

Кинетика и катализ, 2020, T. 61, № 6, стр. 864-872
Влияние промотора M (M = Au, Ag, Cu, Ce, Fe, Ni, Co, Zn) на активность Pd–M/Al2O3-катализаторов конверсии этанола в α-спирты
С. А. Николаев a, *, М. В. Цодиков b, А. В. Чистяков b, П. А. Чистякова b, Д. И. Эзжеленко a, И. Н. Кротова a
a ФГБОУ ВО Московский государственный университет имени М.В. Ломоносова, Химический факультет
119991 Москва, Ленинскиегоры, 1, стр. 3, Россия
b ФГБУН Институт нефтехимического синтеза имени А.В. Топчиева РАН
119991 Москва, Ленинский просп., 29, Россия
* E-mail: serge2000@rambler.ru
Поступила в редакцию 17.02.2020
После доработки 23.04.2020
Принята к публикации 04.05.2020
Аннотация
С помощью ионного обмена и пропитки получены катализаторы Pd/Al2O3 и Pd–M/Al2O3 (M = Au, Ag, Cu, Ce, Fe, Ni, Co, Zn). Установлено, что Pd/Al2O3 обладает высокой начальной активностью в конверсии этанола в α-спирты, однако спустя 10 ч работы катализатор теряет 90% своей активности за счет дезактивации, которая обусловлена хемосорбцией побочного продукта (CO) на атомах Pd. Модификация Pd золотом или серебром приводит к росту скорости дезактивации Pd. В результате системы Pd–Au и Pd–Ag менее активны и стабильны. Напротив, системы Pd–Fe, Pd–Co, Pd–Ni, Pd–Cu, Pd–Zn и Pd–Ce проявляют бóльшую, чем у Pd, устойчивость к отравлению CO и демонстрируют высокую активность и стабильность. Наблюдаемые закономерности каталитического действия моно- и биметаллических систем объясняются в рамках модели d-зоны, предложенной Хаммером и Норсковым. Наиболее эффективен в целевом процессе Pd–Cu/Al2O3, который не отравляется CO и позволяет проводить конверсию этанола в α-спирты c селективностью 95%, при этом время его стабильной работы составляет не менее 100 ч. Структура каталитической системы Pd–Cu изучена с помощью ПЭМ, ЭДА, РФЭС, ТПВ-H2, ТПД-NH3. Предложена модель активных центров катализатора.
ВВЕДЕНИЕ
Разработка технологий конверсии биомассы для производства энергоносителей и синтетических углеводородов давно привлекает внимание ученых [1, 2]. Наиболее отработанным процессом в этом направлении можно считать получение из биомассы этанола (биоэтанол), который применяется в качестве добавки к моторному топливу [3]. По данным Министерства Энергетики США мировой профицит производства биоэтанола в 2016 г. составил 10 млн тонн в год и продолжает расти. Таким образом, биоэтанол можно рассматривать в качестве перспективного сырья для разработки на его основе новых технологий синтеза ценных продуктов.
Одним из перспективных процессов конверсии этанола в ценные продукты является каталитическая реакция, приводящая к формированию линейных α-спиртов (бутанол, гексанол, октанол) путем конденсации углеводородного скелета этанола при 250–300○С [1, 2]. Согласно данным [1–4] образование бутанола протекает через цепочку реакций: дегидрирования этанола в этаналь; конденсации двух молекул этаналя в 2-бут-2-эналь; гидрирования 2-бут-2-эналь в бутаналь с последующим гидрированием бутаналя в бутанол (схема 1 ). Формирование гексанола и октанола протекает по аналогичному механизму. В ходе конверсии этанола в α-спирты также образуются побочные продукты: этоксиэтан, метан и моноксид углерода (схема 1 ).
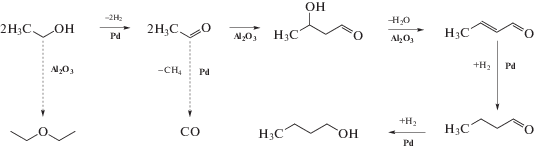
Схема 1 . Конденсация этанола в бутанол-1 на бифункциональном катализаторе Pd/Al2O3 при 275○С (сплошные стрелки). Побочные процессы: декарбонилирование на Pd-центрах и дегидратация этанола на кислых центрах Al2O3 (пунктирные стрелки). По данным [1–5, 8, 11].
Наибольшую активность в каталитической конверсии этанола в α-спирты проявляют бифункциональные системы M0/Al2O3 (M = Pd, Pt, Ni, Cu и др.), в которых металл является катализатором гидрирования–дегидрирования интермедиатов реакции, а кислотно-оснóвные центры оксида алюминия катализируют конденсацию этаналя в 2-бут-2-эналь. Эффективность работы таких систем рассмотрена в обзорных работах [2–5]. Некоторые представительные данные приведены ниже. Marcu и др. изучали активность систем M/MgO–Al2O3 (M = Pd, Ag, Cu, Fe и Sm) в конверсии этанола при 200°C [6]. Лучшие показатели процесса были достигнуты при использовании катализатора 5%Pd/MgO–Al2O3 (конверсия этанола – 12%; селективность по α-спиртам – 72%). Riittonen и др. исследовали активность систем M/Al2O3 (M = Pd, Pt, Ru, Rh и Ni) в конверсии этанола при 250°C [7]. Наиболее эффективным оказался катализатор 20%Ni/Al2O3 (конверсия этанола – 25%; селективность по α-спиртам – 80%). Однако при этом авторы работы [7] отмечали, что в ходе длительных экспериментов системы Ni/Al2O3 проявляли низкую стабильность работы из-за дезактивации в ходе реакции.
Быстрая дезактивация монометаллических катализаторов конверсии этанола в α-спирты была выявлена и при изучении кинетики процесса на 0.1%Pd/Al2O3 [8]. Установлено, что в первые 2 ч реакции образец Pd/Al2O3 проявлял высокую активность в целевом процессе, но в последующие 3 ч она резко падала и становилась сопоставима с активностью немодифицированного Al2O3. Следовательно, с течением времени активные центры Pd переставали участвовать в катализе. Также было обнаружено, что в присутствии Pd/Al2O3 целевой процесс сопровождался побочной реакцией декарбонилирования (схема 1 ) с выделением CO, хемосорбция которого на Pd приводила к формированию прочных комплексов Pd–CO [9, 10]. Очевидно, при этом должна была сокращаться доля активных центров Pd, доступных для сорбции этанола, в результате чего скорость целевого процесса снижалась. Авторы работы [8] выдвинули гипотезу о вероятной дезактивации Pd-компоненты катализатора конверсии этанола в α-спирты за счет хемосорбции CO.
Целью настоящей работы являлась разработка новых катализаторов конверсии этанола в α‑спирты, стабильно работающих в среде CO. Для достижения поставленной задачи было изучено влияние промоторов M на активность Pd в модельных катализаторах 0.1%Pd–M/Al2O3 (M = = Au, Ag, Cu, Ce, Fe, Ni, Co, Zn).
Выбор промоторов не случаен и обусловлен двумя причинами. Во-первых, M/Al2O3 являются менее активными катализаторами синтеза α‑спиртов по сравнению с Pd/Al2O3 [11, 12], что позволяет связать активность Pd–M/Al2O3 именно с активностью атомов Pd в составе биметаллических систем. Во-вторых, металлы M представляют собой модификаторы электронной структуры Pd, которые согласно [13–15] смещают положение центра d-зоны Pd относительно уровня Ферми: Pd–Co (–3.2 эВ) < Pd–Fe (–3.1 эВ) < Pd–Ni (‒2.8 эВ) < Pd–Cu (–2.5 эВ) < Pd–Zn (–2.45 эВ) < < Pd–Ce (–2.3 эВ) < Pd (–1.8 эВ) < Pd–Au (–1.6 эВ) < < Pd–Ag (–1.5 эВ). Известно, что сдвиг центра d‑зоны Pd от уровня Ферми приводит к снижению энергии связывания CO с Pd, и, наоборот, в результате смещения центра d-зоны Pd к уровню Ферми энергия связывания CO с Pd растет [13, 16]. Таким образом, системы Pd–Fe/Al2O3, Pd–Co/Al2O3, Pd–Ni/Al2O3, Pd–Cu/Al2O3, Pd–Zn/Al2O3 и Pd–Ce/Al2O3 должны иметь бóльшую устойчивость к отравлению CO и демонстрировать стабильную и высокую активность в конверсии этанола в α-спирты, а Pd–Au/Al2O3 и Pd–Ag/Al2O3, наоборот, быть менее устойчивы к отравлению CO и проявлять пониженную активность по сравнению с Pd/Al2O3.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Предшественник катализаторов ${\text{Pd/A}}{{{\text{l}}}_{2}}{\text{O}}_{3}^{*}$ (0.1 вес. % Pd) готовили осаждением из раствора нитрата Pd [17]. Для этого навеску Pd(NO3)2 ⋅ 2H2O, содержащую 4.7 × 10–4 моль Pd, растворяли в 150 мл воды. С помощью 0.1 M NaOH доводили pH раствора до 7.0 и добавляли 50 г прокаленного при 350°C носителя (γ-Al2O3, Sуд = 160 м2/г, размер гранул 0.5 мм). Полученную суспензию перемешивали при 70°C в течение 1 ч. В ходе перемешивания раствор обесцвечивался, а гранулы Al2O3 окрашивались в коричневый цвет, что свидетельствовало об осаждении Pd на поверхность носителя. Далее гранулы промывали 5 л воды, сушили и прокаливали при 350°C в течение 1 ч.
Катализатор Pd/Al2O3 (0.1 вес. % Pd) получали прокаливанием ${\text{Pd/A}}{{{\text{l}}}_{2}}{\text{O}}_{3}^{*}$ при 350°C в течение 2 ч c последующим восстановлением H2 при 200°C в течение 2 ч. Катализаторы Pd–M/Al2O3 (M = Au, Ag, Cu, Ce, Fe, Ni, Co, Zn) готовили пропиткой ${\text{Pd/A}}{{{\text{l}}}_{2}}{\text{O}}_{3}^{*}$по влагоемкости [17, 18]. В типовом синтезе к 5 г ${\text{Pd/A}}{{{\text{l}}}_{2}}{\text{O}}_{3}^{*}$ добавляли 4.5 мл водного раствора нитрата M, содержащего эквимольное палладию количество M. После пропитки Pd–M/Al2O3 сушили и прокаливали при 350°C в течение 2 ч. Перед тестированием катализаторы Pd–M/Al2O3 восстанавливали H2 при 200°C в течение 2 ч.
Содержание металлов в восстановленных образцах определяли методом атомной абсорбционной спектрометрии (ААС) на приборе Thermo iCE 3000 (“Thermo Fisher Scientific”, США). Относительная погрешность измерения содержания металлов этим методом не превышает 1% [19, 20]. Фактическое содержание Pd в катализаторах составило 0.1 вес. %. Мольное отношение Pd : M в биметаллических катализаторах было близко к 1 : 1.
Микрофотографии восстановленных образцов получали с помощью метода просвечивающей электронной микроскопии (ПЭМ) на приборе JEM 2100F/UHR (”JEOL”, Япония) с разрешающей способностью 0.1 нм. Средний размер частиц определяли обработкой данных по 200–250 частицам [21, 22]. Идентификацию состава частиц проводили методом энергодисперсионного анализа (ЭДА) на приборе JED–2300 (”JEOL”, Япония).
Рентгеновские фотоэлектронные спектры металлов в восстановленных образцах регистрировали на спектрометре Axis Ultra DLD (“Kratos Analytical Limited”, Великобритания), монохроматическое AlKα-излучение (1486.6 эВ). Съемку производили с применением электронной пушки для компенсации статического заряда на гранулах катализаторов. Спектры записывали с энергией пропускания анализатора 40 эВ и шагом 0.1 эВ. Для калибровки шкалы энергии использовали внешний стандарт – золотую фольгу с энергией связывания электронов Есв(Au4 f5/2) = 83.96 ± 0.03 эВ.
Исследование невосстановленных катализаторов с помощью температурно-программированного восстановления водородом (ТПВ-Н2) осуществляли на анализаторе хемосорбции УСГА-101 (“УНИСИТ”, Россия). Навеску образца массой 0.1 г помещали в кварцевый реактор и прокаливали в токе аргона (скорость подачи – 20 мл/мин) при температуре 400○С в течение 1 ч. Образец охлаждали до 60○С, замещали ток аргона на смесь 5% H2 + 95% Ar (скорость подачи – 30 мл/мин) и нагревали образец со скоростью 10○С/мин. Поглощение водорода регистрировали детектором по теплопроводности.
Кислотность восстановленных образцов измеряли с помощью температурно-программированной десорбции NH3 (ТПД-NH3) на анализаторе хемосорбции УСГА-101 [23]. Для этого 0.2 г образца помещали в кварцевый реактор и прокаливали в токе He (скорость подачи – 20 мл/мин) при температуре 400°С в течение 1 ч. Реактор охлаждали до 25°C и насыщали образец парами аммиака в течение 30 мин. Адсорбированные физически формы аммиака удаляли прокаливанием в токе He при 100°C в течение 1 ч. Затем проводили линейный нагрев образца со скоростью 8°С/мин до 750°С в потоке гелия (скорость подачи – 30 мл/мин). Выделяющийся аммиак регистрировали с использованием детектора по теплопроводности.
Каталитические тесты осуществляли на установке автоклавного типа Parr 5000 Series (“Parr Instrument Company”, США) при подобранной ранее оптимальной температуре 275°С [11, 24]. В стандартном цикле тестирования в реактор помещали 25 мл этанола и 5 г восстановленного катализатора. Реактор нагревали до 275°С и проводили интенсивное перемешивание реакционной среды в течение 5 ч. Далее реактор охлаждали до 25°С, вскрывали и проводили качественный и количественный анализ продуктов реакции.
Продукты реакции анализировали методом газовой хроматографии: газообразные углеводороды С1–С5 – на хроматографе Кристалл-4000М (“Мета-хром”, Россия, пламенно-ионизационный детектор, колонка HP-PLOT); СО, СО2 и Н2 – на хроматографе Кристалл-4000 (“Мета-хром”, Россия), Россия, детектор по теплопроводности, колонка СКТ). Качественный состав жидких органических продуктов определяли методом хромато-масс-спектрометрии на приборах MSD 6973 (“Agilent Technologies”, США, пламенно-ионизационный детектор, колонка HP-5MS) и Automass-150 (“Delsi Nermag”, Франция, пламенно-ионизационный детектор, колонка CPSil-5) с EI = 70 эВ. Количественное содержание жидких органических веществ находили методом газо-жидкостной хроматографии на приборе Varian 3600 (“Varian”, США, колонка “Хроматэк” SE-30, 0.25 × 250 см, Df = 0.3 мм, 50°С (5 мин), 10 град/мин, 280°С, tинж = 250°С, деление потока 1/200, ПИД, внутренний стандарт – н-октан).
Конверсию этанола (α) определяли по формуле:
где (C2H5OH)кон – количество этанола в продуктах реакции, моль; (C2H5OH)исх – количество исходного этанола, моль.
Количество газообразных продуктов (ni, моль) рассчитывали по формуле:
Количество жидких продуктов (li, моль) рассчитывали по формуле:
где Ci – это массовая доля i-го компонента в жидкой фазе; mж – суммарная масса жидких продуктов, г; Mi – молекулярная масса i-го компонента.Селективность образования i-го компонента (Si) определяли по формуле:
Суммарный выход продуктов (ω, %) рассчитывали по формуле:
где α − конверсия этанола: S1 – селективность по бутанолу; S2 – селективность по гексанолу; S3 – селективность по октанолу.РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЯ
Активность Pd/Al2O3
Перед тем как переходить к обсуждению эффективности биметаллических катализаторов необходимо кратко остановиться на закономерностях конверсии этанола в присутствии немодифицированного катализатора Pd/Al2O3.
Из данных табл. 1 видно, что в 1-м цикле тестирования Pd/Al2O3 суммарная селективность по α-спиртам составляет 68.8% при конверсии этанола 24%. Соответственно выход α-спиртов на Pd/Al2O3 равен 16.5%. Однако уже во 2-м цикле тестирования выход продуктов на Pd/Al2O3 резко падает до 1.1%. Стоит отметить, что снижение выхода продуктов происходит преимущественно за счет уменьшения селективности образования α‑спиртов. Этот результат указывает на ингибирование скорости целевого процесса (схема 1 ).
Таблица 1.
Каталитические показатели конверсии этанола в α-спирты в присутствии катализаторов, содержащих 0.1% Pd*
| Катализатор | Цикл | α, % | S1, % | S2, % | S3, % | ω, % |
|---|---|---|---|---|---|---|
| Pd/Al2O3 | 1 | 24 | 65 | 3.6 | 0 | 16.5 |
| Pd/Al2O3 | 2 | 11.4 | 9.5 | 0.3 | 0 | 1.1 |
| Pd/Al2O3 + CO** | 1 | 12 | 8 | 0.5 | 0 | 1.0 |
| Pd–Ag/Al2O3 | 1 | 11.5 | 30 | 3 | 2 | 4.0 |
| Pd–Ag/Al2O3 + CO** | 1 | 7.4 | 2 | 0.1 | 0 | 0.2 |
| Pd–Au/Al2O3 | 1 | 16 | 50 | 2.5 | 0 | 8.4 |
| Pd–Ce/Al2O3 | 1 | 32 | 64 | 4 | 0 | 21.8 |
| Pd–Zn/Al2O3 | 1 | 59 | 53 | 10 | 1 | 37.8 |
| Pd–Cu/Al2O3 | 1 | 45.6 | 69.5 | 19 | 0 | 40.4 |
| Pd–Cu/Al2O3 + CO** | 1 | 41 | 73 | 19 | 2 | 38.5 |
| Pd–Cu/Al2O3 | 20 | 40.4 | 72 | 20 | 3 | 38.8 |
| Pd–Ni/Al2O3 | 1 | 29 | 73 | 10 | 2 | 24.7 |
| Pd–Fe/Al2O3 | 1 | 33 | 70 | 9.6 | 2 | 26.9 |
| Pd–Co/Al2O3 | 1 | 32 | 70 | 9 | 5 | 26.9 |
| Pd–Co/Al2O3 + CO** | 1 | 31.4 | 69.3 | 9 | 4 | 25.8 |
Примечание. α – конверсия этанола; S1, S2 и S3, – селективность образования бутанола, гексанола и октанола соответственно; ω – суммарный выход продуктов. Условия стандартного цикла тестирования: 275○С; 5 ч; 5 г катализатора; 30 мл этанола. *При 275○С выход продуктов в присутствии катализаторов 0.1%M/Al2O3 менее 1% [11]. **В автоклав с катализатором и этанолом добавляли 1 × 10–4 моль CO.
Из данных [8] следует, что каталитическим ядом для Pd-содержащих катализаторов конверсии этанола в линейные α-спирты может быть CO, который образуется в результате декарбонилирования этаналя (схема 1 ) и прочно связывается с активными центрами Pd. Для проверки применимости гипотезы об отравлении катализатора CO в наших условиях был проведен анализ газовой среды после 1-го цикла тестирования Pd/Al2O3. Оказалось, что CO действительно присутствует в составе продуктов, при этом его количество, образовавшееся в 1-м цикле, составляет 5.2 × 10–5 моль, что сопоставимо с общим количеством палладия в навеске Pd/Al2O3. Полученный результат согласуется с механизмом отравления катализатора в результате хемосорбции CO.
Для прямой проверки влияния CO на каталитическую конверсию этанола в α-спирты был выполнен эксперимент с “отравленным катализатором”. Суть эксперимента состояла в следующем. В автоклав с исходным Pd/Al2O3 и этанолом добавляли 1 × 10–4 моль CO. Далее проводили стандартный цикл тестирования и анализировали состав продуктов. Из данных табл. 1 видно, что уже в 1-м цикле выход продуктов на отравленном до реакции Pd-катализаторе был ниже, чем при конверсии этанола в присутствии исходного Pd-образца (табл. 1, Pd/Al2O3 + СO, цикл 1; Pd/Al2O3, цикл 1). Таким образом, CO можно считать каталитическим ядом для Pd-содержащих катализаторов.
Активность Pd–M/Al2O3 (M = Au, Ag, Ce, Cu, Fe, Ni, Co)
Модификация Pd/Al2O3 добавками Au или Ag не приводит к формированию высокоактивных катализаторов. Этот вывод следует из низких значений выхода продуктов на Pd–Au/Al2O3 и Pd–Ag/Al2O3 (табл. 1, цикл 1).
На первый взгляд, малая активность Pd–Au/Al2O3 и Pd–Ag/Al2O3 может быть связана с изменением структуры биметаллических частиц под действием CO, который выделяется в ходе побочного процесса декарбонилирование этаналя. Известно, что адсорбция молекул CO на Pd–Au- и Pd–Ag-частицах приводит к их перестройке и обогащению поверхности биметаллических частиц палладием [25–27]. Поэтому в условиях каталитической конверсии этанола частицы Pd–Au-и Pd–Ag-сплавов способны трансформироваться в частицы со структурой ядро(PdM)–оболочка(Pd). В результате Pd–Au/Al2O3 и Pd–Ag/Al2O3 можно рассматривать в качестве структурных аналогов катализатора Pd/Al2O3, который, как было показано выше, малоактивен в реакции превращении этанола в α-спирты.
Однако стоит отметить, что наблюдаемая в настоящей работе активность Pd–Au- и Pd–Ag-катализаторов существенно ниже, чем у Pd-образца. Это следует из значений выхода продуктов на Pd–Au/Al2O3, Pd–Ag/Al2O3 и на Pd/Al2O3 в стандартном цикле тестирования (табл. 1, цикл 1; ω(Pd–Au) = 8.4%, ω(Pd–Ag) = 4%, ω(Pd) = 16.5%). При этом скорость дезактивации Pd–Au и Pd–Ag в атмосфере CO выше, чем таковая для монометаллического Pd-образца. Например, после добавки 1 × 10–4 моль CO в реактор с этанолом и катализатором были получены следующие значения выхода продуктов в стандартном цикле тестирования: 0.2% для Pd–Ag/Al2O3 и 1% для Pd/Al2O3.
Таким образом, описанная в [25–27] сегрегация может приводить к формированию структур ядро(PdM)–оболочка(Pd), но в рамках этой модели сложно объяснить экспериментально наблюдаемый рост скорости дезактивации Pd–Ag- и Pd–Au-катализаторов в присутствии СO и более низкую, в сравнение c Pd/Al2O3, активность Pd–Au/Al2O3 и Pd–Ag/Al2O3.
Полученные результаты можно интерпретировать в рамках теории d-зоны (d-band theory), рассмотренной в работе [13]. Так, из данных [13] следует, что взаимодействие Pd с Au (Ag) приводит к положительному смещению центра d-зоны Pd на +0.2 (+0.3) эВ. В результате палладий в Pd–Au- и Pd–Ag-системах становится более реакционноспособным, и энергия связывания CO с Pd возрастает [13]. Очевидно, что при конверсии этанола в α-спирты повышение энергии связывания CO с атомами палладия в составе металлсодержащей фазы должен приводить к более быстрой дезактивации катализатора и, как следствие, к более низкой скорости накопления α-спиртов, что и наблюдается в наших экспериментах (табл. 1, рис. 1).
Рис. 1.
Зависимость выхода продуктов реакции от положения центра d-зоны палладия в катализаторах. εd(Pd) – энергия центра d-зоны палладия, εF(Pd) – энергия уровня Ферми палладия.
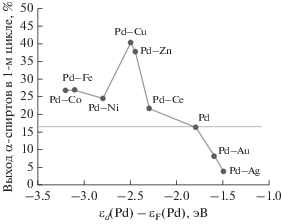
Модификация Pd/Al2O3 добавками Ce, Zn или Cu способствует росту выхода α-спиртов (табл. 1), что указывает на устойчивость катализаторов Pd–Ce/Al2O3, Pd–Zn/Al2O3 и Pd–Cu/Al2O3 к отравлению CO. Этот факт, вероятно, снова связан со сдвигом центра d-зоны Pd. Известно, что относительно чистого палладия центр d-зоны Pd в Pd–Ce-, Pd–Zn- и Pd–Cu-системах сдвинут на –0.5, –0.65 и –0.7 эВ соответственно [13–16]. В результате отрицательного сдвига центра d-зоны Pd его реакционная способность уменьшается [13], что должно приводить к снижению сорбции CO на Pd и, как следствие, возрастанию выхода α-спиртов. Как видно из данных табл. 1 и рис. 2, скорость целевого процесса на катализаторах Pd–Ce/Al2O3, Pd–Zn/Al2O3 и Pd–Cu/Al2O3 действительно увеличивается.
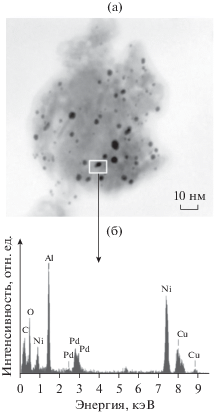
Рис. 2.
Типичная микрофотография ПЭМ восстановленного образца Pd–Cu/Al2O3 (а) и идентификация Pd–Cu-частиц с помощью спектров ЭДА (б).
В 1-м цикле тестирования выход α-спиртов на Pd–Cu/Al2O3 составляет 40%, что примерно в два раза выше, чем при конверсии этанола на Pd/Al2O3, и в течение 20-и последовательных циклов практически не меняется. Добавление избытка CO в смесь до начала реакции также не приводит к дезактивации катализатора (табл. 1). Наблюдаемая в присутствии Pd–Cu/Al2O3 эффективность конверсии этанола в α-спирты значительно превосходит таковую для описанных в литературе катализаторов целевого процесса [1–7].
Следует отметить, что Pd–Fe-, Pd–Co-, Pd–Ni-катализаторы с относительно большим отрицательным сдвигом центра d-зоны Pd (от –1 до ‒1.4 эВ [13]) также не теряют активность в присутствии выделяемого в реакции CO и демонстрируют стабильность в повторных циклах (табл. 1, рис. 1). Однако, в отличие от наиболее эффективного Pd–Cu-катализатора, Pd–Fe-, Pd–Co-, Pd–Ni-системы характеризуются пониженным выходом целевого продукта, что в большей степени обусловлено уменьшением конверсии этанола, а не изменением селективности в образовании α‑спиртов (табл. 1). Вероятно, в случае Pd–Fe-, Pd–Co-, Pd–Ni-систем реакционная способность Pd падает настолько сильно, что это приводит к снижению сорбции и активации не только CO, но и этанола и/или интермедиатов реакции.
Структура активных центров оптимального Pd–Cu-катализатора
В ходе работы было установлено, что оптимальным катализатором конверсии этанола в α‑спирты является Pd–Cu/Al2O3, поэтому представляло интерес изучить структуру его поверхности и провести реконструкцию активных центров.
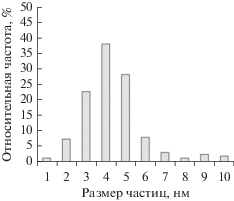
Морфологические особенности нанесенных фаз восстановленного катализатора были изучены с помощью ПЭМ. Типичная микрофотография Pd–Cu/Al2O3 приведена на рис. 2а. На фотографии видны темные частицы округлой формы, контрастирующие с серой поверхностью носителя. Полученный из локации с единичной частицей спектр ЭДА содержит линии C, Al, O, Ni, а также линии Pd и Cu. Линии Ni и C обусловлены наличием этих элементов в используемой для анализа сетке ПЭМ, а линии Al и O – присутствием в катализаторе носителя Al2O3. Оставшаяся комбинация элементов (Pd и Cu) однозначно указывает на биметаллический состав анализируемой частицы (рис. 2б). Гистограмма распределения частиц по размерам, имеющая мономодальную форму, приведена на рис. 3. Из гистограммы видно, что размер детектируемых частиц лежит в интервале от 1 до 10 нм. Средний размер частиц равен 4 ±1 нм.
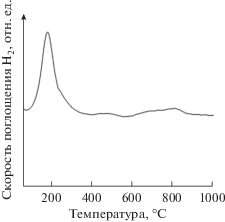
Профиль ТПВ-H2 невосстановленного образца PdO–CuO/Al2O3 приведен на рис. 4 и содержит интенсивный пик в области 180°С и два слабых пика в области 500 и 750°С. Сравнение его с полученным ранее профилем носителя позволяет сделать вывод о том, что высокотемпературные пики относятся к восстановлению примесей оксидов переходных металлов, содержащихся в Al2O3. Пик в области 180°С (рис. 4) характеризует восстановление осажденных на поверхность Al2O3 оксидов палладия и меди, находящихся в тесном контакте друг с другом [28, 29]. Таким образом, данные ТПВ-H2, как и результаты ПЭМ-ЭДА, указывают на присутствие биметаллических структур в составе Pd–Cu-катализатора. Из анализа профиля ТПВ-H2 следует, что используемая в настоящем исследовании температура восстановление катализатора водородом перед реакцией (200°С) является достаточной для полного восстановления нанесенных фаз Pd–Cu-катализаторе. Для того чтобы в этом убедиться, были проведены дополнительные РФЭ-исследования.
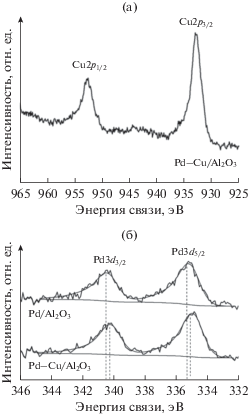
РФЭ-спектры Cu2p и Pd3d, полученные для восстановленного Pd–Cu/Al2O3 катализатора, приведены на рис. 5. Отсутствие “shake-up” сателлитов в области 944 и 963 эВ в спектре Cu2p и интенсивных пиков с энергиями связи электронов 337.4 и 342.7 эВ в спектре Pd3d указывает на то, что в восстановленном Pd–Cu-образце нет катионов прекурсоров меди (Cu+2) и палладия (Pd+2). В РФЭ-спектрах меди присутствует пик Cu2p3/2 с Есв = 932.8 эВ, что свидетельствует о наличие в образце соединений ноль-валентной меди [30]. В РФЭ-спектрах палладия имеется пик Pd3d5/2 с энергией связи электронов 335.1 эВ, относящийся к соединениям ноль-валентного палладия [17, 31]. Таким образом, данные по химическому составу нанесенных металлов в Pd–Cu-образце хорошо согласуются с обсужденными ранее результатами ТПВ-H2 (рис. 4). Отдельно стоит сказать, что в сравнение с образцом Pd/Al2O3 пики Pd3d в спектре Pd–Cu-катализатора сдвинуты в сторону меньших энергий связи на ~0.2 эВ (рис. 5). Согласно [28, 32] такой эффект можно объяснить модификацией электронной структуры Pd в частицах Pd–Cu-сплава. Отметим, что по данным ПЭМ-ЭДА биметаллические частицы действительно присутствуют в восстановленном Pd–Cu-катализаторе.
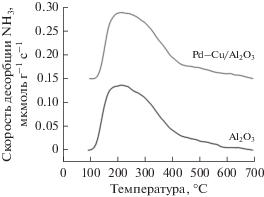
Профиль ТПД-NH3 восстановленного катализатора Pd–Cu/Al2O3 представлен на рис. 6. Он идентичен спектру ТПД-NH3 чистого носителя (рис. 6) и содержит широкий пик десорбции аммиака в области 200–300°C, который согласно данным [6, 33] указывает на присутствие поверхностных кислых центров средней и слабой силы.
На основании приведенных выше результатов можно сделать вывод о том, что поверхность Pd–Cu/Al2O3 состоит из осажденных на поверхности Al2O3 частиц Pd–Cu размером 4 ± 1 нм. Из данных работ [1–5] следует, что эффективный катализатор синтеза α-спиртов должен содержать как металлические центры дегидрирования/гидрирования, так и кислые центры (схема 1 ), при этом для максимальной скорости реакции необходимо, чтобы центры двух типов располагались вблизи друг от друга [5]. В катализаторе Pd–Cu/Al2O3 таким условиям удовлетворяют бифункциональные пары Cu0–Al2O3 и Pd0–Al2O3, которые образуются на периферии нанесенных Pd–Cu-частиц. Однако, как показали авторы работы [11], пары Cu0–Al2O3 проявляют невысокую активность в рассматриваемом процессе из-за низкой реакционной способности Cu в гидрировании. Поэтому к наиболее активным центрам катализатора Pd–Cu/Al2O3 следует отнести пары Pd0–Al2O3, которые образуются в зоне контакта Pd–Cu-частиц и носителя.
ЗАКЛЮЧЕНИЕ
Установлено, что монометаллический катализатор Pd/Al2O3 обладает высокой начальной активностью в конверсии этанола в линейные α‑спирты. Спустя 10 ч работы Pd/Al2O3 теряет примерно 90% своей активности за счет дезактивации, которая вызвана хемосорбцией побочного продукта CO на активных центрах палладия.
Модификация Pd/Al2O3 добавками Au или Ag приводит к формированию менее активных катализаторов, что согласуется с положительным смещением центра d-зоны Pd в Pd–Au и Pd–Ag системах и, следовательно, большей реакционной способностью палладия к взаимодействию с CO. При модификации Pd/Al2O3 добавками Cu, Ce, Zn, Fe, Ni, Co формируются более активные катализаторы, что коррелирует с отрицательным смещением центра d-зоны Pd в системах Pd–Cu, Pd–Ce, Pd–Zn, Pd–Fe, Pd–Ni и Pd–Co, и, следовательно, меньшей реакционной способностью Pd в сорбции CO.
Среди изученных биметаллических катализаторов наибольшую эффективность в целевом процессе проявляет Pd–Cu-образец. Он устойчив к отравлению CO и позволяет получать из этанола линейные α-спирты c селективностью 95%. Время стабильной работы катализатора составляет не менее 100 ч. На основе известного механизма реакции и данных ПЭМ, ЭДА, РФЭС, ТПВ-H2 и ТПД-NH3 проведена реконструкция активных центров Pd–Cu/Al2O3. Установлено, что наиболее активные центры Pd–Cu-катализатора представляют собой модифицированные медью металлические центры Pd0, расположенные вблизи кислотно-оснóвных центров Al2O3.
Список литературы
Wu L., Moteki T., Gokhale A.A., Flaherty D.W., Toste F.D. // Chem. 2016. V. 1. P. 32.
Angelici C., Weckhuysen B.M., Bruijnincx P.C.A. // ChemSusChem. 2013. V. 6. P. 1595.
Sun J., Wang Y. // ACS Catal. 2014. V. 4. P. 1078.
Gabriëls D., Hernández W.Y., Sels B., Van Der Voort P., Verberckmoes A. // Catal. Sci. Technol. 2015. V. 5. P. 3876.
Kozlowski J.T., Davis R.J. // ACS Catal. 2013. V. 3. P. 1588.
Marcu I.-C., Tanchoux N., Fajula F., Tichit D. // Catal. Lett. 2013. V. 143. P. 23.
Riittonen T., Toukoniitty E., Madnani D.K., Leino A.-R., Kordas K., Szabo M., Sapi A., Arve K., Wärnå J., Mikkola J.-P. // Catalysts. 2012. V. 2. P. 68.
Nikolaev S.A., Tsodikov M.V., Chistyakov A.V., Zharova P.A., Ezzhelenko D.I., Shilina M.I. // Catal. Tod. 2020. In press. https://doi.org/10.1016/j.cattod.2020.06.061
Royer S., Duprez D. // ChemCatChem. 2011. V. 3. P. 24.
Эллерт О.Г., Цодиков М.В., Николаев С.А., Новоторцев В.М. // Успехи Химии. 2014. Т. 83. № 8. С. 718.
Nikolaev S.A., Tsodikov M.V., Chistyakov A.V., Zharova P.A., Ezzgelenko D.I. // J. Catal. 2019. V. 369. P. 501.
Николаев С.А., Чистяков А.В., Жарова П.А., Цодиков М.В., Кротова И.Н., Эзжеленко Д.И. // Нефтехимия. 2016. Т. 56. № 5. С. 502.
Hammer B., Nørskov J.K. // Adv. Catal. 2000. V. 45. P. 71.
Che J., Li Y., Lu N., Tian C., Han Zh., Zhang L., Fang Y., Qian B., Jiang X., Cui R. // J. Mater. Chem. A. 2018. V. 6. P. 23560.
Chen Z.-X., Neyman K.M., Gordienko A.B., Rösch N. // Phys. Rev. B. 2003. V. 68. P. 075417.
Lopez N., Nørskov J.K. // Surf. Sci. 2001. V. 477. P. 59.
Nikolaev S.A., Golubina E.V., Shilina M.I. // Appl. Catal. B: Environ. 2017. V. 208. P. 116.
Наумкин А.В., Васильков А.Ю., Волков И.О., Смирнов В.В., Николаев С.А. // Неорганические материалы. 2007. Т. 43. № 4. С. 445.
Zanaveskin K.L., Lukashev R.V., Makhin M.N., Zanaveskin L.N. // Ceramics International. 2014. V. 40. P. 16577.
Tjurina L.A., Smirnov V.V., Potapov D.A., Nikolaev S.A., Esipov S.E., Beletskaya I.P. // Organometallics. 2004. V. 23. P. 1349.
Николаев С.А., Голубина Е.В., Кустов Л.М., Тарасов А.Л., Ткаченко О.П. // Кинетика и Катализ. 2014. Т. 55. № 3. С. 326.
Занавескин К.Л., Лукашев Р.В., Маслеников А.Н., Терехов А.В., Махин М.Н., Занавескин Л.Н. // Неорганические материалы. 2016. Т. 52. № 8. С. 858.
Dik P.P., Danilova I.G., Golubev I.S., Kazakov M.O., Nadeina K.A., Budukva S.V., Pereyma V.Yu, Klimov O.V., Prosvirin I.P., Gerasimov E.Yu., Bok T.O., Dobryakova I.V., Knyazeva E.E., Ivanova I.I., Noskov A.S. // Fuel. 2019. V. 237. P. 178.
Chistyakov A.V., Zharova P.A., Nikolaev S.A., Tsodikov M.V. // Catal. Today. 2017. V. 279. P. 124.
Mamatkulov M., Yudanov I.V., Bukhtiyarov A.V., Prosvirin I.P., Bukhtiyarov V.I., Neyman K.M. // J. Phys. Chem. C 2019. V. 123. P. 8037.
Bukhtiyarov A.V., Prosvirin I.P., Saraev A.A., Klyushin A.Yu., Knop-Gericke A., Bukhtiyarov V.I. // Faraday Discuss. 2018. V. 208. P. 255.
Smirnova N.S., Markov P.V., Baeva G.N., Rassolov A.V., Mashkovsky I.S., Bukhtiyarov A.V., Prosvirin I.P., Panafidin M.A., Zubavichus Y.V., Bukhtiyarov V.I., Stakheev A.Yu. // Mendeleev Commun. 2019. V. 29. P. 547.
Di L., Xu W., Zhan Z., Zhang X. // RSC Adv. 2015. V. 5. P. 71854.
Cai F., Yang L., Shan S., Mott D., Chen B.H., Luo J., Zhong C.-J. // Catalysts. 2016. V. 6. P. 96.
Biesinger M.C., L.W.M. Lau, Gerson A.R., Smart R.St.C. // Appl. Surf. Sci. 2010. V. 257. P. 887.
Ivanova A.S., Slavinskaya E.M., Gulyaev R.V., Zaikovskii V.I., Stonkus O.A., Danilova I.G., Plyasova L.M., Polukhina I.A., Boronin A.I. // Appl. Catal. B: Environ. 2010. V. 97. P. 57.
Sheng J., Kang J., Ye H., Xie J., Zhao B., Fu X.-Z., Yu. Y., Sun R., Wong C.-P. // J. Mater. Chem. A. 2018. V. 6. P. 3906.
Hu Z., Li W.-Z., Sun K.-Q., Xu B.-Q. // Catal. Sci. Technol. 2013. V. 3. P. 2062.
Дополнительные материалы отсутствуют.
Инструменты
Кинетика и катализ