

Кинетика и катализ, 2019, T. 60, № 6, стр. 776-781
Катализ разложения гидропероксида кумила солями и комплексами кальция: кинетический эксперимент и квантово-химическое моделирование
Н. М. Нуруллина a, *, Н. Н. Батыршин a, Ю. Х. Усманова a, А. Н. Маслий a, Х. Э. Харлампиди a
a ФГБОУ ВО Казанский национальный исследовательский технологический университет
420015 Казань, ул. К. Маркса, 68, Россия
* E-mail: nyryllina@mail.ru
Поступила в редакцию 11.03.2019
После доработки 02.07.2019
Принята к публикации 09.07.2019
Аннотация
Экспериментально получены кинетические и термодинамические параметры процесса разложения гидропероксида кумила в среде хлорбензола в присутствии 2-этилгексаноата кальция и комплекса CaCl2 с краун-эфиром. Кинетически установлено образование промежуточных комплексов в системе катализатор–гидропероксид. С помощью квантово-химического моделирования подтверждено формирование промежуточных комплексов, оптимизирована их структура и найдены термодинамические характеристики реакций с их участием. Предложен механизм постадийного распада интермедиата.
ВВЕДЕНИЕ
В развившемся процессе окисления алкилароматических углеводородов молекулярным кислородом соединения непереходных металлов, разлагая небольшую часть гидропероксида, заметно ускоряют процесс [1–3]. Недостатком использования таких соединений является их малая растворимость в алкилароматических углеводородах и склонность к образованию коллоидных растворов [1].
Необходимость сравнить каталитическую активность различных металлов в идентичных условиях и связать ее с атомными свойствами металлов привела нас к поиску лиганда, придающего растворимость и одинаково подходящего для всех непереходных металлов. Для металлов IIb подгруппы таким лигандом оказался остаток 2-этилгексановой кислоты [4]. Однако было обнаружено, что этилгексаноаты не всех интересующих нас металлов растворимы в хлорбензоле – стандартном растворителе для изучения реакций разложения гидропероксидов. Был найден более универсальный лиганд – краун-эфир, одним из главных преимуществ которого является возможность перевода первоначально нерастворимых в органической среде солей в раствор [5–7].
Это позволило нам подробно изучить распад гидропероксида третичного бутила в присутствии комплексов хлоридов металлов IIa подгруппы периодической системы [8].
В настоящей статье представлены результаты работы по исследованию каталитического разложения гидропероксида кумила (ГПК) в присутствии 2-этилгексаноата кальция (Са(ЭГ)2) и комплекса хлорида кальция с дибензо-18-краун-6 эфиром (СаСl2 · ДБК).
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Во избежание уноса растворителя каталитический распад ГПК изучали ампульным методом [8] при [ГПК]0 = 0.1–1.5 моль/л, [Кт]0 = (0.5–7) × 10–3 моль/л в температурном интервале 100–130°С. За концентрацией гидропероксида следили йодометрическим методом [9]. Кинетические кривые аппроксимировали кубическим полиномом и дифференцировали его по времени. Начальные скорости распада определяли как производную при τ → 0.
Гидропероксид кумила очищали через натриевую соль [10]. Концентрация ГПК составляла 6.98 моль/л (99.8%). Растворитель (хлорбензол) подвергался очистке кислотно-щелочным методом [11]. Подробная методика получения 2-этилгексаноатов описана в работе [12]. В отличие от нафтенатов и стеаратов этилгексаноаты не склонны к самоассоциации и гомогенны в органических растворах в широком диапазоне концентраций и температур.
Квантово-химические расчеты осуществляли в программном пакете Priroda [13] с применением метода теории функционала плотности PBE [14]. Для описания атомных орбиталей использовали базисный набор 6-31G(d,p) [15]. Была проведена оптимизация всех геометрических параметров, после чего рассчитаны частоты нормальных колебаний. Отсутствие мнимых мод свидетельствовало о достижении минимума энергии. С помощью стандартной процедуры термохимических расчетов в рамках программного пакета Priroda были вычислены значения энтальпии, энтропии и свободной энергии Гиббса для исследуемых соединений.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
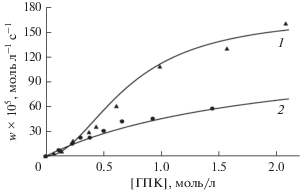
Как видно из рис. 1, соединения кальция ускоряют распад рассматриваемого гидропероксида. При увеличении концентрации гидропероксида [ГПК]0 зависимость скорости распада стремится к предельному значению. В представленном интервале концентраций кривая, характеризующая скорость процесса в присутствии СaCl2 · ДБК, имеет S-образный характер и выходит на плато первой. Выход зависимостей на плато (рис. 1) является кинетическим признаком образования промежуточного комплекса гидропероксида с катализатором [16].
Рис. 1.
Зависимость скорости распада гидропероксида кумила от концентрации ГПК в присутствии различных катализаторов ([Кт]0 = 5 × 10–3 моль/л, Т = = 130°С, хлорбензол): 1 – CaCl2 ⋅ ДБК; 2 – Са(ЭГ)2.
Порядок реакций по катализатору для рассматриваемых систем из линейности зависимостей в координатах ln(w0)–ln([Кт]0) определен как близкий к первому.
Формально-кинетическую схему распада гидропероксида можно представить следующим образом:
где n – число молекул гидропероксида, связанных в комплекс; K – константа равновесия образования комплекса, k – константа скорости распада комплекса.Обработка формально-кинетической схемы в квазиравновесном приближении приводит для скорости брутто-распада ГПК (w0) к уравнению типа Михаэлиса–Ментен:
(1)
${{w}_{0}} = \frac{{kK{{{[{\text{Кт}}]}}_{0}}[{\text{ГПК}}]_{0}^{n}}}{{1 + K[{\text{ГПК}}]_{0}^{n}}}.$Как видно из этого уравнения, скорость разложения в зависимости от концентрации гидропероксида описывается гиперболическим законом и стремится к предельному значению.
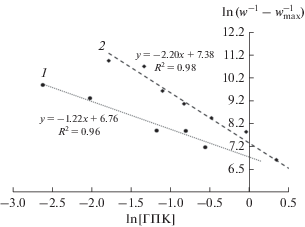
Для определения стехиометрии комплексов воспользовались уравнением [17]:
(2)
$\ln \left( {\frac{1}{w} - \frac{1}{{{{w}_{{\max }}}}}} \right) = \ln \left( {\frac{1}{{{{w}_{{\max }}}K}}} \right) - n \times \ln {{\left[ {{\text{ГПК}}} \right]}_{0}},$Из тангенсов угла наклона соответствующих прямых в координатах уравнения (2) найдено число молекул ГПК, входящих в промежуточный комплекс. Оказалось, что Са(ЭГ)2 удерживает только одну молекулу ГПК. В состав комплекса с СaCl2 · ДБК входят две молекулы гидропероксида (рис. 2).
При формировании комплексов органического гидроперокида с солями металлов II группы периодической системы возможно образование водородной связи по атомам кислорода кислотного остатка. Теоретически возможно присоединение до четырех молекул гидропероксида к одной молекуле карбоксилата кальция. Однако, опираясь на приведенные выше кинетические данные, можно предположить, что при образовании комплексов органического гидроперокида с солями металлов II группы периодической системы сначала возникает водородная связь гидропероксида кумила с кислородом кислотного остатка, а после этого замыкается шестичленный цикл с формированием связи металл–кислород. В результате образуется комплекс Са(ЭГ)2 : ГПК = 1 : 1 (схема 1 ):
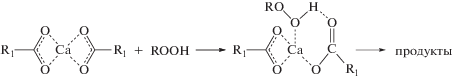
Схема 1 .
Формирование комплексов с участием краун-эфиров должно быть облегчено возможностью образования многочисленных водородных связей с атомами короны эфира и координационных связей с центральным атомом металла.
Кинетические параметры брутто-распада (k, K) определяли линеаризацией уравнения (1) по методу Лайнуивера–Берка [18]:
(3)
$\frac{1}{w} = \frac{1}{{{{w}_{{\max }}}}} + \frac{1}{{{{w}_{{\max }}}K}} \times \frac{1}{{\left[ {{\text{ГПК}}} \right]_{0}^{n}}}.$Далее по температурной зависимости константы скорости разложения комплекса определяли активационные параметры распада.
Ниже приведены полученные аррениусовские выражения для константы скорости в среде хлорбензола для Са(ЭГ)2:
(4)
$k = 1.82 \times {{10}^{{10}}} \times \exp \left( { - \frac{{81.55 \times {{{10}}^{3}}}}{{RT}}} \right)\,\,({{{\text{с}}}^{{ - 1}}})$(5)
$k = 5.44 \times {{10}^{{15}}} \times \exp \left( { - \frac{{125.26 \times {{{10}}^{3}}}}{{RT}}} \right)\,\,({{{\text{с}}}^{{ - 1}}}).$Значения термодинамических характеристик реакции комплексообразования представлены в виде соответствующих уравнений Вант-Гоффа для Са(ЭГ)2:
(6)
$\ln K = \frac{{65.09}}{{RT}} - \frac{{172.37}}{R}\,\,({{\text{л}} \mathord{\left/ {\vphantom {{\text{л}} {{\text{моль}}}}} \right. \kern-0em} {{\text{моль}}}})$(7)
$\ln K = \frac{{74.28}}{{RT}} - \frac{{179.78}}{R}\,\,({{{{{\text{л}}}^{2}}} \mathord{\left/ {\vphantom {{{{{\text{л}}}^{2}}} {{\text{мол}}{{{\text{ь}}}^{2}}}}} \right. \kern-0em} {{\text{мол}}{{{\text{ь}}}^{2}}}}).$Отметим, что, как и для других металлов II группы периодической системы, для соединений кальция комплексообразование – реакция экзотермическая [16], поэтому с повышением температуры равновесие смещается в обратную сторону и концентрация комплекса уменьшается.
Молекулы гидропероксида связаны в комплексе с атомами кислорода катализатора водородными связями. Для всех исследованных соединений теплота комплексообразования значительно превышает прочность водородных связей в гидропероксидах (8–13 кДж/моль). Следовательно, один из атомов кислорода О–О-группы гидропероксида координируется еще и с центральным атомом металла.
Образование водородных связей ослабляет связь О–О в гидропероксиде и облегчает ее гомолитический разрыв. Отметим, что для всех рассматриваемых катализаторов энергия активации разложения Ea значительно меньше прочности связи О–О в гидропероксидах (167 кДж/моль) [19], и такое снижение Ea не может быть объяснено лишь возникновением водородных связей. Этот факт является еще одним свидетельством координации ГПК с металлом.
Разложение гидропероксида в присутствии краун-эфирного катализатора протекает последовательно, в системе происходит сложная цепь реакций распада и формирования комплексов различной стехиометрии, которую можно выразить следующей схемой (схема 2 ) (для упрощения образование продуктов не показано):
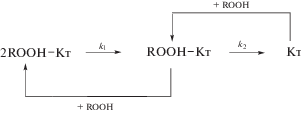
k1 – константа скорости распада комплекса 2ROOH‒Кт; k2 – константа скорости распада комплекса ROOH‒Кт. Схема 2 .
Из схемы 2 видно, что в случае использования краун-эфирного катализатора одновременно могут распадаться два комплекса различной стехиометрии – ROOH–СaCl2·ДБК и 2ROOH–СaCl2 · ДБК, что приводит к более высоким наблюдаемым значениям констант скоростей распада (рис. 1), чем при катализе Са(ЭГ)2.
В ходе квантово-химических расчетов в первую очередь были оптимизированы структуры реагентов: молекул ГПК, Са(ЭГ)2, СаCl2 · ДБК. На основании анализа структур этих соединений нами были предложены и смоделированы структуры комплексов катализатора с гидропероксидом (интермедиатов).
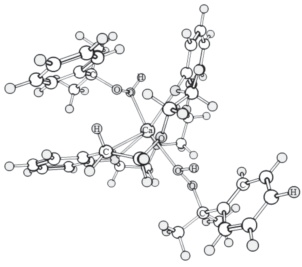
Оптимизированная геометрическая структура промежуточного комплекса ГПК–Са(ЭГ)2 приведена на рис. 3. Как и предполагалось, образуется шестичленный цикл с участием иона кальция. Соответствие этого интермедиата реакции (I) было проверено методом расчета внутренней координаты реакции (IRC). Расчеты спусков в обе стороны привели, соответственно, к структурам реагентов и продуктов реакции, идентичных оптимизированным ранее.
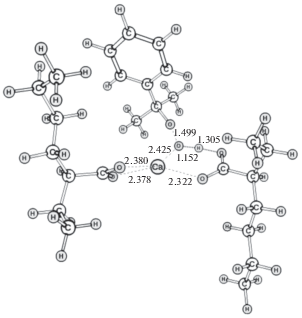
На рис. 4 представлена оптимальная конформация промежуточного комплекса с участием ГПК и катализатора, содержащего краун-эфир. Интересно отметить, что здесь молекулы гидропероксида через свои гидроксильные атомы кислорода непосредственно связываются с атомом металла, не формируя дополнительных водородных связей и циклических структур. Причем одна из молекул ГПК присоединена со стороны атомов кислорода “короны”, а другая – с тыльной стороны краун-эфира, где возможности возникновения водородных связей не имеется.
С помощью специальных расчетов показано, что сам краун-эфир удерживает в водородносвязанном комплексе две молекулы ГПК и обе – со стороны атомов кислорода “короны”. Таким образом, введение иона кальция (вероятно, и любого другого металла) во внутреннюю полость краун-эфира в силу больших размеров центрального иона и разветвленности молекулы ГПК создает стерические препятствия для образования водородных связей гидропероксида с кислородными атомами краун-эфира.
По данным квантово-химических расчетов для всех катализаторов изменение энтальпии в реакции образования промежуточного комплекса (реакция (I)) отрицательное, что согласуется с экспериментальными данными. Так, вычисленная энтальпия образования равна –117.88 кДж/моль для комплекса ГПК–Са(ЭГ)2 и –103.40 кДж/моль для комплекса 2ГПК–СаСl2 ⋅ ДБК.
Принимая во внимание эффект сольватации, в рамках проведенного исследования нами были получены значения ΔGs сольватации компонентов реакционной смеси, на основе которых было найдено изменение свободной энергии Гиббса с учетом сольватации молекул в хлорбензоле. В табл. 1 представлены значения ΔG реакций образования и распада комплекса (для 2-этилгексаноата кальция).
Таблица 1.
Сравнение свободной энергии Гиббса основных реакций в газовой фазе и в растворителе
| Условия проведения реакции | Реакция | Параметр | |
|---|---|---|---|
| ΔG системы в газовой фазе, кДж/моль | ΔG системы в хлорбензоле, кДж/моль | ||
| Без катализатора | ${\text{ROOH}} \to {\text{R}}{{{\text{O}}}^{\centerdot }} + {\text{О}}{{{\text{Н}}}^{\centerdot }}$ | 160.41 | 158.40 |
| Ca(ЭГ)2 | ${\text{ROOH}} + {\text{Kт}}~\, \rightleftarrows {\text{ROOH}}--{\text{Кт}}$ | –64.79 | –60.60 |
| ${\text{ROOH}}--{\text{Кт}} \to {\text{R}}{{{\text{O}}}^{\centerdot }} + {\text{О}}{{{\text{Н}}}^{\centerdot }} + {\text{Кт}}$ | 225.20 | 219.00 | |
Таким образом, растворитель (хлорбензол) не оказывает существенного влияния на ход основных стадий процесса распада. Расхождение значений термодинамического потенциала основных стадий исследуемого процесса в газовой фазе и в хлорбензоле составляет 3–7%.
Распад промежуточного комплекса состава nROOH–Кт протекает с образованием радикалов, затем молекул продуктов распада ГПК и восстановлением первоначальной структуры катализатора. Время жизни промежуточного комплекса, пути его распада и энергия активации распада гидропероксида зависят от многих факторов: природы катализатора, концентрации компонентов, природы растворителя, условий проведения эксперимента и т.д.
Нами были оптимизированы различные структуры продуктов распада, включающие катализатор, а также радикалы RO• и OH•.
В качестве исследуемой реакции был рассмотрен акт первичного распада гидропероксида:
(III)
$n{\text{ROOH}} - {\text{Кт}} \to n{\text{R}}{{{\text{O}}}^{\centerdot }} + n{\text{O}}{{{\text{H}}}^{\centerdot }} + {\text{Кт}}.$Очевидно, что распад протекает ступенчато, поскольку процесс требует больших энергетических затрат. Причем данный механизм справедлив для всех катализаторов (этилгексаноата и металлокомплекса краун-эфира):
(V)
$[{\text{ROOH}} - {\text{Кт}}] \to [{\text{O}}{{{\text{H}}}^{\centerdot }} \cdot {\text{Кт}}] + {\text{R}}{{{\text{O}}}^{\centerdot }},$(VI)
$[{\text{O}}{{{\text{H}}}^{\centerdot }} \cdot {\text{Кт}}] \to {\text{O}}{{{\text{H}}}^{\centerdot }} + {\text{Кт}}.$В этой схеме первая стадия – образование комплекса, вторая – разрыв О–О-связи с отрывом радикала RO•, третья – регенерация катализатора с выходом в объем гидроксильного радикала. По результатам квантово-химического расчета единовременное отщепление от промежуточного комплекса и RO•-, и OH•-радикалов с регенерацией катализатора намного энергозатратнее, чем последовательное (табл. 2).
Таблица 2.
Термодинамические характеристики основных стадий процесса распада ГПК
| Реакция | Параметр | |
|---|---|---|
| ΔН, кДж/моль | ΔG, кДж/моль | |
| ${\text{ROOH}} + {\text{Кт}} \rightleftarrows [{\text{ROOH}} - {\text{Кт}}]$ | –117.88 | –57.87 |
| $[{\text{ROOH}} - {\text{Кт}}] \to {\text{R}}{{{\text{O}}}^{\centerdot }} + {\text{O}}{{{\text{H}}}^{\centerdot }} + {\text{Кт}}$ | 327.71 | 225.20 |
| $[{\text{ROOH}} - {\text{Кт}}] \to [{\text{R}}{{{\text{O}}}^{\centerdot }} \cdot {\text{Кт]}} + {\text{O}}{{{\text{H}}}^{\centerdot }}$ | 231.10 | 185.11 |
| ${\text{[R}}{{{\text{O}}}^{\centerdot }} \cdot {\text{Кт]}} \to {\text{R}}{{{\text{O}}}^{\centerdot }} + {\text{Кт}}$ | 96.61 | 40.09 |
| $[{\text{ROOH}} - {\text{Кт}}] \to [{\text{O}}{{{\text{H}}}^{\centerdot }} \cdot {\text{Кт]}} + {\text{R}}{{{\text{O}}}^{\centerdot }}$ | 202.55 | 135.70 |
| ${\text{[O}}{{{\text{H}}}^{\centerdot }} \cdot {\text{Кт]}} \to {\text{O}}{{{\text{H}}}^{\centerdot }} + {\text{Кт}}$ | 125.16 | 89.50 |
На основании расчетных значений энтальпии каждой описанной ступени для исследуемых каталитических систем установлено, что лимитирующей стадией является реакция (VI). Последующая ступень распада требует затрат меньшего количества энергии. Значения ΔH лимитирующей стадии процесса распада в присутствии Са(ЭГ)2 составляет 202.50 кДж/моль, в присутствии CaCl2 · ДБК – 117.88 кДж/моль.
ЗАКЛЮЧЕНИЕ
Таким образом, на основании анализа полученных двумя различными способами (экспериментальным и теоретическим) данных можно отметить, что образование промежуточных активированных комплексов возможно и энергетически выгодно. Оба метода подтверждают каталитические свойства исследуемых каталитических систем: соединения кальция ускоряют распад ГПК, снижая энергию активации разрыва связи О–О в гидропероксиде.
Результаты квантово-химических расчетов подтверждают предположенный нами механизм образования интермедиата.
В ходе теоретического исследования выявлен постадийный механизм образования и распада промежуточного интермедиата. Определена лимитирующая стадия разложения ГПК.
Список литературы
Stojanova L.F., Batyrshin N.N., Kharlampidi Kh.E. // Oxid. Commun. 2000. V. 23. № 2. P. 187.
Нестеров М.В., Иванов В.А., Дмитриева Г.Е., Потехин В.М. // Кинетика и катализ. 1982. Т. 23. № 6. С. 1466.
Цысковская И.В., Потехин В.М., Овчинников В.И., Хачатуров А.С. // Журн. прикл. химии. 1980. Т. 54. № 4. С. 783.
Нуруллина Н.М., Батыршин Н.Н., Харлампиди Х.Э. // Кинетика и катализ. 2007. Т. 48. № 5. С. 695.
Матиенко Л.И., Мосолова Л.А., Заиков Г.Е. // Вест. МИТХТ. 2009. Т. 4. № 6. С. 3.
Мельник Ю.Р. Дис. … канд. техн. наук. Львов, Нац. университет “Львiвська полiтехнiка”, 2001.
Матиенко Л.И., Мосолова Л.А., Скибида И.П. // Кинетика и катализ. 1990. Т. 31. № 6. С. 1377.
Нуруллина Н.М., Батыршин Н.Н., Харлампиди Х.Э. // Кинетика и катализ. 2016. Т. 57. № 4. С. 441.
Антоновский В.Л., Бузланова М.М. Аналитическая химия органических пероксидных соединений. Москва: Химия, 1978. 308 с.
Беляев В.А., Немцов М.С. // ЖОХ. 1961. Т. 31. Вып. 12. С. 3855.
Цепалов В.Ф., Шляпинтох В.Я. // Докл. АН СССР. 1959. Т. 124. № 4. С. 883.
Нуруллина Н.М., Батыршин Н.Н., Харлампиди Х.Э. // Нефтехимия. 2004. Т. 44. № 6. С. 475.
Гришаева Т.Н., Маслий А.Н. // Вест. Казанского технол. университета. 2012. Т. 15. № 12. С. 7.
Perdew J.P., Burke K., Ernzerhof M. // Phys. Rev. Lett. 1996. V. 77. № 18. P. 3865.
Rassolov V., Pople J.A., Ratner M., Windus T.L. // J. Chem. Phys. 1998. V. 109. P. 1223.
Нуруллина Н.М., Батыршин Н.Н., Харлампиди Х.Э. // Кинетика и катализ. 2013. Т. 54. № 6. С. 702.
Успехи химии органических перекисных соединений и аутоокисления / Сб. под ред. Эмануэля Н.М., Иванова К.И., Разуваева Г.А., Юрженко Т.И., Шушунова В.А., Меняйло А.Т., Ерофеева Б.В., Юрженко А.И. Москва: Химия, 1969. С. 152.
Березин И.В., Мартинек К. Основы физической химии ферментативного катализа. Москва: Высшая школа, 1977. 280 с.
Эмануэль Н.М., Денисов Е.Т., Майзус З.К. Цепные реакции окисления углеводородов в жидкой фазе. Москва: Наука, 1965. 375с.
Дополнительные материалы отсутствуют.
Инструменты
Кинетика и катализ