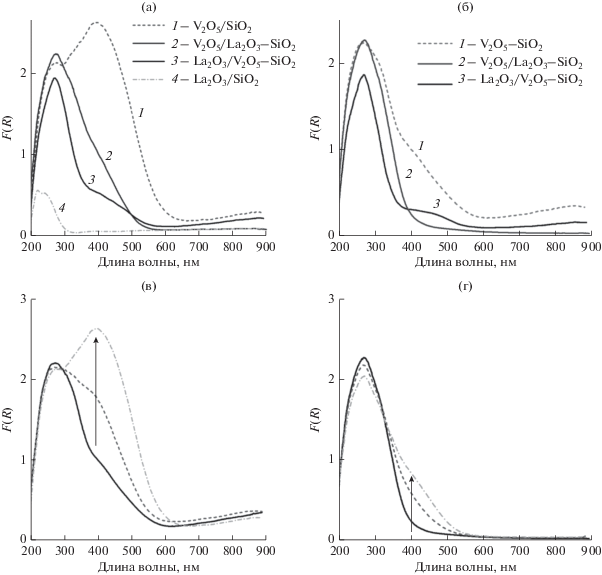
Спектр исходного (гидратированного) образца V2O5/SiO2 характеризуется интенсивным поглощением в интервале 200–600 нм с ярко выраженной полосой при ~400 нм, связанной с ванадием в VO6 и/или VO5 (рис. 4а). Дегидратация приводит к снижению интенсивности вышеуказанной полосы, при этом ярко выраженной становится полоса при 270 нм, характерная для ванадия в изолированных тетраэдрах VO4. В интервале 350–600 нм остаются полосы поглощения, которые могут быть обусловлены ППЗ в VO6/VO5 в поверхностных структурах, присутствующих в образце вследствие его быстрой гидратации в ходе исследования, или в микрокристаллическом V2O5. Отсутствие в КР-спектре полос, характерных для V2O5, позволяет отнести полосы в интервале 350–600 нм в спектре ЭСДО дегидратированного образца к ППЗ в VO6/VO5 в поверхностных структурах. Последующее взаимодействие образца с влажным воздухом приводит к обратному увеличению интенсивности полосы в области 400 нм, что свидетельствует об обратимости процесса гидратации–дегидратации (рис. 4б). Изменение вида спектра в результате гидратации–дегидратации образца говорит о формировании в образце V2O5/SiO2 преимущественно поверхностных ванадийоксидных структур, доступных для взаимодействия с водой [38].
Модифицирование V2O5/SiO2 оксидом лантана приводит к существенному изменению вида спектров ЭСДО. Спектры гидратированного и дегидратированного образца La2O3/V2O5–SiO2 практически идентичны и характеризуются интенсивной полосой при 270 нм, обусловленной V(V) в изолированных тетраэдрах VO4, а также относительно слабым поглощением в области 350–550 нм, связанным с ППЗ V(V) в VO6/VO5. Неизменность спектра в ходе дегидратации–гидратации свидетельствует о присутствии в нем ванадия в стабильных кристаллических фазах, что согласуется с данными РФА и КР-спектроскопии, подтверждающими формирование микрокристаллических фаз ортованадата лантана LaVO4 и оксида ванадия V2O5.
Спектр исходного образца V2O5/La2O3–SiO2 также характеризуется интенсивной полосой при 270 нм, обусловленной ванадием(V) в изолированных тетраэдрах VO4, с плечом при 400 нм, связанным с ППЗ V(V) в VO6. Дегидратация образца приводит к полному исчезновению полосы при 400 нм, однако в ходе последующего его взаимодействия с влажным воздухом в спектре наблюдается ее обратное появление (рис. 4г), что говорит о присутствии ванадия в дегидратированном образце только в изолированных VO4-тетраэдрах, из которых часть остается доступной для взаимодействия с водой. Интенсивная полоса при 270 нм в спектре гидратированного образца свидетельствует о стабилизации в нем части ванадия в изолированных тетраэдрах VO4, что может быть обусловлено наличием лантана во второй координационной сфере ванадия. Однако согласно данным КР-спектроскопии образование фазы ортованадата лантана LaVO4 в образце не происходит. Таким образом, изменение локальной структуры ванадия в ходе взаимодействия с водой указывает на формирование в образце V2O5/La2O3–SiO2 не стабильных трехмерных фаз, а некоторых поверхностных двумерных LaVOx-структур.
Исследование образцов методом ТПВ-Н2
На рис. 5 представлены профили ТПВ-Н2 исследуемых катализаторов. Профили всех образцов характеризуются двумя пиками поглощения водорода при температурах выше 450°C. Профиль ТПВ-Н2 образца V2O5/SiO2 содержит относительно интенсивный ассиметричный пик с максимумом при 613°C и плечом при 550°C, а также небольшой пик с максимумом при 886°C. Низкотемпературный пик можно отнести к восстановлению поверхностных VOx-фаз [39, 40], в то время как высокотемпературный пик, в соответствии с данными КР-спектроскопии, может быть обусловлен восстановлением ванадия из поверхностного силиката ванадия. В целом, количество водорода, поглощенного при восстановлении образца V2O5/SiO2, невысоко по сравнению с другими катализаторами, что, согласно данным ЭСДО, может быть связано с присутствием в образце относительно большого количества ванадия(IV).
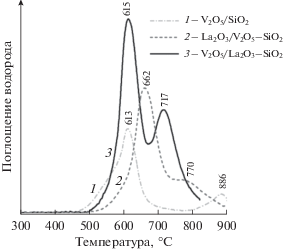
На профиле ТПВ-Н2 образца La2O3/V2O5–SiO2 наблюдаются интенсивный пик при 662°C и менее интенсивный пик при 770°C, которые определяются восстановлением дисперсных кристаллических фаз ортованадата лантана LaVO4 и V2O5 [41–44]. В целом количество водорода, поглощенного при восстановлении La2O3/V2O5–SiO2, выше, чем при восстановлении V2O5/SiO2, что, вероятно, обусловлено стабилизацией ванадия(V) в кристаллических фазах V2O5 и LaVO4.
Профиль ТПВ-Н2 образца V2O5/La2O3–SiO2 характеризуется двумя пиками поглощения водорода с максимумами при 613 и 717°С. Температура максимума первого пика аналогична таковой пика поглощения водорода на профиле V2O5/SiO2. В этой связи вышеуказанный пик может быть отнесен к восстановлению ванадия из поверхностных LaVOx-структур, доступных для гидратации. В отличие от V2O5/SiO2 для образца V2O5/La2O3–SiO2 на профиле ТПВ-Н2 не наблюдается плеча при 550°C, что говорит об отсутствии в нем легковосстанавливающихся VOx-форм. Пик с максимумом при 717°C соответствует восстановлению ванадия из поверхностных LaVOx-структур с лантаном во второй координационной сфере, не доступных для гидратации. При этом количество водорода, поглощенного при восстановлении V2O5/La2O3–SiO2, выше, чем при восстановлении образцов V2O5–SiO2 и La2O3/V2O5–SiO2, что согласуется с данными ЭСДО об отсутствие в этом образце ванадия(IV) и свидетельствует о еще большей стабилизации ванадия(V) в образующихся поверхностных LaVOx-фазах.
Исследование каталитических свойств образцов
На рис. 6 представлены результаты каталитических исследований ванадийсодержащих образцов в реакции окислительного дегидрирования пропана. Согласно полученным данным в присутствии всех катализаторов конверсия пропана была невысокой (менее 2%) в интервале температур 500–600°C, что связано с низким содержанием в них ванадия (рис. 6а). Наибольшей активностью обладает V2O5/SiO2. Скорость реакции для него при 500°C сопоставима с таковой для схожего образца в работах Grant с соавт. [17, 18]. Образцы, модифицированные оксидом лантана, характеризуются пониженной активностью, при этом образование кристаллических фаз в La2O3/V2O5–SiO2 образце приводит к более сильному падению активности, что согласуется с меньшей реакционной способностью образцов в реакции восстановления водородом. Для всех образцов наблюдается рост активности с повышением температуры.
Рис. 6.
Зависимости скорости реакции и конверсии пропана от температуры для различных катализаторов (а); зависимости селективности образования пропилена, констант скорости последовательного превращения пропана и их соотношения от состава катализатора при 600°C (б).
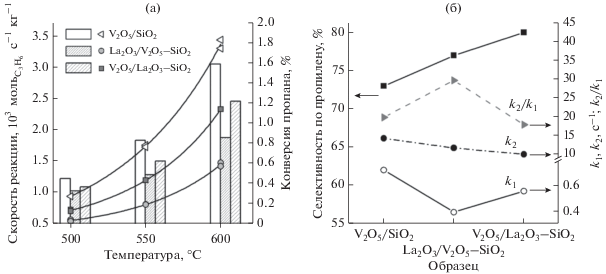
На рис. 6б приведены значения селективности по пропилену для исследуемых катализаторов при 600°C. Ввиду различий конверсии пропана для образцов в 1.3–1.8 раз корректное сравнение полученных значений селективности не представляется возможным. Для анализа вклада неселективного превращения пропана были определены константы скорости последовательных превращений:
Вкладом прямого окисления пропана в продукты глубокого окисления пренебрегали [1]. Значения соответствующих констант скорости и их отношения k2/k1 представлены на рис. 6б. Величина k2/k1 уменьшается в ряду La2O3/V2O5–SiO2$ \gg $ $ \gg $ V2O5/SiO2 > V2O5/La2O3–SiO2, что соответствует росту селективности по пропилену. Из сравнения констант скорости и их отношения k2/k1 следует, что образование кристаллических фаз LaVO4 и V2O5 в образце La2O3/V2O5–SiO2 приводит не только к заметному падению его активности, но и селективности по пропилену. Образование поверхностных фаз LaVOx в катализаторе V2O5/La2O3–SiO2 сопровождается повышением его селективности по пропилену на фоне умеренного снижения активности по сравнению с немодифицированным V2O5/SiO2. Это может быть связано с отсутствием высокоактивных, но не селективных центров на поверхности образца V2O5/La2O3–SiO2, характеризующихся по данным ТПВ-H2 наиболее низкими температурами восстановления.
ЗАКЛЮЧЕНИЕ
Результаты проведенных исследований модифицирования оксидом лантаном катализаторов V2O5/SiO2 при малых поверхностных плотностях ванадия показали, что состав и строение нанесенных фаз, образующихся в модифицированных образцах, а также их каталитические свойства определяются порядком введения наносимых компонентов. Модифицирование нанесенного катализатора V2O5/SiO2 способствует формированию в образце микрокристаллических фаз LaVO4 и V2O5, что приводит к ухудшению каталитических свойств (заметному снижению активности и селективности образца) по сравнению с исходным катализатором. В то же время нанесение оксида ванадия на предварительно модифицированный носитель La2O3/SiO2 способствует образованию в модифицированном образце поверхностных LaVOx-фаз, что сопровождается повышением его селективности по пропилену на фоне умеренного снижения активности по сравнению с немодифицированным образцом. Учитывая достаточно высокое мольное отношение La/V в исследуемых катализаторах, следует ожидать дальнейшего улучшения каталитических свойств образцов, полученных нанесением оксида ванадия на предварительно модифицированный носитель La2O3/SiO2, при снижении мольного отношения La/V, в том числе в результате увеличения поверхностной плотности ванадия.
Таким образом, в настоящей работе показано, что введение лантана в нанесенные катализаторы V2O5/SiO2 путем модифицирования поверхности носителя может рассматриваться в качестве перспективного подхода для формирования активных и селективных ванадийоксидных катализаторов на силикагельном носителе, характеризующихся высокой плотностью покрытия поверхности носителя двумерными VOx-фазами.
БЛАГОДАРНОСТИ
Статья написана в рамках научного проекта (№ НУ 8.2.03.2018 Л), выполненного при поддержке Программы повышения конкурентоспособности ТГУ.
Список литературы
Carrero C.A., Wachs I.E. // Amer. Chem. Soc. 2014. V. 5. P. 3357.
Grant J.T., Venegas J.M., McDermott W.P., Hermans I. // Chem. Rev. 2018. V. 118. P. 2769.
Deo G., Wachs I.E. // J. Catal. 1994. V. 146. P. 323.
Bulushev D.A., Ross J.R.H. // ChemSusChem. 2018. V. 11. P. 821.
Каичев В.В., Попова Г.Я., Чесалов Ю.А., Сараев А.А., Андрушкевич Т.В., Бухтияров В.И. // Кинетика и катализ. 2016. Т. 57. № 1. С. 84.
Андрушкевич Т.В., Чесалов Ю.А. // Успехи химии. 2018. Т. 87. № 6. С. 586.
Autthanit C., Praserthdam P., Jongsomjit B. // J. Env. Chem. Eng. 2018. V. 6. P. 6516.
Andrushkevich T.V., Kaichev V.V., Chesalov Yu.A., Saraev A.A., Buktiyarov V.I. // Catal. Today. 2017. V. 279. P. 95.
Xua J., Chena G., Guoa F., Xie J. // Chem. Eng. J. 2018. V. 353. P. 507.
Zhang J., Li X., Chen P., Zhu B. // Materials. 2018. V. 11. P. 1632.
Liua Z., Zhanga S., Li J. // Appl. Catal. B: Environmental. 2014. V. 158–159. P. 11.
Artiglia L., Agnoli S., Granozzi G. // Coordination Chem. Rev. 2015. V. 301–302. P. 106.
Weckhuysen B.M., Keller D.E. // Catal. Today. 2003. V. 78. P. 25.
Burcham L.J., Wachs I.E. // Catal. Today. 1999. V. 49. P. 467.
Gao X., Wachs I.E. // J. Phys. Chem. B. 2000. V. 104. P. 1261.
Fukudome K., Suzuki T. // Catal. Surv. Asia. 2015. V. 19. P. 172.
Grant J.T., Carrero A.C., Love A.M., Verel R. // Amer. Chem. Soc. 2015. V. 5. P. 5787.
Grant J.T., Carrero A.C. // Topics in Catal. 2016. V. 59. P. 1545.
Hoj M., Kessler T., Beato P., Jensen A.D., Grunwaldt J.-D. // Applied Catalysis A: General. 2014. V. 472. P. 29.
Yang S., Iglesia E., Bell A.T. // J. Phys. Chem. 2005. V. 109. P. 8987.
Kharlamova T., Sushchenko E., Izaak T., Vodyankina O. // Catal. Today. 2016. V. 278. P. 174.
Сущенко Е.Д., Харламова Т.С., Изаак Т.И., Водянкина О.В. // Кинетика и катализ. 2017. Т. 58. № 5. С. 642.
Brussinoa P., Bortolozzia J.P., Dalla Costab B., Banúsa E.D., Ulla M.A. // Appl. Catal. A: General. 2019. V. 575. P. 1.
Wang X., Kang Y., Cui D., Li J., Li D. // Catal. Commun. 2019. V. 118. P. 39.
Ismagilov Z.R., Matus E.V., Ismagilov I.Z., Sukhova O.B., Yashnik S.A., Ushakov V.A., Kerzhentsev M.A. // Catal. Today. 2019. V. 323. P. 166.
Shamskar F.R., Meshkani F., Rezaei M. // J. of CO2 Utilization. 2017. V. 22. P. 124.
Liu B.S., Rui G., Chang R.Z., Au C.T. // Appl. Catal. A: General. 2008. V. 335. P. 88.
Iglesias-Juez A., Martínez-Huerta M.V., Rojas-García E., Jehng J.-M., Bañares M.A. // J. Phys. Chem. C. 2018. V. 122. P. 1197.
Khan M.Y., Al-Ghamdi S., Razzak S.A., Hossain M.M., de Lasa H. // Mol. Catal. 2017. V. 443. P. 78.
Бугрова Т.В., Литвякова Н.Н., Мамонтов Г.В. // Кинетика и катализ. 2015. Т. 56. № 6. С. 746.
Vidal H., Bernal S., Baker R.T., Finol D., P’erez Omil J.A., Pintado J.M., Rodr’ıguez-Izquierdo J.M. // J. Catal. 1999. V. 183. P. 53.
Kharlamova T., Vodyankina O., Matveev A., Stathopoulos V., Ishchenko A., Khabibulin D., Sadykov V. // Ceramics International. 201. V. 41. P. 13393.
Wang Ch.-B., Deo G., Wachs I.E. // Journal of Catalysis. 1998. V. 178. P. 640.
Jia C.-J., Sun L.-D., You L.-P., Jiang X.-Ch., Luo F., Pang Y.-Ch., Yan Ch.-H. // J. Phys. Chem. B. 2005. V. 109. P. 3284.
Antic-Fidancevt E., Holsa J. // J. Phys.: Condens. Matter. 1991. V. 3. P. 682.
Sanchez C., Livage J., Lucazeau G. // J. Raman Spectrometry. 1982. V. 12. P. 68.
Baddour-Hadjean R., Smirnov M.B., Smirnov K.S., Kazimirov V.Yu, Gallardo-Amores J.M., Amador U., Arroyo-de Dompablo M.E., Pereira-Ramos J.P. // Inorg. Chem. 2012. V. 51. P. 3194.
Gao X., Bare S.R., Weckhuysen B.M. // J. Phys. Chem. 1998. V. 102. P. 10842.
Gao X., Wachs I.E. // J. Catal. 1999. V. 192. P. 18.
Gao X., Bare S.R., Fierro J.L.G., Wachs I.E. // J. Phys. Chem. B. 1999. V. 103. P. 618.
Arena F., Giordano N., Parmaliana A. // J. Catal. 1997. V. 167. P. 66.
Koranne M.M., Goodwin J.G., Marcelin G. // J. Catal. 1994. V. 148. № 1. P. 369.
Hou T., Yang H., Fan X., Zhang X. // Catal. Lett. 2011. V. 141. P. 1215.
Varma S., Ambekar B.R., Wani B.N., Gupta N.M. // Thermochim. Acta. 2003. V. 405. P. 279.
Дополнительные материалы отсутствуют.
Инструменты
Кинетика и катализ