

Кинетика и катализ, 2020, T. 61, № 3, стр. 334-342
Прогностическая модель связи антирадикальной активности с потенциалом ионизации молекул и ионов флавоноидов
Н. И. Белая a, *, А. В. Белый a, И. Н. Щербаков b
a ГОУ ВПО Донецкий национальный университет
283001 Донецк, ул. Университетская, 24, Украина
b ФГАОУ ВПО Южный федеральный университет
344006 Ростов-на-Дону, ул. Большая Садовая, 105/42, Россия
* E-mail: nat.iv.belaya@gmail.com
Поступила в редакцию 23.07.2019
После доработки 21.12.2019
Принята к публикации 24.12.2019
Аннотация
Методами линейного однофакторного регрессионного анализа установлена зависимость между антирадикальной активностью и молекулярными дескрипторами флавоноидов, связанными с механизмом их антирадикального действия в виде полуэмпирического линейного однофакторного уравнения. Проведена оценка прогностической способности установленной модели на контрольной выборке, средняя ошибка аппроксимации составила не более 8.5%. Полученная взаимосвязь дескриптор–активность является основой для прогнозирования антирадикальных свойств флавоноидов и подобных им структур в водных растворах с физиологическим значением рН среды.
Флавоноиды (FlavOH) являются одной из наиболее многочисленных групп вторичных метаболитов растений, способных проявлять антиоксидантную, капилляроукрепляющую, противовоспалительную, противоопухолевую и детоксикационную активности [1, 2]. Из-за широкой распространенности этих соединений в растительной пище, фармацевтической и парфюмерной продукции возникает необходимость в отработке модели для оценки и прогнозирования их реакционной способности. Факторами, стимулирующими развитие прогностических методологий, являются как экономический эффект, так и возможность исключения веществ с нежелательными свойствами на ранних этапах доклинических исследований.
Особое место занимают полуэмпирические модели прогнозирования биологической активности на основе количественной взаимосвязи структура–реакционная способность вещества QSAR/QSPR (Quantitative Structure–Activity/Property Relationships) [3, 4] в виде линейных регрессионных уравнений. Для их разработки используют большое количество дескрипторов, характеризующих электронное и пространственное строение, полярность, гидрофильность молекул антиоксидантов и т.д. Однако, исходя из возможного механизма антиоксидантной активности в выбранной модельной реакции, применение большого числа дескрипторов затруднено, что часто приводит к установлению лишь качественной связи структура–активность [5, 6]. Для получения количественных зависимостей необходим поиск молекулярных дескрипторов, связанных непосредственно с механизмом действия природных фенолов в реакции со свободными радикалами.
Целью данной работы являлось проведение факторного анализа зависимости антирадикальной активности (АРА) от молекулярных дескрипторов флавоноидов, связанных с механизмом их действия в реакции с 2-амидинопропан-2-пероксильными радикалами (APOO•) в водном растворе при физиологическом значении рН среды, а также оценка прогностической способности полученной линейной однофакторной модели с использованием контрольной выборки.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Реактивы
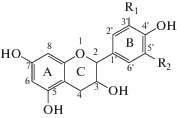
В качестве объектов исследования использовали FlavOH, относящиеся к группе флаванов, флавонолов и их гликозидов, а также соединения группы гидроксиацетофенонов:
| Флаванолы-3 (катехины) | ||
| I – катехин | R1 = OH, R2 = H; | |
| II – галлокатехин | R1 = R2 = OH. | |
| Флавонолы | ||
| III – кемпферол | R1 = R2 = R3 = R4 = H; | |
| IV – кверцетин | R2 = OH, R1 = R4 = R3 = H; | |
| V – изорамнетин | R2 = ОСН3, R1 = R3 = R4 = H; | |
| VI – морин | R1 = OH, R2 = R3 = R4 = H; | |
| VII – мирицетин | R2 = R3 = OH, R1 = R4 = H; | |
| VIII – кверцитрин | R2 = OH, R1 = R3 = H, R4 = L-рамноза; | |
| IX – рутин | R2 = OH, R1 = R3 = H, R4 = рутиноза. | |
| Гидроксиацетофеноны | ||
| X – 2-гидроксиацетофенон | R1 = OH, R2 = R3 = R4 = H; | 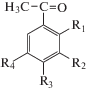 |
| XI – 3-гидроксиацетофенон | R2 = OH, R1 = R3 = R4 = H; | |
| XII – 4-гидроксиацето-фенон | R3 = OH, R1 = R2 = R4 = H; | |
| XIII – 2,4-дигидроксиаце-тофенон | R1 = R3 = OH, R2 = R4 = H; | |
| XIV – 2,5-дигидроксиацетофенон | R1 = R4 = OH, R2 = R3 = H; | |
| XV – 3,4-дигидроксиацетофенон | R2 = R3 = OH, R1 = R4 = H; | |
| XVI – 3-метокси-4-гидроксиацетофенон | R2 = ОСН3, R3 = OH, R1 = R4 = H. | |
Флавоноиды (I–IX) (“Fluka”, “Merck”, “Panreac”) и гидроксиацетофеноны (“Merck”, “Acros Organics”) использовали без предварительной очистки. Генератором свободных радикалов служило гидрофильное азосоединение 2,2'-азо-бис-(2-амидинопропан) дигидрохлорид (AAPH, “Sigma-Aldrich”). В качестве активатора свечения применяли родамин 6Ж (Rd6G, “Sigma-Aldrich”). Для задания рН водных растворов использовали солянокислую (рН 2), фосфатную (рН 7.35) и борно-щелочную (рН 9) буферные системы, приготовленные по методике [7]. Значение точного рН приготовленных буферных растворов контролировали с помощью иономера И-160МИ (“ООО “Измерительная техника”, Россия). Диметилсульфоксид (ДМСО) очищали по методике [8].
Кинетические исследования
Окисление флавоноидов пероксирадикалами APOO•, генерируемыми при термическом распаде AAPH в водной среде, изучали методом хемилюминесценции (ХЛ) [9]. Флавоноиды растворяли в ДМСО, а затем аликвоту этого раствора (0.1 мл) вводили в буферный раствор (4.9 мл), содержащий AAPH и Rd6G. Окисляемую смесь помещали в термостатированную кювету хемилюминометра и барботировали воздухом для насыщения ее кислородом и перемешивания. Температура проведения эксперимента 323 ± 2 К соответствовала температуре, при которой используемый инициатор распадается на радикалы APOO• с постоянной скоростью. Значения константы скорости реакции распада AAPH на радикалы (kd= 7.1 × 10–6 c–1) и выхода радикала из клетки (e = 0.48) при pH 7.4 и T = 323 К взяты из работ [10, 11]. Скорость инициирования (генерирования) пероксирадикалов (Wi) рассчитывали по формуле ${{W}_{i}} = 2e{{k}_{{\text{d}}}}\left[ {{\text{AAPH}}} \right]$ [9]. Измерения проводили с помощью хемилюминесцентной установки (фотоумножитель ФЭУ-38), принципиальная схема которой описана в работе [9, 12], с цифровой обработкой сигнала посредством аналого-цифрового преобразователя (“ЗАО “Л‑КАРД”, Россия). Активатор Rd6G усиливает хемилюминесценцию (ХЛ), но не влияет на кинетику ее затухания. Область свечения, определенная с помощью граничных светофильтров, совпадает с областью флуоресценции Rd6G λ = 550–650 нм [13]. Таким образом, электронно-возбужденный краситель является эмиттером наблюдаемой ХЛ. В спектрах поглощения Rd6G, снятых после реакции, не наблюдалось сколько-нибудь заметных изменений, что является косвенным аргументом в пользу того, что краситель не вовлекался в реакцию, а проявлял себя как физический активатор ХЛ.
Квантово-химический расчет
Квантово-химические расчеты проводили с использованием программного пакета Gaussian 09 [14] в рамках теории функционала плотности (DFT) с гибридным функционалом B3LYP, выбор которого обусловлен опубликованными данными об успешном его применении для соединений фенольного типа [15, 16]. Поиск стабильных конформеров предварительно осуществляли полуэмпирическим методом PM6. Структуры с минимальной энергией использовали в качестве начального приближения для расчетов на уровне B3LYP/6-311++G(d,p) для систем с закрытыми оболочками (синглетное основное состояние) и UB3LYP/6-311++G(d,p) для систем с открытыми оболочками (дублетное основное состояние). Геометрию всех структур реагентов и продуктов реакций оптимизировали по всем независимым переменным и без ограничений по симметрии для водной среды. Влияние растворителя учитывали в рамках модели поляризуемого континуума РСМ [17]. Для построения полости растворенного вещества задавали радиусы атомных сфер из модели силового поля UFF [18]. Характер всех стационарных точек определяли расчетом матрицы Гессе. Частоты нормальных колебаний (в гармоническом приближении) и тепловые поправки к свободной энергии (с использованием немасштабированных частот) рассчитывали теми же методами. Все полученные результаты относили к нормальным условиям (T = 298 К, Р = 1 атм) в водной среде.
Расчет методом QSPR
Расчет рKа веществ и распределение их молекулярной и ионных форм в зависимости от рН водной среды при 323 К проводили в программе Marvin 18.14 [19]. Модуль для расчета Protonation интегрирован в MarvinSketch и способен предсказывать рKа для органических молекул, смоделированных в MarvinSketch. Величины рKа оценивали путем моделирования методом QSPR [20].
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Согласно классическим представлениям метода ХЛ [9] и современным исследованиям [21–23], в реакции рекомбинации пероксирадикалов (АРOO•), генерируемых при распаде азосоединения (в данной работе гидрофильный ААРН), может возникать ХЛ, которая в случае необходимости усиливается активатором свечения Rd6G:
(I)
$\begin{gathered} {\text{ААРН}}\xrightarrow{{{{k}_{{\text{d}}}}}}2{\text{А}}{{{\text{Р}}}^{\centerdot }}{\text{\;}},\,\,\,\,{\text{А}}{{{\text{Р}}}^{\centerdot }} + {{{\text{O}}}_{2}}\xrightarrow{{{{k}_{{\text{i}}}}}}{\text{АРO}}{{{\text{O}}}^{\centerdot }}{\text{\;}}, \\ {\text{АРO}}{{{\text{O}}}^{\centerdot }} + {\text{АРO}}{{{\text{O}}}^{\centerdot }}\xrightarrow{{{{k}_{6}}}}{\text{М*}}, \\ {\text{М*}} + {\text{Rd}}6{\text{G}} \to {\text{М}} + {\text{Rd}}6{\text{G*}}, \\ {\text{Rd}}6{\text{G*}} \to {\text{Rd}}6{\text{G}} + h{\nu }, \\ \end{gathered} $Если считать, что ХЛ (I0) возбуждается лишь в реакции рекомбинации ${\text{АРO}}{{{\text{O}}}^{ \bullet }}{\text{,}}$ то интенсивность ХЛ должна быть пропорциональна квадрату концентрации перекисных радикалов, т.е. I0 ~ [APOO•]2. При добавлении в систему флавоноидов как акцепторов пероксирадикалов наблюдается уменьшение интенсивности ХЛ-свечения (I) по реакции:
(II)
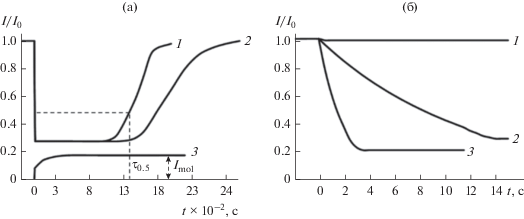
${\text{АРO}}{{{\text{O}}}^{\centerdot }} + {\text{FlavOH}}\xrightarrow{{{{k}_{7}}}}{\text{APOOH}} + {\text{Flav}}{{{\text{O}}}^{\centerdot }}{\text{\;}}.$На рис. 1а видно, что относительная интенсивность (I/I0) ХЛ сразу после введения антиоксиданта резко снижается, а затем, по мере его расходования, восстанавливается до начального уровня. При высоких концентрациях наблюдается период индукции (${{{\tau }}_{{0.5}}}$) – время полувосстановления ХЛ, когда I становится равной половине I0 [23]. Однако в периоде индукции ХЛ не ослабляется до нуля. Это, вероятно, связано с возбуждением ХЛ в каких-либо нерадикальных (молекулярных) реакциях (Imol), на которые перехватчик радикалов FlavOH не влияет. В отдельном опыте было показано, что появление молекулярной ХЛ связано с влиянием активатора свечения. Относительную интенсивность Imol определяли, когда в реакционной смеси присутствовал только растворитель и Rd6G, и ее величина не превышала 0.18 (рис. 1а).
Рис. 1.
а – Кинетика ХЛ при окислении ААРН в фосфатном буфере с рН 7.35 после введения катехина, моль/л: 1 – 9 × 10–6, 2 – 1.1 × 10–5, 3 – без добавок катехина и ААРН. б – Начальные участки кинетических кривых ХЛ при окислении ААРН после введения катехина ([FlavOH] = 9 × 10–6 моль/л) в буферных растворах с разными значениями рН: 1 – 2, 2 – 7.35, 3 – 9. Т = 323 К, [ААРН] = 7.3 × 10–4 моль/л, Wi = 5 × 10–9 моль л–1 c–1, [Rd6G] = 3 × 10–3 моль/л.
Константу скорости реакции (II) k7 определяли в фосфатном буфере при рН 7.35 методом, предложенным в работе [9], при условии низких скоростей инициирования (${{W}_{{\text{i}}}}$) и достаточно высоких концентрациях антиоксиданта. Расчет k7 проводили в начальный момент времени, когда концентрация FlavО• крайне мала и реакциями (III) и (IV) пероксирадикалов с продуктами превращения FlavOH можно пренебречь:
(III)
${\text{АРO}}{{{\text{O}}}^{\centerdot }} + {\text{Flav}}{{{\text{O}}}^{\centerdot }}\xrightarrow{{{{k}_{8}}}}{\text{продукты\;}},$(IV)
${\text{Flav}}{{{\text{O}}}^{\centerdot }} + {\text{Flav}}{{{\text{O}}}^{\centerdot }}\xrightarrow{{{{k}_{9}}}}{\text{продукты}}.$С учетом реакций (I)–(III) изменение концентрации перекисных радикалов описывается дифференциальным уравнением:
(1)
$\begin{gathered} \frac{{{\text{d}}\left[ {{\text{APO}}{{{\text{O}}}^{\centerdot }}} \right]}}{{{\text{d}}t}} = {{W}_{{\text{i}}}} - {{k}_{6}}{{\left[ {{\text{APO}}{{{\text{O}}}^{\centerdot }}} \right]}^{2}} - \\ - \,\,{{k}_{7}}\left[ {{\text{APO}}{{{\text{O}}}^{\centerdot }}} \right]\left[ {{\text{FlavOH}}} \right] - {{k}_{8}}\left[ {{\text{APO}}{{{\text{O}}}^{\centerdot }}} \right]\left[ {{\text{Flav}}{{{\text{O}}}^{\centerdot }}} \right]. \\ \end{gathered} $В начальный момент, когда [FlavO•] → 0, последним членом в уравнении (1) можно пренебречь. Тогда его решение при условии ${{k}_{7}}{{\left[ {{\text{FlavOH}}} \right]}_{0}} \gg $ $ \gg \sqrt {{{k}_{6}}{{W}_{{\text{i}}}}} $ и после преобразований будет иметь вид [9]:
где k7[FlovOH]0 = k' – угловой параметр линейной зависимости (1), равный константе скорости реакции первого порядка (с–1), [FlovOH]0 – начальная концентрация флавоноида (моль/л), t – время (с).Начальный участок ХЛ-кривой (рис. 2а) спрямляется (рис. 2б) в полулогарифмических координатах кинетического уравнения первого порядка (1а). Величина k' полученных анаморфоз пропорциональна начальной концентрации флавоноида (рис. 3а), что позволяет рассчитать значение $~{{k}_{7}}$ с использованием линейного регрессионного анализа в системе Statistica Demo 6.0. Представленный метод расчета констант ${{k}_{7}}$ [9] крайне удобен, поскольку позволяет исключить влияние на кинетику реакции продуктов превращения флавоноидов, а также не требует знания величины константы скорости ${{k}_{6}},$ определение которой возможно только после детального изучения ХЛ реакций самого инициатора и образующихся при его распаде радикалов [22].
Рис. 2.
а – Кинетика ХЛ при окислении ААРН в фосфатном буфере (рН 7.35) после введения катехина, моль/л: 1 – 1.1 × 10–5, 2 – 9 × 10–6, 3 – 7 × 10–6. б – Полулогарифмические анаморфозы кинетических кривых ХЛ (рис. а). Т = 323 К, [ААРН] = 7.3 × 10–4 моль/л, Wi = = 5 × 10–9 моль л–1 c–1, [Rd6G] = 3 × 10–3 моль/л.
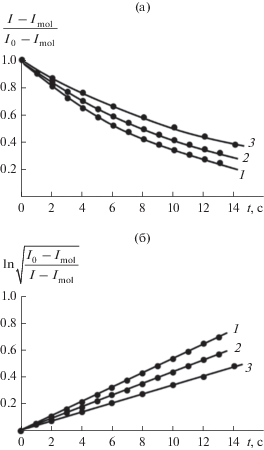
Рис. 3.
а – Определение константы скорости реакции k7 из зависимости углового параметра (k') линейных анаморфоз (рис. 2б) от начальной концентрации катехина [FlavOH]. б – Распределение доли ионных форм кемпферола (W, %) в зависимости от рН среды (вода, 323 К), рассчитанное методом QSPR в программе Marvin 18.14: 1 – FlavOН, 2 – FlavO– (7-ОН), 3 – FlavO2– (5,7-ОН), 4 – FlavO– (5-ОН). Представлено распределение ионов, доля которых превышает 5%.
Анализ полученных констант k7 (табл. 1), которые являются характеристикой АРА флавоноидов, показал, что соединения группы флаванолов‑3,4 (катехинов, I и II) проявили низкую антирадикальную активность, что может быть связано с непродуктивным расходованием вещества в реакциях полимеризации и поликонденсации, к которым склонны катехины [1], вследствие их крайней неустойчивости из-за отсутствия двойной связи в положении 2, 3 и карбонильной группы >С=О кольца С. Среди флавонолов, как наиболее устойчивой (окисленной) группы антиоксидантов, фактически все исследуемые соединения III–VII высокореакционноспособны по отношению к пероксирадикалам (k7 > 104–105 л моль–1 с–1). Гликозиды флавоноидов (VIII и IX) – кверцитрин и рутин – проявили более высокую активность, чем их агликон – кверцетин (IV), что, по-видимому, связано с их лучшей растворимостью. Из распределения ионных форм фенолов (рис. 3б) в зависимости от рН среды при 323 К (растворитель – вода), рассчитанного методом QSPR в программе Marvin 18.14.0, следует, что при рН 7.35 (табл. 1) представленные флавоноиды частично диссоциируют с образованием фенолят-ионов (FlavO–), способных реагировать с гидразильным радикалом путем последовательной потери протона с последующей лимитирующей (slow) стадией переноса электрона (Sequential Proton Loss Electron Transfer, SPLET) [25, 26]:
(V)
$\begin{gathered} {\text{FlavOH}} + {{{\text{H}}}_{2}}{\text{O\;}} \rightleftarrows {\text{Flav}}{{{\text{O}}}^{--}} + {{{\text{H}}}_{3}}{{{\text{O}}}^{ + }}, \\ {\text{Flav}}{{{\text{O}}}^{--}} + {\text{APO}}{{{\text{O}}}^{ \bullet }}{\text{\;}}\xrightarrow{{{\text{slow}}}}{\text{Flav}}{{{\text{O}}}^{\centerdot }} + {\text{APO}}{{{\text{O}}}^{--}}, \\ {\text{APO}}{{{\text{O}}}^{--}} + {{{\text{H}}}_{3}}{{{\text{O}}}^{ + }} \rightleftarrows {\text{APOO}}--{\text{H}} + {{{\text{H}}}_{2}}{\text{O}}. \\ \end{gathered} $Таблица 1.
Экспериментальные константы скорости k7, величины рKa флавоноидов и доля образовавшихся ионов FlavO– (W), рассчитанные в программе Marvin 18.14, а также их потенциалы ионизации ($P{{I}_{{{\text{FlavOH}}}}},$ $P{{I}_{{{\text{Flav}}{{{\text{O}}}^{--}}}}}$), вычисленные методом DFT на уровне B3LYP/6-311++G(d,p). Растворитель – вода
| Соединение | k7, л моль–1 с–1 | pKa* (323 K) |
W, % (pH 7.35, 323 К) |
$P{{I}_{{{\text{FlavOH}}}}},$ кДж/моль | $P{{I}_{{{\text{Flav}}{{{\text{O}}}^{--}}}}}$*, кДж/моль |
|---|---|---|---|---|---|
| I | (4.78 ± 0.14) × 103 | 8.64 (3'-ОН) | 0.9 | 571 | 421 (3'-ОН) |
| II | (9.5 ± 0.3) × 103 | 8.37 (4'-ОН) | 2.3 | 569 | 418 (4'-ОН) |
| III | (3.75 ± 0.12) × 104 | 6.22 (7-ОН) | 57.9 | 564 | 407 (7-ОН) |
| IV | (6.9 ± 0.3) × 104 | 6.22 (7-ОН) | 57.5 | 562 | 405 (7-ОН) |
| V | (4.21 ± 0.12) × 104 | 6.22 (7-ОН) | 60.3 | 563 | 406 (7-ОН) |
| VI | (2.92 ± 0.12) × 105 | 6.21 (7-ОН) | 48.1 | 554 | 394 (7-ОН) |
| VII | (1.13 ± 0.04) × 105 | 6.21 (7-ОН) | 51.2 | 559 | 402 (7-ОН) |
| VIII | (7.1 ± 0.3) × 104 | 6.21 (7-ОН) | 58.8 | 560 | 403 (7-ОН) |
| IX | (8.5 ± 0.3) × 104 | 6.37 (7-ОН) | 71.9 | 557 | 404 (7-ОН) |
Недиссоциированные молекулы FlavOH могут дезактивировать APOO• по механизму последовательного переноса электрона с последующей потерей протона (Electron Transfer–Proton Transfer, ЕТ–РТ) [27]:
(VI)
$\begin{gathered} {\text{FlavOH}} + {\text{APO}}{{{\text{O}}}^{\centerdot }}{\text{\;}}\xrightarrow{{{\text{slow}}}} \\ \to {\text{FlavO}}{{{\text{H}}}^{{\centerdot + }}} + {\text{APO}}{{{\text{O}}}^{--}}, \\ {\text{FlavO}}{{{\text{H}}}^{{\centerdot + }}} + {{{\text{H}}}_{2}}{\text{O\;}} \rightleftarrows {\text{\;\;Flav}}{{{\text{O}}}^{\centerdot }} + {{{\text{H}}}_{3}}{{{\text{O}}}^{ + }}, \\ {\text{APO}}{{{\text{O}}}^{--}} + {{{\text{H}}}_{3}}{{{\text{O}}}^{ + }} \rightleftarrows {\text{APOO}}--{\text{H}} + {{{\text{H}}}_{2}}{\text{O}}. \\ \end{gathered} $Механизм переноса атома Н (Hydrogen Atom Transfer, НАТ) с участием FlavOH
Определить, какой из двух параллельных механизмов – SPLET или ET–PT – является доминирующим и определяет скорость реакции, в целом можно путем изменения рН реакционной смеси. На примере катехина показано (рис. 1б), что введение антиоксиданта в солянокислый буфер (рН 1.5) не приводит к падению интенсивности ХЛ, а значит, концентрация пероксирадикалов не изменяется. По сравнению с рН 7.35 (кривая 2) в кислых средах (кривая 1) реакция FlavO– с APOO• подавляется вследствие снижения концентрации ионных форм, более активных в реакции с пероксирадикалом, чем нейтральные молекулы [28]. Напротив, замена кислого буфера на борно-щелочной (рН 9) приводит к резкому увеличению скорости расходования радикалов в присутствии FlavOH (кривая 3). Таким образом, зависимость интенсивности ХЛ исследуемой реакции от рН среды подтверждает, что в нейтральных средах преобладает механизм SPLET.
Квантово-химическим дескриптором, связанным с реализацией механизма SPLET в нейтральных средах, является потенциал ионизации фенолят-иона флавоноида, который непосредственно участвует в лимитирующей стадии реакции (V). Концентрация такого фенолята в реакционной смеси должна быть максимальна по сравнению с другими ионными формами, а значит, он образуется по I ступени диссоциации. Нередко при исследовании механизма SPLET рассчитывается энтальпия ионизации аниона антиоксиданта без учета его доли в системе [31] либо для прогнозирования АРА вещества используется только его константа диссоциации [32], что не позволяет полностью учесть все факторы, влияющие на реакционную способность соединения.
Поиск и расчет структуры фенолята, участвующего в лимитирующей стадии реакции, осуществляли следующим образом: вначале в программе Marvin 18.14 были рассчитаны рKа изученных веществ, по величинам которых определяли OH-группы флавоноидов, диссоциирующие по I ступени, а затем квантово-химическим методом рассчитывались значения адиабатических потенциалов ионизации образующихся ионов в среде воды.
Скорость реакции FlavOH с APOO• (VI), протекающей по механизму ЕТ–РТ, будет зависеть от потенциала ионизации молекулы флавоноида, рассчитанного по формуле:
В нейтральном буфере при физиологическом значении рН 7.35 активные фенолят-ионы FlavO– будут образовываться в количестве (табл. 1, рис. 3б), достаточном для участия в лимитирующей стадии (V) по механизму SPLET. На это указывает как зависимость интенсивности ХЛ исследуемой реакции от рН среды (рис. 1б), так и более высокая корреляция антирадикальной активности (lnk7) изученных флавоноидов с расчетными значениями $P{{I}_{{{\text{Flav}}{{{\text{O}}}^{--}}}}}$ (уравнение (2)) по сравнению с $P{{I}_{{{\text{FlavOH}}}}}$ (уравнение (3)).
(2)
$\begin{gathered} {\text{ln}}{{k}_{{\text{7}}}} = \left( {{\text{72}} \pm {\text{3}}} \right)--\left( {{\text{15}}.{\text{1}} \pm 0.{\text{7}}} \right) \times {\text{1}}{{0}^{{ - {\text{2}}}}}P{{I}_{{{\text{Flav}}{{{\text{O}}}^{--}}}}}, \\ n = {\text{9}},\,\,\,\,r = 0.{\text{992}},\,\,\,\,{{r}^{2}} = 0.{\text{984}},\,\,\,\,F = {\text{425}}, \\ p < 0.0000{\text{1}},\,\,\,\,{{S}_{{{\text{est}}}}} = 0.{\text{17}}, \\ \end{gathered} $(3)
$\begin{gathered} {\text{ln}}{{k}_{{\text{7}}}} = \left( {{\text{1}}.{\text{37}} \pm 0.{\text{11}}} \right) \times {\text{1}}{{0}^{{{\text{2 }}}}}--\left( {{\text{22}}.{\text{4}} \pm {\text{1}}.{\text{9}}} \right) \times \\ \times \,\,{\text{1}}{{0}^{{ - {\text{2 }}}}}P{{I}_{{{\text{FlavOH}}}}}, \\ n = {\text{9}},\,\,\,\,r = 0.{\text{974}},\,\,\,\,{{r}^{2}} = 0.{\text{949}},\,\,\,\,F{\text{ }} = {\text{ 138}}, \\ p < 0.0000{\text{1}},\,\,\,\,{{S}_{{{\text{est}}}}} = 0.{\text{29}}. \\ \end{gathered} $С ростом потенциалов ионизации FlavO– реакционная способность антиоксидантов закономерно падает (табл. 1), что обусловлено с точки зрения доминирования механизма SPLET замедлением переноса электрона с иона на радикал. При этом по величине коэффициента корреляции r видно, что в водной среде влияние потенциала ионизации молекулы FlavOH явно ниже, а значит, механизм ЕТ–РТ можно считать параллельным конкурирующим, но не доминирующим. При этом установить зависимость между $P{{I}_{{{\text{Flav}}{{{\text{O}}}^{ - }}}}},$ PIFlavOH и lnk7 не удалось, поскольку взаимосвязь между независимыми переменными превышает 0.9, а значит, многофакторный анализ не возможен [34].
Анализ взаимосвязи дескриптор–активность показал, что флавонолы (III–IX) с более низкими значениями рKа проявляют наиболее высокую реакционную способность по отношению к АРОО•. Это обусловлено не только их лучшей способностью к диссоциации, но и более низкими потенциалами ионизации их молекул (табл. 1). Особенно низкая АРА характерна для флаванов, потенциалы ионизации фенолят-ионов которых выше, чем у флавонолов, а значит, ионы соединений I и II будут медленнее отдавать электрон в реакции с радикалами АРОО• по механизму SPLET.
При использовании методологии QSAR важно контролировать прогностическую способность построенных моделей. Один из наиболее надежных подходов к решению этой задачи состоит в использовании контрольной выборки. Последняя необходима для независимого контроля предсказательной способности моделей. Такой подход позволяет в большинстве случаев избежать случайных корреляций.
В качестве такой группы использовали гидроксиацетофеноны (AcArOH) (X–XVI) как соединения, относящиеся к природным растительным фенолам [1] и имеющие близкую с ними химическую структуру и одинаковый антирадикальный механизм действия. Гидроксиацетофеноны редко встречаются в растительных тканях [1], но используются при исследовании окислительно-восстановительных свойств флавоноидов [33], поскольку моделируют фрагменты молекулы флавонолов, которые формируют кольца А и С.
По молекулярному дескриптору гидроксиацетофенонов $\left( {P{{I}_{{{\text{AcAr}}{{{\text{O}}}^{--}}}}}} \right),$ рассчитанному по аналогии с флавоноидами, были спрогнозированы константы скорости k7(calc) их реакции с АРОО• по уравнению (2) (табл. 2). Согласно предложенной модели, величина константы k7(calc) > 104–105 л моль–1 с–1 только для 3,4- и 2,5-гидроксиацетофенонов, которые можно выбрать в качестве потенциальных эффективных антиоксидантов. Все остальные соединения по результатам скрининга должны проявлять умеренную или слабую антирадикальную активность.
Таблица 2.
Экспериментальные k7 и расчетные k7(calc) константы скорости, величины рKa гидроксиацетофенонов и доля образовавшихся ионов AcArO– (W), рассчитанные в программе Marvin 18.14, а также их потенциалы ионизации ($P{{I}_{{{\text{AcArOH}}}}},$ $P{{I}_{{{\text{AcAr}}{{{\text{O}}}^{--}}}}}$), вычисленные методом DFT на уровне B3LYP/6-311++G(d,p). Растворитель – вода
| Соединение | k7, л моль–1 с–1 | k7(calc)*, л моль–1 с–1 | Δk, % | $\overline {{{\Delta }_{k}}} ,$ % | pKa** (323 К) |
W, % (pH 7.35, 323 К) |
$P{{I}_{{{\text{AcAr}}{{{\text{O}}}^{--}}}}}$**, кДж/моль |
|---|---|---|---|---|---|---|---|
| X | (3.14 ± 0.12) × 103 | 3.5 × 103 | 12.1 | 8.5 | 8.77 | 2.8 | 424 |
| XI | (1.04 ± 0.04) × 104 | 1.2 × 104 | 13.3 | 8.56 | 2.9 | 416 | |
| XII | (7.2 ± 0.3) × 103 | 7.5 × 103 | 4 | 7.52 | 29.1 | 419 | |
| XIII | (5.7 ± 0.3) × 103 | 5.5 × 103 | 2.9 | 7.6 (4-ОН) | 23.5 | 421 (4-ОН) | |
| XIV | (1.28 ± 0.04) × 105 | 1.1 × 105 | 11.4 | 8.84 (5-ОН) | 0.6 | 401 (5-ОН) | |
| XV | (7.6 ± 0.3) × 104 | 7.2 × 104 | 5.5 | 7.63 (4-ОН) | 22 | 404 (4-ОН) | |
| XVI | (1.74 ± 0.04) × 103 | 1.9 × 103 | 10.6 | 7.97 | 11.8 | 428 |
* Константы скорости реакции рассчитаны по уравнению (2). ** В скобках указано положение фенольной ОН-группы, которая диссоциирует по I ступени.
Чтобы иметь более наглядное суждение о качестве предложенной модели из относительных отклонений для каждой константы $\left( {{{\Delta }_{k}}_{{_{{_{{_{{_{{_{{_{{}}}}}}}}}}}}}}^{{^{{^{{^{{}}}}}}}} = } \right.$ $\left. { = \left| {\frac{{\left( {{{k}_{7}} - {{k}_{{7\left( {{\text{calc}}} \right)}}}} \right)}}{{{{k}_{7}}}}} \right| \times 100\% } \right)$ определяли среднюю ошибку аппроксимации $\left( {{{{\bar {\Delta }}}_{k}}{{ = 1} \mathord{\left/ {\vphantom {{ = 1} {n\sum {{{\Delta }_{k}}} }}} \right. \kern-0em} {n\sum {{{\Delta }_{k}}} }}} \right),$ которая не должна превышать 8–10% [34]. Как было установлено (табл. 2), единичные относительные отклонения предсказанных констант изменяются от 4 до 13%, а средняя ошибка аппроксимации не превышает 8.5%, что свидетельствует о хорошем подборе модели (уравнение (2)). Наибольшие погрешности в расчете характерны для тех соединений (X, XI, XIV и XVI), которые незначительно диссоциируют на ионы, что, по-видимому, объясняется большим влиянием на АРА молекулярных форм, участвующих в конкурирующем механизме ЕТ–РТ. Поскольку k7 и k7(calc) значительно не отличаются, то k7 для FlavOH и AcArOH были объединены в одну выборку данных и скоррелированы с соответствующими потенциалами ионизации ионов (рис. 4).
(4)
$\begin{gathered} {\text{ln}}{{k}_{7}} = \left( {{\text{73}}.{\text{2}} \pm {\text{1}}.{\text{4}}} \right)--\left( {{\text{15}}.{\text{4}} \pm 0.{\text{3}}} \right) \times {\text{1}}{{0}^{{ - {\text{2}}}}}P{{I}_{{{\text{Flav}}{{{\text{O}}}^{--}}}}}, \\ n = {\text{16}},\,\,\,\,r = 0.{\text{996}},\,\,\,\,{{r}^{2}} = 0.{\text{992}},\,\,\,\,F{\text{ }} = {\text{2}}00{\text{3}}, \\ p < 0.0000{\text{1}},\,\,\,\,{{S}_{{{\text{est}}}}} = 0.{\text{13}}. \\ \end{gathered} $Рис. 4.
Линейная однофакторная регрессионная зависимость АРА (ln k7) флавоноидов (⚫) и гидроксиацетофенонов (⚪) от потенциала ионизации их фенолят-ионов ($P{{I}_{{{\text{Flav}}{{{\text{O}}}^{ - }}}}},$ $P{{I}_{{{\text{AcAr}}{{{\text{O}}}^{ - }}}}}$).
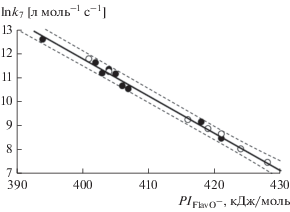
По сравнению с зависимостью для FlavOH (уравнение (2)) видно, что угловой и свободный параметры расширенной регрессии (уравнение (4)) фактически не изменяются, коэффициент корреляции возрастает до 0.996, стандартная ошибка оценки предсказанных величин уменьшается до Sest = 0.13, все точки лежат в границах доверительного интервала (рис. 4), а значит, данные по AcArOH могут быть внесены в общее регрессионное уравнение.
ЗАКЛЮЧЕНИЕ
Подбор молекулярных дескрипторов флавоноидов зависит от механизма их окислительных превращений в реакции с пероксирадикалами. Путем использования контрольной выборки (группы гидроксиацетофенонов) была показана высокая прогностическая способность построенной линейной модели связи химической структуры и антирадикальной активности флавоноидов с относительной ошибкой аппроксимации не более 8.5%. За счет контрольной выборки объем данных может быть увеличен до 16 соединений, что будет способствовать повышению прогностической способности предложенной модели как для флавоноидов, так и для их генетических предшественников. Полученная количественная взаимосвязь дескриптор–активность представляет собой не только готовое полуэмпирические однофакторное уравнение для подбора эффективных антиоксидантов, но и является основой скрининга антирадикальной активности природных фенолов в реакции с пероксирадикалами в разных средах.
Список литературы
Vermerris W., Nicolson R. Phenolic Compound Biochemistry. Dodrecht: Springer, 2006. 276 p.
Костюк В.А., Потапович А.И. Биорадикалы и биоантиоксиданты. Минск: БГУ, 2004. 179 с.
Alov P., Tsakovska I., Pajeva I. // Curr. Top. Med. Chem. 2015. V. 15. P. 85.
Lagunin A., Zakharov A., Filimonov D., Poroikov V. // Mol. Inform. 2011. V. 30. № 2–3. P. 241.
Javan A.J. // Food Chem. 2014. V. 165. P. 451.
Vaganek A., Rimarčik J., Dropkova K., Lengyel J., Klein E. // Comput. Theoret. Chem. 2014. V. 1050. P. 31.
Рабинович В.А., Хавин З.Я. Краткий химический справочник. Л.: Химия, 1991. 432 с.
Armarego W.L.F., Chai C.L.L. Purification of Laboratory Chemicals. Burlington: Elsevier Science, 2003. 608 p.
Шляпинтох В.Я., Карпухин О.Н., Постников Л.М., Захаров И.П., Вичутинский А.А., Цепалов В.Ф. Хемилюминесцентные методы исследования медленных химических процессов. М.: Наука, 1966. 300 с.
Stefek M., Kyselova Z., Rackova L., Krizanova L. // Biochim. Biophys. Acta. 2005. V. 1741. P. 183.
Rackova L., Stefek M., Majekova M. // Redox Report. 2002. V. 7. P. 207.
Беляков В.А., Васильев Р.Ф., Федорова Г.Ф. // Кинетика и катализ. 2004. Т. 45. № 3. С. 355.
Южаков В.И., Пащенко В.З. // Квантовая электроника. 1980. Т. 7. № 3. Р. 613.
Frisch M.J., Trucks G.W., Schlegel H.B., Scuseria G.E., Robb M.A., Cheeseman J.R., Scalmani G., Barone V., Mennucci B., Petersson G.A., Nakatsuji H., Caricato M., Li X., Hratchian H.P., Izmaylov A.F., Bloino J., Zheng G., Sonnenberg J.L., Hada M., Ehara M., Toyota K., Fukuda R., Hasegawa J., Ishida M., Nakajima T., Honda Y., Kitao O., Nakai H., Vreven T., Montgomery J.A., Jr., Peralta J.E., Ogliaro F., Bearpark M., Heyd J.J., Brothers E., Kudin K.N., Staroverov V.N., Keith T., Kobayashi R., Normand J., Raghavachari K., Rendell A., Burant J.C., Iyengar S.S., Tomasi J., Cossi M., Rega N., Millam J.M., Klene M., Knox J.E., Cross J.B., Bakken V., Adamo C., Jaramillo J., Gomperts R., Stratmann R.E., Yazyev O., Austin A.J., Cammi R., Pomelli C., Ochterski J.W., Martin R.L., Morokuma K., Zakrzewski V.G., Voth G.A., Salvador P., Dannenberg J.J., Dapprich S., Daniels A.D., Farkas O., Foresman J.B., Ortiz J.V., Cioslowski J., Fox D.J. Gaussian 09, Revision B.01 Gaussian, Inc., Wallingford CT, 2010.
Weinberg D.R., Gagliardi C.J., Hull J.F., Murphy C.F., Kent C.A., Westlake B., Paul A., Ess D.H., McCafferty G.D., Meyer T.J. // Chem. Rev. 2007. V. 107. № 11. P. 5004.
Белая Н.И., Белый А.В., Заречная О.М., Щербаков И.Н., Михальчук В.М., Дорошкевич В.С. // ЖОХ. 2017. Т. 87. № 4. С. 556.
Tomasi J., Mennucci B., Cammi R. // Chem. Rev. 2005. V. 105. № 8. P. 2999.
Rappe A.K., Casewit C.J., Colwell K.S., Goddard W.A., Skiff W.M. // J. Am. Chem. Soc. 1992. V. 114. № 25. P. 10 024.
Marvin 18.14.0. ChemAxon (https://www.chemaxon.com).
Elyashberg M., Williams A., Blinov K. Contemporary Computer-Assisted Approaches to Molecular Structure Elucidation. Cambridge: RSC Publishing, 2012. 469 p.
Алексеев А.В., Проскурнина Е.В., Владимиров Ю.А. // Вестн. моск. ун-та. Сер. 2. Химия. 2012. Т. 53. № 3. С. 187.
Вепринцев Т.Л., Наумов В.В., Трофимов А.В. // Бутлеров. сообщ. 2011. Т. 25. № 5. С. 96.
Васильев Р.Ф., Вепринцев Т.Л., Долматова Л.С., Наумов В.В., Трофимов А.В., Цаплев Ю.Б. // Кинетика и катализ. 2014. Т. 55. № 2. С. 157.
Рогинский В.А. Фенольные антиоксиданты. Реакционная способность и эффективность. М.: Наука, 1988. 247 с.
Galano A., Mazzone G., Alvarez-Diduk R., Marino T., Alvarez-Idaboy J.R., Russo N. // Annu. Rev. Food Sci. Technol. 2016. V. 7. P. 335.
Mazzone G., Russo N., Toscano M. // Comput. Theoret. Chem. 2016. V. 1077. P. 39.
Litwinienko G., Ingold K.U. // Acc. Chem. Res. 2007. V. 40. № 3. P. 222.
Волков В.А., Мисин В.М. // Кинетика и катализ. 2015. Т. 56. № 1. С. 48.
Mazzone G., Russo N., Toscano M. // Comput. Theoret. Chem. 2016. V. 1077. P. 39.
Белая Н.И., Белый А.В., Заречная О.М., Щербаков И.Н., Дорошкевич В.С. // ЖОХ. 2018. Т. 88. № 7. С. 1057.
Marković Z., Đorović J., Petrović Z.D. // J. Mol. Model. 2015. V. 21. P. 293.
Pérez-González A., Galano A., Alvarez-Idaboy J.R. // New J. Chem. 2014. V. 38. P. 2639.
Черников Д.А., Пальшин В.А., Баженов Б.Н., Сафронов А.В. // Изв. вузов. Химия и хим. технология. 2012. Т. 55. № 8. С. 43.
Дерффель К. Статистика в аналитической химии. М.: Мир, 1994. 268 с.
Дополнительные материалы отсутствуют.
Инструменты
Кинетика и катализ