

Кинетика и катализ, 2020, T. 61, № 1, стр. 134-143
Особенности синтеза метанольного компонента бифункционального катализатора получения диметилового эфира
М. А. Кипнис a, *, Э. А. Волнина a, И. А. Белостоцкий a, И. С. Левин a
a ФГБУН Институт нефтехимического синтеза им. А.В. Топчиева РАН
119991 Москва, Ленинский просп., 29, Россия
* E-mail: kipnis@ips.ac.ru
Поступила в редакцию 21.03.2019
После доработки 17.06.2019
Принята к публикации 10.09.2019
Аннотация
В производстве диметилового эфира из синтез-газа используются бифункциональные катализаторы, на которых протекает образование метанола и его дегидратация. Это обеспечивается присутствием в катализаторе метанольного и дегидратирующего компонентов, которые могут быть получены независимо. Имеющиеся в литературе сведения, касающиеся приготовления метанольного катализатора CuО/ZnО/Al2О3 осаждением из раствора нитратов, противоречивы, в связи с чем нами изучено влияние способа и условий осаждения растворов нитратов Cu, Zn и Al карбонатом натрия на активность вышеуказанного катализатора. На основании данных о фазовом составе осадков после термообработки и активности полученных катализаторов в синтезе метанола можно сделать вывод, что наличие нитратных групп в осадках приводит к снижению каталитической активности. Предложен способ приготовления метанольного катализатора при комнатной температуре путем введения раствора солей Cu, Zn, Al в раствор карбоната натрия (обратное прикапывание). Такой метод обеспечивает активность полученных образцов непосредственно в синтезе метанола на уровне промышленного метанольного катализатора. Изучено образование ДМЭ на бифункциональных катализаторах, метанольным компонентом которых выступали образцы, приготовленные разными способами, а дегидратирующим компонентом являлся промышленный гамма-Al2О3. Селективность образования метанола в присутствии полученных образцов практически одинаковая, однако производительность по оксигенатам и ДМЭ существенно различается.
ВВЕДЕНИЕ
В последние 10–15 лет наблюдается рост интереса к синтезу метанола и диметилового эфира (ДМЭ), что связано с вовлечением в переработку различного углеродсодержащего сырья [1–4]. При этом ДМЭ, как и метанол, может быть использован для производства водорода (см., например, [5, 6]) и ценных химических продуктов [2, 3, 7, 8].
Основной путь промышленного синтеза ДМЭ – дегидратация метанола на среднекислотном катализаторе (в частности, активном оксиде алюминия).
В то же время активно разрабатывается одноступенчатый (прямой) способ получения ДМЭ из синтез-газа, когда производство метанола совмещено с его дегидратацией на бифункциональном гибридном катализаторе, включающем как метанольный, так и дегидратирующий компоненты [9].
Эффективный низкотемпературный метанольный катализатор CuO/ZnO/Al2O3 был разработан специалистами фирмы “ICI” в 60-х годах прошлого века. Согласно [10] для его приготовления предварительно полученную коллоидную взвесь гидроксида алюминия добавляют к раствору нитратов Cu и Zn. Далее при 85°С и рН ~ 7 проводится осаждение смешением потоков двух растворов – нитратного и карбоната натрия. Образовавшийся осадок после фильтрации и отмывки от натрия подвергается термообработке. Катализаторы этого типа и до настоящего времени наиболее часто используют в промышленности [11], хотя различные аспекты их синтеза по-прежнему находятся в поле зрения исследователей.
Подробное изучение влияния рН и температуры осаждения на свойства метанольных катализаторов Cu/ZnO/Al2O3, приготовленных осаждением карбонатом натрия из нитратных солей, было проведено в [12]. Обнаружена сложная параметрическая зависимость характеристик катализатора (состава, активности, удельной поверхности, фазового состава) от температуры (которую изменяли от комнатной до 70°С) и рН осаждения. Большинство прекурсоров, синтезированных при температуре ниже 70°С и слабокислотном, нейтральном или основном рН, представляют собой по данным рентгенографического анализа гидроксосоли: Zn2Al(OH)6NO3 ⋅ 1.9H2O и Cu2(NO3)(OH)3. Положительное влияние повышенной температуры осаждения, по мнению авторов, является следствием более равномерного распределения компонентов в осадке. Фазовый состав прекурсоров в случае активных катализаторов, полученных при 70°С и рН 6–8, отвечает Zn-содержащему малахиту (Cu,Zn)2(OH)2CO3.
Вывод о Zn-содержащем малахите, как предшественнике активного катализатора, сделан и авторами [13] на основе анализа ряда собственных работ.
Исследования кристаллических прекурсоров Cu–Zn-катализаторов позволили получить зависимость их фазового состава (фазы: малахит, аурихальцит, гидроцинкит) от соотношения Cu/Zn [14, 15]. Аурихальцит является оптимальным прекурсором в случае катализаторов реакции водяного сдвига [16, 17].
Соосаждение из нитратных и формиатных солей Cu, Zn, Al карбонатом натрия было изучено в [18]. Нитраты солей растворяли перед осаждением в концентрированной азотной кислоте. Соосаждение вели при рН 6.5 в автоматизированном реакторе при 65°С. Для достижения гомогенности распределения компонентов в осадке предлагается использовать микрокапельную технику осаждения или осаждение из формиатных солей.
Влияние условий прокаливания, типа прекурсора и содержания остаточных карбонатов на характеристики структуры оксидов меди и цинка и дезактивацию в синтезе метанола было рассмотрено в [19]. Образцы получали с применением автоматизированного реактора при рН 6.5 и Т = 65°С при вариации соотношения Cu/Zn. Изучение каталитических свойств приготовленных образцов в синтезе метанола при 230°С и 3 МПа показало, что их активность, независимо от фазового состава прекурсора, заметно снижается уже в первые часы работы.
Влияние типа осаждающего агента (карбонаты Na, K, NH4) на фазовый состав образующихся осадков было исследовано в [20]. Нагретые растворы нитратных солей с концентрацией 0.15 моль/л и соответствующего карбоната с концентрацией 0.18 моль/л с помощью насосов подавали с постоянной скоростью в смеситель. Образовавшиеся осадки накапливались в емкости с водой, постоянство рН (6 или 7) в которой поддерживали прикапыванием раствора солей. По данным рентгенофазового анализа (РФА) в прекурсорах присутствуют различные гидроксокарбонаты Cu, Zn. Независимо от типа осаждающего агента катализаторы, синтезированные по предложенной методике, показали высокую активность.
В то же время осаждение может быть проведено методом “обратного прикапывания”, когда в раствор осадителя вводится раствор соответствующих нитратных солей так, что осаждение различных компонентов происходит одновременно [14, 21–23].
Так, в [21] были рассмотрены два варианта осаждения из нитратных солей Cu, Zn, Al при добавлении их раствора к раствору гидрокарбоната натрия при 70°С. В так называемом “стандартном” варианте использовали тройной раствор солей, в “последовательном” варианте сначала вводили раствор нитратов Cu и Zn до стабилизации рН (6.6–6.9), а затем раствор нитрата Al. В работе [23] был проведен синтез обратным прикапыванием раствора нитратных солей Cu, Zn, Zr в раствор гидрокарбоната натрия при 37–47°С и рН 7–7.5. По данным [24] прекурсоры, полученные осаждением при температуре не выше 40°С, характеризуются высокой активностью, тогда как повышение температуры осаждения до 60°С приводит к ее снижению. Согласно [25] достаточно активны Cu–Zn–Zr-катализаторы, приготовленные при температуре не выше 40°С (имеющие, по данным РФА, рентгеноаморфных предшественников).
Для прямого синтеза ДМЭ требуется бифункциональный катализатор, содержащий как метанольный, так и дегидратирующий компоненты. Такой катализатор может быть получен механическим смешением тонких фракций порошков метанольного катализатора и оксида алюминия с последующим таблетированием [26–28].
Как показывает проведенный обзор литературы, технология синтеза метанольного компонента сложна и в отдельных моментах противоречива, при этом упор делается на нейтральность значений рН при осаждении. Вместе с тем при приготовлении небольших партий или при разработке новых катализаторов синтеза ДМЭ необходим надежный и относительно простой способ получения метанольного компонента, поиск которого и является целью настоящей работы.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Титрование нитратных растворов и синтез образцов
Кривые титрования индивидуальных растворов нитратов и смешанного тройного раствора изучали при комнатной температуре, используя растворы нитратов (0.1 М) и карбоната натрия (0.2 М). В 83 мл соответствующего нитратного раствора при постоянном перемешивании магнитной мешалкой прикапывали по 1 мл раствор карбоната натрия, дожидаясь стабилизации значений рН (примерно 3–5 мин), контролируемых pH-метром Аквилон 40 (“Научно-производственная компания Аквилон”, Россия).
Для получения метанольного компонента катализатора синтеза ДМЭ применяли смешанный раствор нитратов Cu, Zn и Al. Опробованы следующие варианты, проводимые как при комнатной температуре, так и при нагреве: прямое прикапывание (осаждение раствором карбоната натрия), обратное прикапывание (к раствору карбоната натрия добавляли раствор нитратов) и комбинированное осаждение, сочетающее прямое прикапывание до рН 10 с соосаждением.
Осаждение осуществляли при постоянном перемешивании, постепенно вводя требуемые растворы. После прекращения осаждения и старения осадка в течение примерно 15–17 ч, фильтрации и промывки дистиллированной водой до получения рН в сливе не выше 7 (промывка 3–5 раз) полученный осадок сушили при 80°С, а затем, как правило, прокаливали при 300°С в течение 6 ч.
Для обозначения способа и условий приготовления образцов метанольного катализатора далее использованы следующие шифры: КО – комбинированное осаждение, ПП – прямое прикапывание, ОП – обратное прикапывание. При синтезе с нагревом в обозначение дополнительно введена цифра, указывающая на температуру осаждения, и далее (через черту) добавлена цифра, отвечающая мольному соотношению карбонат натрия/нитраты металлов (в расчете на двухвалентный металл). Так, например, обозначению ОП68-1.5 отвечает синтез обратным прикапыванием при 68°С с мольным соотношением карбонат натрия/нитраты металлов, равным 1.5.
Образцы бифункциональных катализаторов синтеза ДМЭ готовили смешением порошков (фракция размером менее 0.1 мм) метанольного компонента и оксида алюминия в весовом соотношении 1 : 1 с последующим прессованием на ручном прессе РП-12 (“Биолент”, Россия). Для обеспечения хорошей прессуемости в шихту перед таблетированием вводили добавку коллоидного графита (~1 мас. %). Полученные таблетки высотой ~1 мм дробили до фракции 0.4–0.63 мм.
В качестве дегидратирующего компонента бифункциональных катализаторов, как и в [26], использовали промышленный гамма-оксид алюминия (ТУ 2163-015-44912618-2003, ЗАО “Промышленные катализаторы”, Рязань) с удельной поверхностью (БЭТ) 210 м2/г, общим объемом пор 0.64 см3/г, средним размером пор 10.1 нм.
При испытаниях в синтезе метанола в качестве образца сравнения применяли промышленный катализатор Мегамакс 507 (артикул 26842627815, “Clariant AG”), представляющий собой таблетки размером 5 × 5 мм и характеризующийся удельной поверхностью (БЭТ) 60 м2/г.
Характеризация образцов
Элементный анализ выполняли на усовершенствованном волновом рентгенофлуоресцентном спектрометре ARL Perform’x (“Thermo Fisher Scientific”, Швейцария). Рентгенофазовый анализ (РФА) проводили с использованием дифрактометра Rotaflex RU-200 (“Rigaku”, Япония), излучение CuKα.
Размер кристаллитов оксида меди CuO оценивали по уширению пика по формуле Шеррера [29] по среднему значению для двух самых интенсивных пиков с угловыми положениями 35.5° и 38.6° по 2θ.
ИК-спектры регистрировали в режиме отражения на ИК-фурье-спектрометре IFS-66 v/s (“Bruker”, Германия), 50 сканирований, кристалл – германий, диапазон 600–4000 см–1.
Каталитические исследования
В экспериментах использовали проточную каталитическую установку, схема которой представлена в [27]. Реактор выполнен из нержавеющей стали и имеет внутренний диаметр 12 мм. По оси реактора расположен термопарный канал (внешний диаметр 6 мм), на котором находится столик с металлической сеткой для размещения навески катализатора. Обогрев реактора проводили с помощью электропечи. Температуру на входе и выходе из слоя контролировали двумя термопарами, расположенными в термопарном канале на соответствующих уровнях.
Поток горячего конвертированного газа на выходе из реактора набором вентилей разделяли на две части. Анализируемый поток направляли на обогреваемую линию хроматографа Кристалл 5000 (“Хроматэк”, Россия), а основной поток – на холодильник-сепаратор и через регулятор давления “до себя” на сброс.
Выход из реактора и соответствующие газовые линии, находящиеся под давлением, поддерживали при температуре не ниже 120°С нагревом от дополнительной электропечи.
Хроматографический анализ проводили с использованием трех независимых линий. На линии ДТП-1 с насадочной колонкой с порапаком Т (газ-носитель – гелий) определяли содержание СО2, ДМЭ, воды и метанола; на линии ДТП-2 с насадочной колонкой с цеолитом NaX (газ-носитель – гелий) – N2 и СО; на линии ДТП-3 с насадочной колонкой с цеолитом NaX (газ-носитель – аргон) – Н2. Отметим, что кроме исходных компонентов синтез-газа и образующихся воды, метанола и ДМЭ других соединений в условиях тестирования не наблюдали.
Показания датчиков расходов, температуры и давления выводили с помощью соответствующей программы (автор Коротков А.С.) на дисплей и сохраняли в виде файла.
Активацию катализатора с целью восстановления оксида меди осуществляли в смеси ~3 об. % Н2/N2 с расходом 2 Нл/ч, поднимая температуру до 220°С со скоростью ~0.5 град/мин и контролируя хроматографически поглощение Н2 и выделение Н2О. Катализатор после активации с целью приработки и сокращения времени на подготовительные процедуры оставляли на ночь в той же смеси при температуре 260°С и давлении 0.2–0.3 МПа. На следующий день в реактор подавали синтез-газ до давления 3 МПа и проводили соответствующие эксперименты.
Стандартную процедуру оценки активности выполняли на навеске катализатора 0.8 г при 260°С, давлении 3 МПа при нескольких значениях расхода (расход изменяли в пределах 5–25 Нл/ч). При каждой величине расхода делали не менее трех хроматографических анализов с интервалом 30–40 мин, используя средние значения для построения зависимостей и расчетов. В конце эксперимента по возможности повторяли измерения при начальном расходе, обычно при объемной скорости 12 000 л ${\text{кг}}_{{{\text{кат}}}}^{{ - 1}}$ ч–1. Повторные данные, как правило, отклонялись в сторону меньших значений, что связано, видимо, с приработкой катализатора. Эти результаты также представлены на графиках.
Погрешность определения содержания компонентов была не более 5–7 отн. %, определения расхода – 1–3% отн. %. Баланс по углероду сходился с погрешностью ≤ 5 отн. %. В проведенных экспериментах градиент температуры по слою не превышал 1°С. Температуру в реакторе поддерживали с погрешностью ≤1°С, а давление – ≤0.02 МПа.
Синтез-газ состава (об. %): СО – 20.6, СО2 – 5.9, N2 – 5.5, Н2 – баланс, готовили из индивидуальных газов в предварительно отвакуумированном баллоне, контролируя давление по показаниям цифрового датчика, поверяемого образцовым манометром. В состав синтез-газа вводили азот, исходя из материального баланса которого рассчитывали расход конвертированного газа на выходе из реактора. Состав синтез-газа уточняли хроматографически перед проведением каталитических испытаний.
Расчетные величины
Расход конвертированного газа после реактора (Vвых, Нл/ч) рассчитывали, исходя из материального баланса по азоту, по формуле:
(1)
${{V}_{{{\text{вых}}}}} = {{{{V}_{0}}~{{С}_{{{\text{вх}}}}}} \mathord{\left/ {\vphantom {{{{V}_{0}}~{{С}_{{{\text{вх}}}}}} {{{С}_{{{\text{вых}}}}}}}} \right. \kern-0em} {{{С}_{{{\text{вых}}}}}}},$Конверсию СО (ХСО, %) находили, используя значения расхода газа на входе и выходе из реактора по формуле:
(2)
${{Х}_{{{\text{СО}}}}} = ({{С}_{0}}{{V}_{0}}--{{С}_{{{\text{СО}}}}}{{V}_{{{\text{вых}}}}}) \times {{100} \mathord{\left/ {\vphantom {{100} {{{С}_{0}}{{V}_{0}}}}} \right. \kern-0em} {{{С}_{0}}{{V}_{0}}}},$Производительность по оксигенатам (метанолу и ДМЭ) в расчете на моль С1 (П, мольС1${\text{кг}}_{{{\text{кат}}}}^{{ - 1}}$ ч–1) рассчитывали по формуле:
(3)
${\text{П}} = {{V}_{{{\text{вых}}}}}\left( {2{{С}_{{{\text{ДМЭ}}}}} + {{С}_{{{\text{МЕ}}}}}} \right){{(22.4 \times m \times 100)}^{{ - 1}}},$Селективность образования метанола (%) вычисляли по формуле:
Выражение (4) отражает соотношение образующегося метанола и СО, превращенного в метанол и СО2.РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Титрование растворов нитратов Cu, Zn и Al карбонатом Na
Для выяснения необходимого при осаждении нитратов соотношения карбонат/нитрат были изучены соответствующие зависимости рН при титровании индивидуальных нитратов Cu, Zn и Al. В случае нитратов Cu и Zn стехиометрии осаждения отвечает мольное соотношение, равное 1, тогда как в случае нитрата Al – 1.5. Для удобства совместного рассмотрения кривых титрования трех солей при построении графиков использовали мольное отношение карбонат натрия/нитрат металла в пересчете на эквивалентный двухвалентный металл (то есть в случае нитрата Al брали мольное соотношение карбонат/нитрат, деленное на 1.5). Соответствующие кривые приведены на рис. 1.
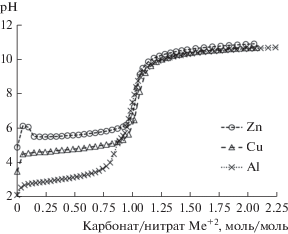
По величине исходного значения рН растворы нитратов Cu, Zn и Al располагаются в ряду: рНAl < рНCu < рНZn. Помутнение раствора, связанное с образованием частиц осадка, для солей Cu и Zn начинается при добавлении первых капель карбоната натрия, тогда как для нитрата Al – после достижения рН ~ 5. Заметный рост значений рН в случае меди и цинка, как видно из рис. 1, происходит при достижении соотношения реагентов, примерно соответствующего стехиометрии реакций нитратов с карбонатом натрия. В то же время в случае алюминия быстрый рост значения рН наблюдается при соотношении карбонат/нитрат, равном ~0.75. Таким образом, осаждение алюмосодержащего осадка начинается при меньших значениях рН, чем для меди и цинка. Это благоприятно с учетом того, что оксид алюминия играет роль носителя.
Характерно, что кривые титрования трех солей качественно аналогичны и могут быть разбиты на 3 участка. Примерно до стехиометрии осаждения (Na2CO3/Me(NO3)2 = 1) рН растет медленно, хотя и отмечается незначительное его увеличение после добавления начального количества карбоната натрия. При достижении стехиометрического соотношения карбонат/нитрат небольшое добавление карбоната натрия приводит к заметному росту рН вплоть до соотношения ~1.3. И, наконец, дальнейшее повышение содержания карбоната натрия в растворе слабо влияет на рН раствора.
Качественно та же картина наблюдается и при титровании тройного раствора солей (весовое соотношение Cu : Zn : Al равно 6 : 3 : 1), при этом в процессе титрования наблюдается изменение цвета раствора. Так, рост рН от 5.3 до 6.46 сопровождается сменой цвета от светло-голубого с зеленоватым отливом к интенсивно-голубому.
Объяснение наблюдаемого S-образного вида кривых титрования, на наш взгляд, кроется в особенностях осаждения нитратных солей. Так, в [30] при изучении осаждения нитрата меди гидроксидом натрия было установлено, что при добавлении щелочи к раствору нитрата меди сначала образуется нитрат-гидроксид меди. Переход к гидроксиду меди за счет обмена осадка с раствором анионами происходит при избытке щелочи относительно стехиометрии осаждения. В то же время в случае добавления раствора нитрата меди к щелочи (“обратное прикапывание”) сразу образуется гидроксид меди. Исходя из данных [30], в работе [12] были обсуждены возможные реакции между катионами меди и цинка и анионами, присутствующими в растворе при осаждении из тройного раствора нитратов. По мнению авторов, в осадке возможно появление соединений Cu(OH)2, Cu(NO3)2 ⋅ 3Cu(OH)2, Cu2(OH)2CO3, (Cu,Zn)2(OH)2CO3.
Отметим, что небольшой рост рН при добавлении первоначальных капель щелочи к раствору нитрата авторы [30] связывают с нейтрализацией свободных анионов нитрата.
Физико-химические данные осадков
Исходя из вышеизложенного, при постепенном введении карбоната натрия в раствор нитратов должны сначала образоваться нитратсодержащие осадки, переходящие при увеличении соотношения карбонат/нитрат существенно выше стехиометрического в безнитратные структуры. Для проверки этого соображения с использованием ИК-спектроскопии были изучены высушенные при 80°С осадки, полученные при осаждении из индивидуальных нитратов при значениях соотношения карбонат/нитрат, отвечающих переходному участку кривых титрования (табл. 1).
Таблица 1.
Характеристики осадков индивидуальных нитратов, отобранных для ИК-спектроскопического исследования
| № п/п | Металл | рН | Карбонат/нитрат Ме+2 |
|---|---|---|---|
| 1 | Cu | 8.07 | 1.06 |
| 2 | Zn | 8.2 | 1.06 |
| 3 | Al | 7.77 | 1.0 |
| 4 | Cu | 8.86 | 1.11 |
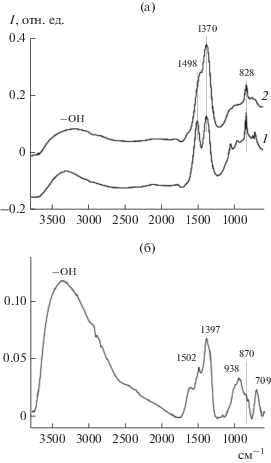
ИК-спектры осадков представлены на рис. 2. Отнесение полос проведено согласно [31]. Как видно из рис. 2, спектры Cu- и Zn-образцов характеризуются наличием полос, отвечающих нитрат-аниону – 1370 и 828 см–1. В случае цинка можно говорить о присутствии карбонат-аниона, которому соответствуют полосы 1498 и 690 см–1. Возможно, расщепление полос происходит за счет наличия разных кристаллогидратных форм. Полоса от ОН-связей в спектре Zn-осадка сдвинута в сторону коротких длин волн, что может служить указанием на образование кристаллогидратов. В случае Al-осадка (рис. 2б) нитрат алюминия идентифицируется по полосам 1397 и 870 см–1. Кроме того, наблюдается небольшое содержание карбонат-иона, о чем свидетельствуют полосы 1502 и 938 см–1. Полосы 3380 (νOH), 1630 см–1 (δHOH) в спектре Al-осадка говорят о присутствии воды. Также обнаружены полосы, характерные для групп ОН (3380 и 1630 см–1) и связи Al–O (709 cм–1).
Рис. 2.
ИК-спектры осадков индивидуальных нитратов после сушки при 80°С (образцы 1–3, табл. 1): а – Cu-осадок (1), Zn-осадок (2); б – Al-осадок.
Результаты ИК-спектроскопии Cu-осадка показали, что при увеличении соотношения карбонат/нитрат меди до 1.11 осадок содержит нитратные группы и представляет собой хорошо окристаллизованный герхардтит Cu2(NO3)(OH)3 (файл JCPDS-ICDD PDF #77-0148) (возможна запись в виде Cu(NO3)2 ⋅ 3Cu(OH)2).
Синтез метанольного компонента катализатора получения ДМЭ
Данные по синтезированным Cu–Zn–Al-образцам приведены в табл. 2, где отмечены способ синтеза, температура осаждения, рН в конце осаждения, мольное соотношение карбонат натрия/нитраты металлов (в расчете на Ме+2, К/Н), мольная концентрация растворов (Ср).
Таблица 2.
Характеристика синтезированных метанольных образцов
| № п/п | Шифр | рН | К/Н* | Ср, М |
|---|---|---|---|---|
| 1 | ОП-1.5 | 8.97 | 1.5 | Нитраты – 0.1, карбонат – 0.15 |
| 2 | ПП-1.5 | 9.7 | 1.5 | Нитраты – 0.11, карбонат – 0.15 |
| 3 | ОП68-1.5 | 8.49 | 1.5 | Нитраты – 0.1, карбонат – 0.15 |
| 4 | КО70-1.5 | ~7.4 | 1.5 | Нитраты – 0.08 карбонат – 0.48 |
| 5 | ПП68-1.5 | 9.47 | 1.5 | Нитраты и карбонат – 1 |
| 6 | ПП-1.1 | 9.41 | 1.1 | Нитраты и карбонат – 0.2 |
| 7 | ПП68-1.1 | ~8 | 1.1 | Нитраты и карбонат – 1 |
Образующийся при осаждении растворов солей осадок имеет голубой цвет, за исключением образцов ОП68-1.5 (№ 3) и ПП68-1.5 (№ 5). Для образца ОП68-1.5 цвет осадка серо-голубой, а для образца ПП68-1.5 – коричневый. Такое изменение окраски возможно связано с так называемым явлением оксоляции [18], когда в осадке появляются частицы оксида меди.
Для образца ПП-1.1 (№ 6), синтезированного при минимальном отношении карбонат/нитраты, был проведен элементный анализ выпаренного в термостате маточного раствора, который показал, что содержание оксидов Al, Cu и Zn незначительно: 0.3, 0.13 и 0.03 вес. % соответственно. Таким образом, наличием металлов в растворе, связанным с неполным осаждением, можно пренебречь. По данным элементного анализа содержание оксида меди в прокаленных образцах в среднем составило 59.9 вес. %, что соответствует расчетному значению при стандартном отклонении 1.9 вес. %.
Дифрактограммы образцов после сушки представлены на рис. 3, нумерация кривых соответствует нумерации образцов в табл. 2.
Рис. 3.
Дифрактограммы синтезированных образцов метанольного компонента после сушки: 1 – ОП-1.5, 2 – ПП-1.5, 3 – ОП68-1.5, 4 – КО70-1.5, 5 – ПП68-1.5, 6 – ПП-1.1.
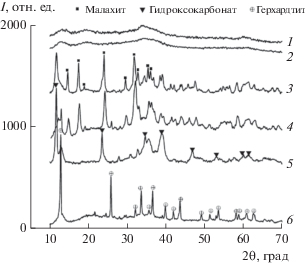
Как видно из рис. 3, образцы ОП-1.5, ПП-1.5 (кривые 1 и 2) после сушки рентгеноаморфны. В случае же образцов ОП68-1.5, КО70-1.5, ПП68-1.5, ПП-1.1 (кривые 3–6) при наличии аморфной фазы также наблюдаются хорошо окристаллизованные фазы, которые можно идентифицировать как малахит Cu2(OH)2CO3 (файл JCPDS-ICDD PDF #75-1163), гидроксокарбонат (или, точнее, гидроталькит, согласно [32]) Cu6Al2(OH)16CO3 ⋅ ⋅ 4H2O (файл JCPDS-ICDD PDF #37-0630) и герхардтит Cu2(NO3)(OH)3 (файл JCPDS-ICDD PDF #77-0148).
Отметим, что по данным ИК-спектроскопии образцы ОП-1.5, ПП-1.5 характеризуются типичными спектрами кристаллогидратов карбонатов металлов и в спектрах отсутствуют полосы нитратов.
Дифрактограммы образцов после прокаливания (образец ПП-1.1 был прокален при 350°С) представлены на рис. 4, нумерация кривых соответствует нумерации образцов в табл. 2.
Рис. 4.
Дифрактограммы синтезированных образцов метанольного компонента после прокаливания: 1 – ОП-1.5, 2 – ПП-1.5, 3 – ОП68-1.5, 4 – КО70-1.5, 5 – ПП68-1.5, 6 – ПП-1.1, 7 – ПП68-1.1.
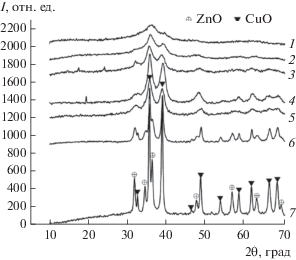
Как видно из рис. 4, дисперсность оксида меди, судя по уширению основных пиков, существенно различается для разных образцов. Так, для образцов ОП-1.5, ПП-1.5 и ОП68-1.5 (кривые 1–3) на дифрактограммах наблюдаются размытые основные пики оксида меди, тогда как для КО70-1.5, ПП68-1.5, ПП-1.1 и ПП68-1.1 (кривые 4–7) размер кристаллитов CuO составил 5, 7, 19 и 23 нм соответственно. В то же время на дифрактограммах образцов ОП-1.5, ПП-1.5, ОП68-1.5, КО70-1.5 и ПП68-1.5 (кривые 1–5) пики, отвечающие оксиду цинка, значительно уширены, что свидетельствует о значительной дисперсности этой фазы.
Каталитические исследования
Предполагая, что образец ОП-1.5 исходя из условий синтеза (обратное прикапывание при комнатной температуре в избытке карбоната натрия) должен быть наиболее активным, мы сопоставили его активность в синтезе метанола с промышленным метанольным катализатором Мегамакс 507.
Как видно из рис. 5 (по оси абсцисс отложены величины m/V0, пропорциональные времени контакта), синтезированный при комнатной температуре обратным прикапыванием катализатор превосходит по активности промышленный метанольный катализатор, что подтвердило наши предположения.
Рис. 5.
Зависимость конверсии СО (а) и содержания метанола (б) в конвертированном газе от m/V0 при синтезе метанола: 1 – ОП-1.5, 2 –катализатор Мегамакс 507.
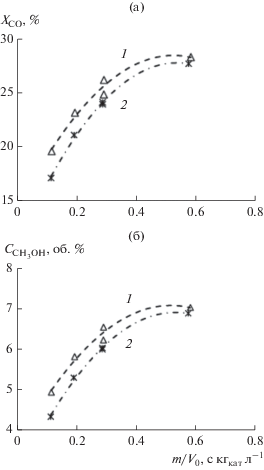
Производительность в расчете на метанол с ростом объемной скорости для обоих катализаторов растет практически линейно. При максимальной объемной скорости 31 000 л ${\text{кг}}_{{{\text{кат}}}}^{{ - 1}}$ ч1 (время контакта 0.115 с кгкат л–1) для Мегамакс 507 удельная производительность (в расчете на метанол) составила 1.8 кг ${\text{кг}}_{{{\text{кат}}}}^{{ - 1}}$ ч–1, а для образца ОП-1.5 – 2 кг ${\text{кг}}_{{{\text{кат}}}}^{{ - 1}}$ ч–1.
Данные об активности в синтезе ДМЭ бифункциональных катализаторов, приготовленных смешением полученных метанольных образцов и гамма-оксида алюминия, представлены на рис. 6 и рис. 7, нумерация кривых на которых соответствует нумерации метанольных образцов в табл. 2.
Рис. 6.
Влияние объемной скорости и типа метанольного компонента на производительность бифункциональных катализаторов по оксигенатам: 1 – ОП-1.5, 2 – ПП-1.5, 3 – ОП68-1.5, 4 – КО70-1.5, 5 – ПП68-1.5, 6 – ПП-1.1.
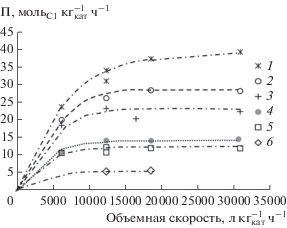
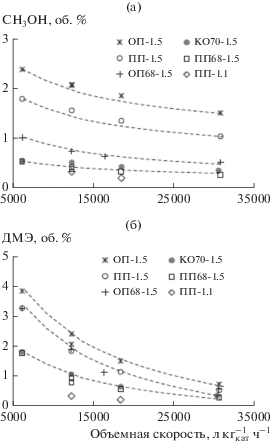
Рис. 7.
Влияние объемной скорости и типа метанольного компонента на содержание метанола (а) и ДМЭ (б) в конвертированном газе при синтезе ДМЭ.
Вид зависимостей производительности по оксигенатам от объемной скорости (рис. 6) характерен для одностадийного синтеза ДМЭ на катализаторах такого типа [26–28]: на начальном участке наблюдается линейный рост, а далее кривые практически выходят на плато.
Наиболее активным оказался катализатор, метанольный компонент которого синтезирован при комнатной температуре обратным прикапыванием (ОП-1.5, кривая 1). В то же время активность катализаторов с метанольными компонентами КО70-1.5, ПП68-1.5 и ПП-1.1 была невысока (кривые 4–6). Катализаторы, содержащие метанольные компоненты ПП-1.5 и ОП68-1.5 (кривые 2 и 3), занимают в ряду активности промежуточное положение.
В той же последовательности располагаются катализаторы при рассмотрении данных по содержанию метанола и ДМЭ в конвертированном газе при синтезе ДМЭ (рис. 7, нумерация кривых соответствует нумерации образцов в табл. 2), а также по конверсии СО (не показано).
По селективности образования метанола при синтезе ДМЭ катализаторы с разными метанольными компонентами достаточно близки (табл. 3). С ростом объемной скорости селективность увеличивается. Для образцов КО70-1.5, ПП68-1.5 и ПП-1.1 (№ 4–6) при высокой объемной скорости анализ баланса СО2 показал образование СО из СО2 (отмечено *), что свидетельствует о протекании обратной реакции водяного газа. Соответственно, расчеты по формуле (4) в этом случае неприменимы, селективность принята равной 100%.
Таблица 3.
Селективность образования метанола в синтезе ДМЭ на катализаторах с разными метанольными компонентами
| m/V0, с кгкат л–1 | Объемная скорость, л ${\text{кг}}_{{{\text{кат}}}}^{{ - {\text{1}}}}$ ч–1 | Метанольный компонент (нумерация по табл. 2) | |||||
|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | ||
| 0.58 | 6200 | 82 | 80 | 79 | 83 | 81 | – |
| 0.29 | 12 300 | 83 | 82 | 80 | 88 | 88 | 88 |
| 0.19 | 18 500 | 89 | 90 | 88 | 99 | 97 | 100* |
| 0.12 | 31 000 | 100 | 100 | 100 | 100* | 100* | – |
Сопоставление данных по активности (рис. 6, 7) и вида дифрактограмм прокаленных катализаторов (рис. 4) позволяет говорить о том, что чем выше активность катализатора, тем шире на дифрактограмме пики, отвечающие оксиду меди, и, соответственно, выше его дисперсность. Одной из причин снижения дисперсности при вариации условий синтеза образцов может быть присутствие в осадках нитратных групп, которые способствуют спеканию кристаллитов оксида меди при разложении нитратов в процессе прокаливания [33]. Отрицательное влияние наличия в прекурсорах нитратных групп было отмечено в [34, 35].
Синтез при повышенной температуре при прочих равных условиях также приводит к образованию более крупных кристаллитов (сравни данные РФА для прокаленных образцов ОП-1.5 и ОП68-1.5). Возможно, причина этого связана с тем, что при осаждении критический размер зародыша твердой фазы тем больше, чем выше температура. Так, после сушки осадок ОП-1.5 рентгеноаморфен, тогда как осадок ОП68-1.5 соответствует хорошо окристаллизованному малахиту. Дисперсность образующегося после прокалки при 300°С оксида меди, по-видимому, коррелирует с дисперсностью фазы высушенного осадка.
Полученные результаты позволяют сделать следующие выводы. Распространенное в литературе мнение о необходимости поддержания при синтезе метанольных компонентов величины рН, близкой к 7, не подтверждается экспериментальными данными. При таких значениях рН в свежем осадке присутствуют нитратные группы. Возможно, при старении осадка в растворе они полностью или частично обмениваются на гидроксилы или карбонаты. Однако проще получать осадок, подавляя возможность перехода нитратов в осадок при использовании избытка осадителя. В этом плане эффективно обратное прикапывание, обеспечивающее быстрое и одновременное осаждение солей Cu, Zn и Al, что способствует гомогенному распределению этих компонентов в осадке. По-видимому, важны как концентрации в растворе карбоната и нитрата, так и их соотношение. Так, уменьшение соотношения до 1.1 (ПП-1.1) приводит к снижению активности и росту размеров кристаллитов оксида меди (рис. 4).
Таким образом, обратное прикапывание при комнатной температуре с избытком карбоната натрия является эффективным способом синтеза метанольного катализатора.
ЗАКЛЮЧЕНИЕ
Рассмотрены варианты синтеза метанольного CuО/ZnО/Al2О3-катализатора осаждением из раствора нитратов. С учетом информации о фазовом составе осадков после термообработки и активности катализаторов можно сделать вывод, что наличие нитратных групп в осадках приводит к снижению каталитической активности.
Сопоставление данных по активности (синтез метанола, ДМЭ) и результатов рентгенофазового анализа прокаленных метанольных катализаторов позволяет говорить о том, что чем выше дисперсность оксида меди, тем более активен катализатор. Величина рН при синтезе не обязательна должна быть близка к 7. Более того, для ухода от появления нежелательных нитратных групп в осадке необходим избыток карбоната натрия при осаждении. Обратное прикапывание более благоприятно для синтеза активного катализатора, чем прямое. Приготовленный при комнатной температуре обратным прикапыванием образец более активен в синтезе метанола, чем промышленный метанольный катализатор Мегамакс 507. Бифункциональный катализатор синтеза ДМЭ на основе образца метанольного компонента, полученного обратным прикапыванием (дегидратирующий компонент – гамма-оксид алюминия), обладает наибольшей активностью по сравнению с образцами, синтезированными в иных условиях.
Список литературы
Mondal U., Yadav G.D. // J. CO2 Util. 2019. V. 32. P. 299.
Fleisch T.H., Basu A., Sills R.A. // J. Natural Gas Sci. Eng. 2012. V. 9. P. 94.
Haro P., Trippe F., Stahl R., Henrich E. // Appl. Energy. 2013. V. 108. P. 54.
Xu X., Liu Y., Zhang F., Di W., Zhang Y. // Catal. Today. 2017. V. 298. P. 61.
Ramos E., Davin L., Angurell I., Ledesma C., Llorca J. // ChemCatChem. 2015. V. 7. P. 2179.
Takeishi K., Akaike Y. // Appl. Catal. A: Gen. 2016. V. 510. P. 20.
Tan E.C.D., Talmadge M., Dutta A., Hensley J., Snowden-Swan L.J., Humbird D., Schaidle J., Biddy M. // Biofuels, Bioprod. Bioref. 2016. V. 10. P. 17.
Волнина Э.А., Кипнис М.А., Хаджиев С.Н. // Нефтехимия. 2017. T. 57. C. 243.
Saravanan K., Ham H., Tsubaki N., Bae J.W. // Appl. Catal. B: Env. 2017. V. 217. P. 494.
Pat. 1159035 UK. 1965.
Busca G. Heterogeneous catalytic materials. Solid state chemistry, surface chemistry and catalytic behavior. Ch. 9. Elsevier B.V., 2014. 463 p.
Baltes C., Vukojević S., Schüth F. // J. Catal. 2008. V. 258. P. 334.
Behrens M., Schlögl R. // Z. Anorg. Allg. Chem. 2013. V. 639. P. 2683.
Bems B., Schur M., Dassenoy A., Junkes H., Herein D., Schlögl R. // Chem. Eur. J. 2003. V. 9. P. 2039.
Khassin A.A., Minyukova T.P., Yurieva T.M. // Mend. Comm. 2014. V. 24. P. 67.
Budiman A., Ridwan M., Kim S.M., Choi J.-W., Yoon C.W., Ha J.-M., Suh D.J., Suh Y.-W. // Appl. Catal. A: Gen. 2013. V. 462–463. P. 220.
Galván C.Á., Schumann J., Behrens M., García Fierro J.L., Schlögl R., Frei E. // Appl. Catal. B: Env. 2016. V. 195. P. 104.
Behrens M., Brennecke D., Girgsdies F., Kißner S., Trunschke A., Nasrudin N., Zakaria S., Idris N.F., Abd Hamid S.B., Kniep B., Fischer R., Busser W., Muhler M., Schlogl R. // Appl. Catal. A: Gen. 2011. V. 392. P. 93.
Schumann J., Tarasov A., Thomas N., Schlögl R., Behrens M. // Appl. Catal. A: Gen. 2016. V. 516. P. 117.
Simson G., Prasetyo E., Reiner S., Hinrichsen O. // Appl. Catal. A: Gen. 2013. V. 450. P. 1.
Jeong C., Ham H., Bae J.W., Kang D.-C., Shin C.-H., Baik J.H., Suh Y.-W. // ChemCatChem 2017. V. 9. P. 4484.
Arena F., Italiano G., Barbera K., Bordiga S., Bonura G., Spadaro L., Frusteri F. // Appl. Catal. A: Gen. 2008. V. 350. P. 16.
Arena F., Barbera K., Italiano G., Bonura G., Spadaro L., Frusteri F. // J. Catal. 2007. V. 249. P. 185.
Wu J., Luo S., Toyir J., Saito M., Takeuchi M., Watanabe T. // Catal. Today. 1998. V. 45. P. 215.
Frei E., Schaadt A., Ludwig T., Hillebrecht H., Krossing I. // ChemCatChem. 2014. V. 6. P. 1721.
Кипнис М.А., Самохин П.В., Белостоцкий И.А., Туркова Т.В. // Катализ в промышленности. 2017. Т. 17. С. 442.
Кипнис М.А., Белостоцкий И.А., Волнина Э. А., Лин Г.И., Маршев И.И. // Кинетика и катализ. 2018. Т. 59. С. 715.
Кипнис М.A., Белостоцкий И.А., Волнина Э.А., Лин Г.И. // Катализ в промышленности. 2018. Т. 18. С. 12.
Гинье А. Рентгенография кристаллов. Теория и практика. Под ред. акад. Белова Н.В. Москва: Физматгиз, 1961. 604 с.
Вассерман И.М., Силантьева Н.И. // Журн. Неорг. Химии. 1968. Т. 13. С. 2008.
Накамото К. Инфракрасные спектры неорганических и координационных соединений. Под ред. Пентина Ю.А. Москва: Мир, 1991. 536 с.
Yamaoka T., Abe M., Tsuji M. // Mater. Res. Bull. 1989. V. 24. P. 1183.
Prieto G., De Jong K.P., De Jongh P.E. // Catal. Today. 2013. V. 215. P. 142.
Chinchen G.C., Denny P.J., Jennings J.R., Spencer M.S., Waugh K.C. // Appl. Catal. 1988. V. 36. P. 1.
Li J.-L., Inui T. // Appl. Catal. A: Cen. 1996. V. 137. P. 105.
Дополнительные материалы отсутствуют.
Инструменты
Кинетика и катализ