

Кинетика и катализ, 2021, T. 62, № 1, стр. 55-76
Гидродехлорирование хлорбензола в паровой фазе на никелевых катализаторах, нанесенных на оксид алюминия: влияние структуры носителя и его модификации гетерополикислотой HSiW
Д. А. Рябошапка a, *, Е. С. Локтева a, **, Е. В. Голубина a, А. Н. Харланов a, К. И. Маслаков a, А. О. Камаев a, А. В. Шумянцев a, И. А. Липатова b, Е. И. Школьников b
a ФГБОУ ВО Московский государственный университет им. М.В. Ломоносова, Химический факультет
119991 Москва, Ленинские горы, 1, стр. 3, ГСП-1, Россия
b ФГБУН Объединенный институт высоких температур РАН
125412 Москва, ул. Ижорская, 13, стр. 2, Россия
* E-mail: ryaboshapkada@gmail.com
** E-mail: e.lokteva@rambler.ru
Поступила в редакцию 23.03.2020
После доработки 27.05.2020
Принята к публикации 17.06.2020
Аннотация
Проведено исследование физико-химических и каталитических свойств катализаторов 6% Ni/Al2O3 в парофазном гидродехлорировании хлорбензола (ХБ). Катализаторы синтезировали нанесением нитрата никеля на два типа оксида алюминия: А (полученный гидролизом изопропилата алюминия) и Е (производства “Engelhard”), различающихся морфологией и текстурой, немодифицированных и модифицированных нанесением 20% гетерополикислоты H8Si(W2O7)6 ⋅ nH2O (ГПК). Для предотвращения разложения ГПК термообработку на воздухе и восстановление модифицированных материалов проводили при относительно низких температурах (250 и 330°С соответственно). С целью корректного сравнения катализаторы, не содержащие ГПК, обрабатывали таким же образом. Методом ТПВ показано, что термообработка на воздухе при 250°С не обеспечивает полного превращения исходного нитрата никеля в оксид; в составе прекурсоров катализаторов присутствуют нитраты и гидроксинитраты никеля, их содержание снижается в результате модификации ГПК. Различия в составе и силе ЛКЦ на поверхности двух типов Al2O3 приводят к неодинаковой координации нитрата и отличиям в способности никеля к восстановлению, выявленным методами ТПВ, ИК ДО спектроскопии с адсорбцией СО и РФЭС in situ. Никель в составе Ni/Al2O3(E) восстанавливается несколько легче, чем в Ni/Al2O3(А), однако использованные в работе условия приводят к восстановлению лишь небольшой его части в обоих катализаторах. По данным РФЭС in situ, ТПВ и ИК ДО с адсорбцией СО, модификация Ni/Al2O3 гетерополикислотой обеспечивает дополнительное изменение кислотных свойств и способа координации нитрата никеля в ходе пропитки, повышение способности никеля к восстановлению, предотвращение ухода никеля с поверхности в объем образца, а также образование новых активных центров в результате сильного взаимодействия никеля и вольфрама в составе ГПК. В зависимости от природы носителя модификация ГПК приводит к улучшению (Ni/ГПК/Al2O3(А)) или ухудшению (Ni/ГПК/Al2O3(E)) каталитической эффективности образцов. При высоких температурах селективность модифицированных ГПК катализаторов по бензолу снижается вследствие образования циклогексана. Эффективность катализаторов увеличивается в ряду Ni/ГПК/Al2O3(E) < Ni/Al2O3(А) < Ni/Al2O3(E) < < Ni/ГПК/Al2O3(А). Самый активный катализатор Ni/ГПК/Al2O3(А) проявляет наибольшую стабильность в ходе продолжительных испытаний при повышении и последующем снижении температуры. Способность никеля к восстановлению оказывает более существенное влияние на эффективность катализатора в гидродехлорировании ХБ, чем различия в текстуре и в содержании никеля.
ВВЕДЕНИЕ
Хлорароматические соединения широко применяют в промышленности в качестве растворителей, при изготовлении полимерной продукции, красителей, химикатов для сельского хозяйства. Однако эти соединения являются ксенобиотиками, многие из них токсичны, оказывают пагубное воздействие на иммунную и репродуктивную системы животных и человека; они практически не подвергаются разложению в природе. Поэтому разрабатываются разнообразные способы утилизации хлорорганических отходов: сжигание, каталитическое окисление, радиолиз, фотокаталитические процессы и др. [1–4]. Хорошие перспективы имеет процесс каталитического гидродехлорирования (ГДХ) [1, 5–10]. Главное достоинство этого метода состоит в обеспечении возможности низкоэнергетического превращения хлорированных органических соединений в углеводороды без образования еще более вредных побочных продуктов – фосгена, хлора, диоксинов, чего практически невозможно избежать в ходе окислительной переработки. Кроме того, образующиеся в реакции углеводороды можно выделить и повторно использовать в других процессах [11].
В качестве катализаторов ГДХ хорошо зарекомендовали себя благородные металлы – Pd, Rh, Pt, проявляющие активность в жидко- и парофазных реакциях [7, 12–14]. Чтобы снизить затраты на проведение процесса, благородные металлы в составе катализаторов заменяют на более дешевые, например, Ni, Co, Fe, которые, как правило, менее активны и стабильны [6, 9, 15, 16].
Выбор носителей играет важную роль для обеспечения высокой эффективности катализаторов ГДХ [7, 17–19]. Подходящий носитель способен заметно снизить дезактивацию активного компонента в результате спекания [7, 20, 21]. Устойчивостью к воздействию HCl, образующегося в качестве продукта в ГДХ, отличаются углеродные носители [22]. Однако для таких систем характерны низкая плотность и механическая прочность , поэтому в промышленности предпочтение отдается оксидным носителям [23]. Так, описано применение в составе катализаторов ГДХ оксидов титана [24], кремния [25, 26], циркония [27], алюминия [9, 28]. Привлекательным носителем считается Al2O3 по причине его термостабильности, доступности и низкой стоимости [29]. В многочисленных работах ГДХ хлорированных соединений проводили на металлсодержащих катализаторах, нанесенных на Al2O3 [30–32].
Реакция ГДХ является структурно-чувствительной, то есть активность катализатора зависит от размера частиц активного компонента [33]; для активации водорода и связи С–Cl благоприятно присутствие металла в восстановленном и частично окисленном состояниях [34]. Существенно улучшить свойства катализаторов ГДХ позволяют различные модификаторы, причем модифицировать можно как активный компонент, так и носитель [9, 35]. Повышенной активностью за счет синергических эффектов часто обладают биметаллические системы на основе переходных металлов [28, 36]. В ряде работ было показано, что перспективным модификатором катализаторов ГДХ являются гетерополикислоты, в частности, со структурой аниона типа Кеггина (${\text{X}}{{{\text{M}}}_{{12}}}{\text{O}}_{{40}}^{{n - }}$), где X – гетероатом (P, Si), M – металл (W, Mo) [9, 35, 37]. Воздействуя на кислотные свойства и морфологию поверхности носителя, они могут влиять на дисперсность наносимого металла и изменять природу активных центров катализатора. Предполагается, что нанесение ГПК помогает предотвратить образование трудно восстанавливаемых поверхностных алюминатов активного металла [38–41]. Вольфрамсодержащие ГПК могут также служить прекурсорами для формирования активной в гидрировании фазы WOx [42]. В работе [9] в результате модификации γ-Al2O3 ГПК возросла активность никельсодержащего катализатора в ГДХ хлорбензола. Разбавление никеля палладием в таких системах не привело к существенному изменению активности, но увеличило селективность образования бензола. В работе [43] добавление ГПК типа Уэллса–Доусона способствовало повышению стабильности Al2O3. Детальные механизмы воздействия ГПК при модификации катализаторов ГДХ пока исследованы недостаточно. В частности, важно знать, сохраняется ли структура ГПК под влиянием высокотемпературных обработок в ходе приготовления катализатора или происходит ее разложение на соответствующие оксиды.
В настоящей работе никелевые катализаторы приготовлены с применением двух типов Al2O3 – коммерчески доступного производства фирмы “Engelhard” и полученного в лабораторных условиях гидролизом изопропилата алюминия с последующим прокаливанием. Оба носителя модифицировали нанесением ГПК H8Si(W2O7)6 ⋅ nH2О. Температурные обработки модифицированных катализаторов проводили при относительно низких температурах с целью сохранения структуры Кеггина модифицирующей ГПК (термообработка на воздухе при 250°С, восстановление водородом при 330°С) и выявления ее влияния на физико-химические свойства и каталитическую эффективность в реакции парофазного ГДХ хлорбензола. Для корректного сравнения немодифицированные материалы обрабатывали в тех же условиях.
1. ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
1.1. Приготовление катализаторов
В качестве носителей использовали два типа γ‑Al2O3. Первый (далее обозначен Al2O3(А)) готовили в лабораторных условиях прокаливанием при 600°С бемита γ-AlO(OH), полученного осаждением из изопропропилата алюминия (99.999 + %, ООО “Прима”). Второй носитель фирмы “Engelhard” далее обозначен Al2O3(E). Модификатором служила гетерополикислота (ГПК) состава H8Si(W2O7)6 ⋅ nH2O (“ДИА-М”, ч. д. а.), ее наносили методом влажной пропитки из раствора в дистиллированной воде в таком расчете, чтобы содержание ГПК на Al2O3 составляло 20 мас. %. Растворитель выпаривали на водяной бане при перемешивании, полученные прекурсоры сушили на воздухе 12 ч при комнатной температуре, затем 3 ч при 110°С и 3 ч при 250°С.
Источником никеля служил Ni(NO3)2 ⋅ 6H2O (“Химмед”, ГОСТ 4055-78, ч. д. а.). Его наносили на носитель методом влажной пропитки, перемешивали в течение 2 ч и выпаривали растворитель на водяной бане. Сушку и термообработку на воздухе проводили так же, как при нанесении ГПК. Целевое содержание никеля во всех катализаторах составляло 6 мас. %. Восстановление катализаторов осуществляли непосредственно перед каталитическим экспериментом в токе H2 (10 мл/мин) при 330°С в течение 3 ч. Далее немодифицированные образцы обозначены Ni/Al2O3(А) и Ni/Al2O3(Е), модифицированные – Ni/ГПК/Al2O3(А) и Ni/ГПК/Al2O3(Е).
1.2. Физико-химические исследования катализаторов
Текстуру каталитических систем изучали методом низкотемпературной адсорбции-десорбции азота на анализаторе сорбции Autosorb-1 (“Quantachrome”, США), после вакуумирования навески образца в течение 3 ч при 200°C. Расчет удельной поверхности, объема и среднего диаметра пор образцов проводили методами БЭТ и Баррета–Джойнера–Халенды (БДХ, BJH) по изотермам адсорбции и десорбции азота с помощью программного обеспечения анализатора.
Электронные микрофотографии образцов получали с помощью сканирующего электронного микроскопа JSM–6390 (“JEOL Ltd.”, Япония). Образец закрепляли на медно-цинковом столике с помощью двухстороннего углеродного электропроводящего скотча. Ускоряющее напряжение при съемке составляло 10 кВ, рабочая дистанция 8–10 мм. Спектры и карты энергодисперсионного рентгеновского микроанализа регистрировали при помощи приставки энерго-дисперсионного анализа (ЭДА) при ускоряющем напряжении 15 кВ.
Исследование методом рентгенофазового анализа (РФА) проводили на автоматических порошковых дифрактометрах STOE Stadi P (“STOE Gmbh”, Германия) в диапазоне углов съемки 2θ от 5° до 80° с шагом 0.1° и скоростью накопления 2°/мин, и Ultima IV (“Rigaku”, Япония) в диапазоне углов съемки 2θ от 5° до 85° с шагом 0.01° и скоростью накопления 1°/мин. Фазовый состав анализировали с использованием программы WinXpow 1.04 сравнением с данными библиотеки JCPDS PDF-2 (база данных ICDD).
Спектры рентгеновской фотоэлектронной спектроскопии (РФЭС) получали на спектрометре Axis Ultra DLD (“Kratos Analytical”, Великобритания) с применением источника монохроматического излучения AlKα (hν = 1486.7 эВ, 150 Вт). Предварительную калибровку шкалы прибора проводили по пикам основных уровней чистых золота и меди Au4f7/2 (83.96 эВ) и Cu2p3/2 (932.62 эВ). Энергия пропускания анализатора составляла 160 эВ для обзорных спектров и 40 эВ для спектров высокого разрешения. Образцы наносили на двустороннюю адгезионную ленту. Для компенсации зарядки образцов использовали нейтрализатор. Спектры высокого разрешения раскладывали на компоненты с помощью программного обеспечения CasaXPS 2.3.22.
Калибровку спектров РФЭС систем, включающих оксид никеля или ГПК на оксидных носителях, часто проводят по пику C1s при 284.5–285 эВ [44–46], хотя в их составе углерод не содержится, а, следовательно, положение его пика обусловлено химическим состоянием примесей или адсорбированных функциональных групп произвольного состава. Кроме того, положение и интенсивность пика C1s может изменяться при термической обработке катализаторов (на воздухе и при восстановлении).
В нашей работе калибровку спектров проводили по линии Al2p, положение которой принимали равным 74.6 эВ, что более правильно с точки зрения состава образцов и ожидаемой стабильности матрицы носителя в условиях восстановительной обработки, но затрудняет сравнение полученных данных с литературными.
In situ восстановительную обработку катализаторов проводили в каталитической ячейке, соединенной с РФЭ-спектрометром. Спрессованную таблетку катализатора продували смесью 10% Н2 + 90% N2, нагревали со скоростью 5°С/мин до заданной температуры (сначала 330°С, затем 450°C) и обрабатывали при ней 1 ч, а потом охлаждали в этой смеси до комнатной температуры, перемещали в ячейку спектрометра и регистрировали спектры РФЭС.
Содержание никеля в катализаторах определяли методом атомно-абсорбционной спектроскопии (ААС) на приборе iCE 3000 Series AA (“Thermo Scientific”, США), с использованием атомизации в пламени. Данные обрабатывали с помощью программного обеспечения Thermo Scientific SOLAAR, входящего в комплектацию прибора. Для перевода никеля в раствор навеску катализатора растворяли в небольшом количестве царской водки при перемешивании и нагревании в течение 1 ч. Полученный раствор разбавляли дистиллированной водой до содержания металла ~4 мг/л. Непосредственно перед определением содержания металла в анализируемых растворах проводили калибровку прибора с применением свежеприготовленных стандартных растворов.
Температурно-программированное восстановление (ТПВ) осуществляли на установке УСГА-1 (“Унисит”, Россия) в кварцевом реакторе при линейном нагреве (10°С/мин) от 30 до 800°С при постоянном потоке смеси 5% H2 + 95% Ar (30 мл/мин). Перед анализом образец выдерживали при 30°С в течение 30 мин в потоке аргона. Изменение состава газовой смеси во время анализа фиксировали с помощью детектора по теплопроводности (температура детектора 60°С). Сигнал детектора калибровали по восстановлению NiO. Количество поглощенного H2 рассчитывали, исходя из площади пиков поглощения водорода в профиле ТПВ.
Исследование методом термического анализа проводили с помощью термоанализатора STA 449C Jupiter (“Netzsch”, Германия) с одновременной регистрацией данных термогравиметрии (ТГ) и дифференциальной сканирующей калориметрии (ДСК). Использовали тигли из Al2O3 массой 250 мг. Пиролиз проводили в смеси воздуха (80 мл/мин) и аргона (40 мл/мин) при нагревании со скоростью 10°C/мин от 40 до 900°C.
ИК-спектры диффузного отражения (ИК ДО) регистрировали на ИК-Фурье-спектрометре EQUINOX 55/S (“Bruker”, США) при комнатной температуре в диапазоне волновых чисел 4000–800 см–1 с разрешением 4 см–1 и усреднением по 250 сканам. Порошкообразную фракцию исследуемого образца помещали в кварцевую ампулу с окошком из CaF2. Образец прокаливали при температуре 550°С в течение 1 ч на воздухе и 2 ч под вакуумом не хуже 5 × 10–5 мм рт. ст. Затем температуру снижали до 330°С и напускали водород до давления 180 мм рт. ст. Образец выдерживали в атмосфере водорода при этой температуре 1 ч, после чего температуру снижали до 130°С и далее охлаждали до комнатной при непрерывном вакуумировании. Газообразные Н2 и СО очищали пропусканием через ловушку с жидким азотом и длительным выдерживанием над прокаленным цеолитом.
1.3. Тестирование в каталитической реакции ГДХ хлорбензола
Тестирование в каталитической реакции гидродехлорирования хлорбензола (ХБ) (“Acros Organics”, 99.5%) проводили в проточном вертикально расположенном кварцевом реакторе с неподвижным слоем катализатора, оснащенном трубчатой печью и термопарой. В реактор загружали катализатор массой 0.1 г, восстанавливали его, как описано выше, охлаждали до реакционной температуры и начинали подавать смесь ХБ и H2 (молярное соотношение 1 : 50), которую получали путем пропускания водорода через барботер, содержащий ХБ. Реагенты поступали в реактор снизу. Скорость подачи H2 составляла 12 мл/мин. В ходе каталитических испытаний температуру повышали от 150 до 350°С с шагом в 50°С и затем снижали в том же интервале. Нагревание или охлаждение реактора осуществляли в токе водорода; после достижения заданной температуры на катализатор подавали реакционную смесь и анализировали продукты на выходе из реактора; подачу продолжали до тех пор, пока три-четыре последовательных анализа не показывали одинаковую конверсию ХБ и селективность по продуктам. Эти значения принимали как определяющие стационарную эффективность катализатора при данной температуре. Затем устанавливали следующее значение температуры при подаче в реактор водорода. Пробы реакционной смеси для анализа отбирали из верхней части реактора с помощью шприца.
Продукты анализировали методом газо-жидкостной хроматографии на хроматографе Agilent 6890 (“Agilent”, США) с использованием капиллярной колонки DBWax (длина 30 м) и пламенно-ионизационного детектора. Для точного определения состава продуктов в начале опыта и при появлении на хроматограмме иных пиков, кроме относящихся к бензолу и хлорбензолу, состав продуктов анализировали также на хромато-масс-спектрометре Trace DSQ II (“Thermo Fisher Scientific”, США) (80 эВ, колонка DB-5 длиной 15 м).
2. ПОЛУЧЕННЫЕ РЕЗУЛЬТАТЫ
2.1. Исследование стабильности ГПК
Поскольку в процессе приготовления предшественники катализаторов подвергаются термической обработке, методом термического анализа исследовали возможность протекания процессов разложения ГПК (рис. 1). В ходе нагревания ГПК на воздухе на кривой ДСК (рис. 1а) наблюдаются два интенсивных эндотермических пика при 102 и 227°С, сопровождающиеся потерей массы, которые связаны с удалением слабо и сильно адсорбированной воды. Снижение массы в интервале температур 300–525°С также связано с удалением воды, которое сопровождается частичным разложением ГПК с сохранением структуры Кеггина. Полностью ГПК разлагается при 527°С, о чем свидетельствует узкий экзотермический пик на ДСК-кривой, не сопровождающийся изменением массы образца. Этот результат согласуется с данными о трансформации структуры H4SiW12O40/Al2O3 в ходе прокаливания, полученными методом EXAFS [47]: при температурах 350–450°С образец терял воду с образованием ${\text{WO}}_{4}^{ - }$ и лакунарных полианионов [SiW11O39]8−, в которых структура Кеггина сохранялась. Полное разложение происходило при 550–650°С с образованием изолированных частиц WO6 и небольшого количества наночастиц WO3.
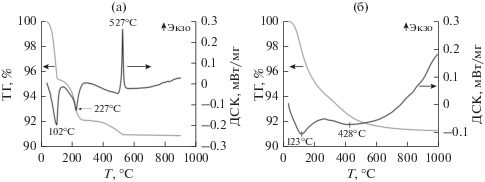
При термическом анализе ГПК, нанесенной на Al2O3 (рис. 1б), картина кардинально меняется: на кривой ТГ наблюдается практически равномерное падение массы образца до температуры 650°С. На кривой ДСК отчетливо выделяются два эндотермических эффекта: при 123°С, вызванный десорбцией физически адсорбированной воды, и широкий пик при 428°С, который связан с удалением структурных молекул воды. Важно отметить, что в случае ГПК/Al2O3 отсутствует экзотермический пик, который присутствовал на термограмме ГПК, поскольку, согласно данным РФА, в нанесенном состоянии ГПК распределена по поверхности Al2O3 в виде небольших частиц. Разложение частиц разного размера, возможно, химически связанных с поверхностью Al2O3, сопровождается малоинтенсивными экзотермическими эффектами, которые перекрываются с эндотермическим эффектом удаления воды.
Результаты ДСК показывают, что разложение нанесенной на Al2O3 ГПК со структурой Кеггина начинается при температуре выше 350°С с потери структурной воды. Поскольку основная задача настоящей работы состояла в изучении влияния на каталитические свойства ГПК со структурой Кеггина, термообработки прекурсоров катализаторов проводили в максимально щадящих условиях: термообработку на воздухе – при 250°С, восстановление водородом – при 330°С, каталитические испытания – в интервале от 150 до 350°С.
2.2. Исследование текстуры и морфологии прекурсоров катализаторов
Поскольку малые частицы никеля в катализаторах легко окисляются на воздухе, то физико-химическими методами в основном исследовали прекурсоры катализаторов, т.е. образцы после термообработки на воздухе при 250°С, но до восстановления. Текстурные свойства использованных в работе носителей и прекурсоров катализаторов изучали методами низкотемпературной адсорбции–десорбции N2 и СЭМ. Величины удельной поверхности и параметры пористой системы приведены в табл. 1. Видно, что значение SБЭТ и радиус пор для Al2O3(А) примерно в два раза, a общий объем пор в 4.6 раза выше по сравнению с Al2O3(Е); увеличение всех трех параметров объясняется широким распределением пор по размерам, имеющим признаки бимодальности в случае Al2O3(А). Текстурные параметры носителя Al2O3(А) с преобладанием мезопор со средним радиусом ~8 нм подтверждены также адсорбционным методом лимитированного испарения бензола [48, 49].
Таблица 1.
Текстурные характеристики носителей и невосстановленных прекурсоров катализаторов
| Прекурсоры катализаторов | SБЭТ, м2/г | Rпор, нм | Vпор, см3/г |
|---|---|---|---|
| Al2O3(A) | 309 ± 31 | 8.1 | 1.899 |
| Al2O3(E) | 155 ± 15 | 3.8 | 0.409 |
| ГПК/Al2O3(E) | 174 ± 17 | 4.1 | 0.363 |
| Ni/Al2O3(A) | 264 ± 26 | 5.5 | 0.921 |
| Ni/Al2O3(E) | 174 ± 17 | 4.1 | 0.409 |
| Ni/ГПК/Al2O3(A) | 241 ± 24 | 6.8 | 0.817 |
| Ni/ГПК/Al2O3(E) | 178 ± 18 | 4.1 | 0.269 |
При нанесении никеля на Al2O3(А) величина SБЭТ незначительно, а Vпор существенно уменьшаются. Нанесение никеля на Al2O3(E) не оказывает почти никакого влияния на эти параметры.
Модификация никелевых катализаторов на обоих носителях добавлением ГПК не приводит к существенному изменению SБЭТ, однако объем пор в образце Ni/ГПК/Al2O3(E) сокращается в 1.5 раза при сохранении значения среднего радиуса пор. Степень снижения Vпор в результате модификации ГПК меньше в случае носителя Al2O3(А) (12% по сравнению с катализатором без ГПК), чем в случае Al2O3(Е) (34%).
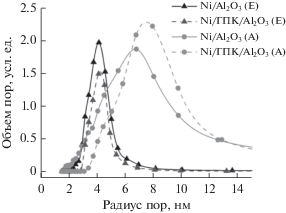
Распределение пор по размерам (рис. 2) для Ni/Al2O3(A) заметно шире, чем для Ni/Al2O3(E), и сдвинуто к бóльшим размерам. Модификация обоих образцов ГПК приводит к небольшому сужению распределения пор по размерам (рис. 2) за счет сокращения доли пор радиусом менее 3 нм для образцов на носителе Al2O3(Е) и менее 4 нм – на носителе Al2O3(А). По-видимому, частицы ГПК закрывают устья мелких пор. Учитывая размер структурных единиц Кеггина (примерно 3.8 нм) [46], такое предположение представляется логичным. При этом в образце Ni/ГПК/Al2O3(A) повышается доля пор радиусом ~8 нм. Несущественные изменения в распределении пор по размерам с сохранением значительной доли относительно более крупных пор в каждом из образцов косвенно свидетельствуют о небольшом размере частиц нанесенной фазы ГПК.
Микрофотографии СЭМ и карты распределения элементов приведены на рис. 3. Видно, что существенных различий в морфологии между прекурсорами катализаторов на носителях Al2O3(А) и Al2O3(Е) не наблюдается. Поверхность частиц образцов, модифицированных ГПК, более рыхлая, по-видимому, из-за высокой дисперсности ГПК. Согласно данным ЭДА вольфрам, кремний и никель равномерно распределены по поверхности всех образцов. Места расположения Ni и W в модифицированных катализаторах совпадают, не обнаружено участков, на которых присутствовал бы только один из этих элементов. Равномерное распределение Ni по слою ГПК может способствовать снижению степени его взаимодействия с Al2O3 и затруднять образование алюминатов никеля. Однако в модифицированных образцах возникают условия для химического взаимодействия никеля с ГПК.
Рис. 3.
Микрофотографии СЭМ и ЭДА-карты распределения Ni и W в немодифицированных и модифицированных ГПК прекурсорах катализаторов.

Метод ПЭМ мало информативен для изучения Ni/Al2O3 из-за низкой контрастности NiO по отношению к Al2O3, поэтому такое исследование в настоящей работе не проводили. Однако в [41] приведены снимки ПЭМ палладий содержащих катализаторов на тех же носителях (А) и (Е), в том числе модифицированных той же ГПК H8Si(W2O7)6 · nH2O, которую применяли и мы. Из них очевидна различная структура двух типов Al2O3: частицы Al2O3(Е) имеют округлую форму, носитель Al2O3(А), наряду с круглыми, содержит палочкообразные частицы длиной до 50 нм и толщиной до 5 нм, что характерно для γ-Al2O3, приготовленного гидролизом изопропилата алюминия. Распределение частиц палладия по размерам на носителе Al2O3(Е) более широкое (2–20 нм), чем на носителе Al2O3(А) (4–8 нм).
2.3. Исследование методом РФА
Результаты РФА Al2O3(Е), ГПК и 75% ГПК/Al2O3(E) приведены на рис. 4а. На дифрактограмме ГПК наиболее интенсивные рефлексы наблюдаются при 2θ 10.2° (110), 25.3° (222) и 34.5° (332), что совпадает с литературными данными для структуры Кеггина (PDF № 00-001-0559, база данных PDF – ICDD). ГПК имеет объемно центрированную кубическую структуру (пространственная группа Pn3m) с параметром решетки a = 12.16 Å, который соответствует гексагидрату [50]. Согласно расчетам по формуле Шеррера размер частиц ненанесенной ГПК составляет примерно 22.8 нм.
Рис. 4.
а – Дифрактограммы образцов после термической обработки на воздухе при 250°С: 1 – Al2O3(Е), 2 – 75%ГПК/Al2O3(E), 3 – ГПК; б – дифрактограммы прекурсоров катализаторов: 1 – дифрактограмма NiO (PDF № 01-089-5881), 2 – Ni/Al2O3(E), 3 – Ni/Al2O3(А), 4 – Ni/ГПК/Al2O3(E), 5 – Ni/ГПК/Al2O3(А).
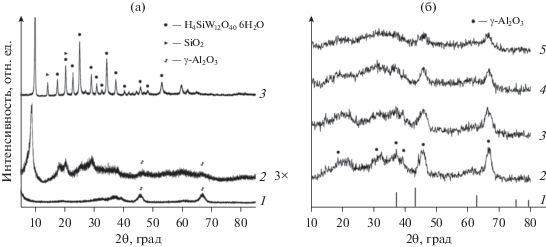
После нанесения ГПК на Al2O3 интенсивность рефлексов существенно уменьшается, и они становятся сильно уширенными. По-видимому, это вызвано распределением ГПК по поверхности преимущественно в виде рентгеноаморфной фазы. Снижение кристалличности может происходить вследствие сильного взаимодействия ГПК с Al2O3 [51]. Похожие изменения вида дифрактограмм наблюдали для НPW/SiO2–Al2O3 в [52, 53], где их связывали с образованием неупорядоченных гидратированных полианионов Кеггина или кластеров из нескольких анионов размером порядка 2 нм в ходе взаимодействия ГПК с носителем.
На дифрактограммах невосстановленных предшественников никелевых катализаторов (рис. 4б) присутствуют только рефлексы, характерные для γ-Al2O3 (PDF № 00-029-0063). Отсутствие рефлексов NiO и ГПК может быть обусловлено их рентгеноаморфностью и малой концентрацией. В работе [54] отмечалось, что рефлексы ГПК в нанесенных системах 5% Ni/HSiW/SiO2 наблюдали лишь при ее содержании выше 50 мас. %. В статьях [55, 56] установлено, что при нанесении на Al2O3 в количествах до 20–25% ГПК присутствует на поверхности в высокодисперсном рентгеноаморфном состоянии.
2.4. Исследование процессов восстановления образцов методами РФЭС и ТПВ
Для определения электронного состояния металлов и их способности к восстановлению образцы исследовали методами ТПВ и РФЭС.
Температуры восстановления, определяемые методом ТПВ, чувствительны к химическому составу, дисперсности и степени взаимодействия нанесенных частиц с носителем. Профили ТПВ носителей и невосстановленных прекурсоров катализаторов приведены на рис. 5. В профиле ТПВ исходных носителей не наблюдается поглощения водорода во всем исследованном интервале температур (в качестве примера на рис. 5 приведен профиль Al2O3(Е)). На профилях ГПК/Al2O3(А) и ГПК/Al2O3(Е) имеются очень широкие и малоинтенсивные пики, т.е. заметного вклада в профили никелевых катализаторов присутствие ГПК не вносит.
Рис. 5.
Профили ТПВ носителя Al2O3(Е), невосстановленных прекурсоров катализаторов и катализатора Ni/Al2O3(А) после каталитических испытаний. Вверху на вставке показана область от 100 до 250°С для трех образцов в большем увеличении.
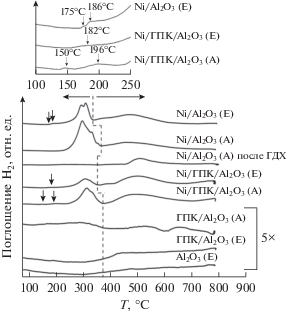
Пики поглощения водорода в профилях ТПВ всех прекурсоров катализаторов можно объединить в две группы: 1) в низкотемпературной области от 250 до 370–390°С и 2) в высокотемпературной области от 390 до 700°С. Они разделены на рис. 5 пунктирной линией и обозначены стрелками. Количественная оценка поглощения водорода в каждой из областей для никельсодержащих прекурсоров приведена в табл. 2.
Таблица 2.
Поглощение H2 прекурсорами катализаторов в ходе ТПВ и данные, рассчитанные на основании содержания никеля, определенного методом ААС
| Прекурсоры катализаторов | Ni, мас. % (ААС) | Поглощение H2, мкмоль/г | Σ(ТПВ)/ Σ(ААС) |
|||
|---|---|---|---|---|---|---|
| по данным ТПВ | расчет по данным ААС | |||||
| T < 350°C | T > 350°C | Σ | Σ | |||
| Ni/Al2O3(A) | 6.45 | 1199 | 982 | 2181 | 1100 | 1.98 |
| Ni/Al2O3(E) | 5.26 | 721 | 1291 | 2012 | 896 | 2.25 |
| Ni/ГПК/Al2O3(A) | 6.70 | 503 | 837 | 1340 | 1140 | 1.18 |
| Ni/ГПК/Al2O3(E) | 6.11 | 272 | 833 | 1105 | 1049 | 1.05 |
Низкотемпературная группа пиков включает две сильно перекрывающиеся компоненты, причем во всех образцах, кроме Ni/Al2O3(Е), преобладает компонента с более низкой температурой восстановления.
На профиле ТПВ прекурсора Ni/Al2O3(E) дополнительно наблюдается малоинтенсивный пик с максимумом около 186°С, который может соответствовать восстановлению легко доступной поверхностной формы Ni2+, наименее прочно связанной с поверхностью носителя (вставка на рис. 5). Похожие пики еще более низкой интенсивности присутствуют также на профилях обоих модифицированных образцов.
В низкотемпературном интервале может происходить как восстановление NiO, не связанного химически с поверхностью Al2O3 [57, 58], так и восстановление солей никеля – нитрата и гидроксинитратов. Согласно литературным данным разложение нитрата никеля происходит при термообработке на воздухе при температурах до 310°С [59, 60], а нанесение на Al2O3 приводит к снижению этой температуры на 30°С [59]. Поэтому в образцах, обработанных при 250°С, может присутствовать как неразложившийся Ni(NO3)2, так и продукты его частичного (xNi(NO3)2 ⋅ yNi(OH)2 [60, 61]) и полного разложения (NiO), а также никель-алюминиевая шпинель.
Низкотемпературный пик в профилях ТПВ прекурсоров катализаторов, модифицированных ГПК, имеет заметно меньшую интенсивность по сравнению немодифицированными аналогами (табл. 2), причем максимальный вклад этой группы характерен для прекурсора катализатора Ni/Al2O3(A). По-видимому, стабильность нитрата никеля на поверхности Al2O3, модифицированного ГПК, снижается аналогично тому, как это наблюдали для систем NiO/WO3/Al2O3 в работе [62]. Следовательно, как мы и ожидали, нанесение ГПК способствует ослаблению взаимодействия никеля с Al2O3. Наименее стабилен нитрат никеля на поверхности носителя ГПК/Al2O3(E), поскольку в профиле прекурсора Ni/ГПК/Al2O3(E) низкотемпературный пик имеет минимальную интенсивность.
Оцененное из профилей ТПВ немодифицированных образцов суммарное поглощение водорода примерно в два раза превышает количество, необходимое для полного восстановления содержащегося в них Ni2+, рассчитанное из данных ААС о содержании никеля (табл. 2). Это может быть вызвано гидрированием углеродсодержащих функциональных групп на поверхности или присутствием неразложившегося или частично разложившегося нитрата никеля. Восстановление нитрогруппы сопровождается выделением аммиака и дает дополнительный вклад в поглощение Н2 при относительно низких температурах [62, 63]. Стабилизация нитрата никеля на поверхности Al2O3 отмечалась в литературе [62, 64].
Широкий пик в высокотемпературном интервале с максимумом около 470°С на профилях ТПВ образцов Ni/Al2O3(А) и Ni/Al2O3(E) можно отнести к восстановлению Ni2+, сильно взаимодействующего с Al2O3 в составе поверхностных псевдошпинельных структур NiAlOx [62, 65]. Восстановление нестехиометрических никель-алюминиевых шпинелей может происходить при температурах от 300 до 700°С [66, 67] в зависимости от состава, размера частиц и расположения в объеме или на поверхности образца. Взаимодействие никеля с Al2O3 затрудняет нуклеацию в ходе обработки водородом и приводит к поляризации связи Ni–O, что замедляет восстановление нанесенного NiO по сравнению с массивным и сдвигает соответствующий пик к высоким температурам [62].
Отсутствие поглощения водорода при температурах >600°С для прекурсоров немодифицированных катализаторов свидетельствует о том, что фаза стехиометрической шпинели NiAl2O4 в них не присутствует [68]. Это согласуется с данными РФА. В то же время на профилях ТПВ модифицированных ГПК прекурсоров наблюдается поглощение H2 в области температур >700°С, которое вызвано образованием NiAlOx или NiWOx.
Интенсивность высокотемпературного пика на профилях ТПВ модифицированных образцов возрастает, и его максимум смещается в сторону бóльших температур. Сдвиг температуры максимума с 470 до 510°С может быть связан с увеличением дисперсности никельсодержащих частиц [69], изменением состава нестехиометрических псевдошпинельных форм NiAlOx, восстановлением оксидов вольфрама, протекание которого вероятно при относительно низких температурах при каталитическом действии никеля [62], а также с появлением новых соединений при взаимодействии никеля с ГПК или продуктами ее разложения [54, 70–72] состава NiWO4 или NiWOAl [73].
Образование вольфрамата никеля в ходе прокаливания происходит при температуре 1000°С, в восстановительной атмосфере – при 800оС [74], однако формирование поверхностных нестехиометрических вольфраматов не исключено и при меньших температурах. Возникновение в модифицированных ГПК образцах фаз NiWOAl и NiWO4 вместо NiAl2O4 ожидаемо вследствие большего отрицательного значения энтальпии смеси NiWO4 + + Al2O3 по сравнению с NiAl2O4 + WO3 [62].
Восстановление NiO, химически связанного с модифицированной поверхностью, может приводить к появлению дефектов, обеспечивающих повышение прочности связывания кластеров и наночастиц никеля, и способствовать большей их устойчивости к спеканию [75].
Таким образом, данные ТПВ свидетельствуют о снижении термической устойчивости нитрата никеля, нанесенного на Al2O3, в результате модификации ГПК, и о сильном взаимодействии никеля с вольфрамсодержащим модификатором, которое проявляется в разной степени для образцов на двух типах носителей. Судя по большей относительной площади высокотемпературного пика, в составе Ni/ГПК/Al2O3(E) взаимодействие никеля с носителем существенно сильнее, чем в составе Ni/ГПК/Al2O3(А).
РФЭС-исследование невосстановленных прекурсоров катализаторов 6%Ni/Al2O3(Е), полученных методом пропитки, проведено ранее в работе [76]. В спектрах наблюдали линии Ni2p3/2 с Есв = = 856.0 эВ, соответствующие поверхностной нестехиометрической шпинели или Ni(OH)2. Шпинельные формы образуются вследствие химического взаимодействия на границе раздела Ni–Al2O3.
С целью изучения процессов восстановления никеля и вольфрама на поверхности катализаторов проведено РФЭС-исследование с in situ обработкой водородом в каталитической ячейке при 330 и 450°С. Обзорные спектры РФЭС, а также спектры высокого разрешения Ni2p и W4f приведены на рис. 6.
Рис. 6.
Спектры РФЭС образцов, восстановленных in situ водородом при 330 (а, в) и 450°С (б, г): обзорные (а, б) и высокого разрешения Ni2p (в, г) и W4f (д).
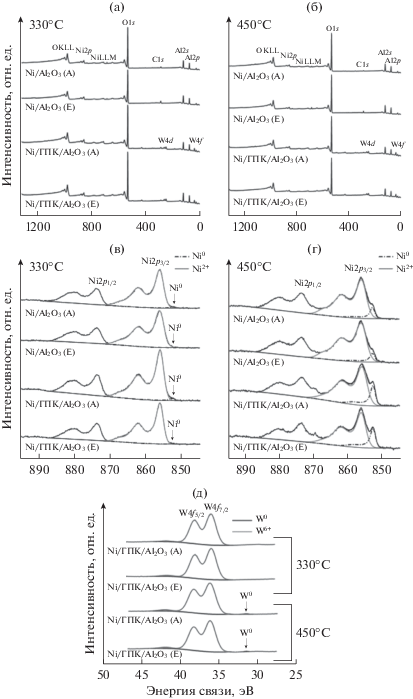
Спектры РФЭС Ni2p3/2 восстановленных in situ катализаторов характеризуются интенсивными shake-up сателлитами с Есв = 855.8–856.3 эВ, что близко к энергии связи Ni2p3/2 в составе шпинели NiAlO4 (856.2 эВ [77]) или Ni(OH)2 (диапазон 855.6–856.2 эВ [78]) и значительно выше, чем в NiO (854.4 эВ [77]). Это согласуется с результатами, полученными методом ТПВ, которые показали присутствие гидроксинитратов, гидроксидов никеля, а также химически связанного никеля в виде поверхностных нестехиометрических шпинелей. Во всех образцах наблюдается небольшое уменьшение Есв основного пика Ni2+ при переходе к температуре восстановления 450°С: с 856.2 до 855.9 эВ в Ni/Al2O3(Е); с 856.3 до 856.0 эВ в Ni/Al2O3(А); с 856.1 до 855.9 эВ в Ni/ГПК/Al2O3(Е); с 856.1 до 855.8 эВ в Ni/ГПК/Al2O3(А).
В спектрах Ni2p3/2 восстановленных образцов можно выделить компоненту с Есв = 852.3–852.8 эВ, соответствующую Ni0 [79, 80]. Значения энергии связи слегка варьируются при изменении температуры восстановления и при переходе от образца к образцу: повышаются с 852.3 до 852.6 эВ в Ni/Al2O3(Е), понижаются с 852.8 до 852.5 эВ в Ni/ГПК/Al2O3(E), однако почти не меняются в Ni/Al2O3(A) и Ni/ГПК/Al2O3(A), составляя 852.6 и 852.5 эВ соответственно. В образцах, восстановленных при 330°С, доля Ni0 не превышает 2% (табл. 3). Хотя содержание Ni0 находится на грани погрешности определения, все же можно отметить, что оно несколько больше в Ni/Al2O3(А) по сравнению с Ni/Al2O3(Е) и в модифицированных ГПК системах.
Таблица 3.
Содержание элементов на поверхности и доля атомов никеля и вольфрама в различных химических состояниях по данным РФЭС in situ после восстановления водородом
| Образец | Tвосст., °С | Доля элемента в соответствующем окислительном состоянии, % | Содержание на поверхности, % |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Ni | O | C | Al | W | ||||||
| Ni2p3/2 | W4 f7/2 | |||||||||
| Ni0 | Ni2+ | W0 | W6+ | |||||||
| Ni/Al2O3(А) | 330 | 1 | 99 | – | – | 3.0 | 61.6 | 5.6 | 29.8 | – |
| 450 | 9 | 91 | – | – | 2.4 | 62.9 | 2.6 | 32.1 | – | |
| Ni/Al2O3(E) | 330 | 0 | 100 | – | – | 2.9 | 55.5 | 11.1 | 30.5 | – |
| 450 | 8 | 92 | – | – | 2.1 | 57.6 | 10.7 | 29.6 | – | |
| Ni/ГПК/Al2O3(А) | 330 | 2 | 98 | 0 | 100 | 3.5 | 62.5 | 5.2 | 27.6 | 1.2 |
| 450 | 17 | 83 | 1 | 99 | 3.2 | 62.3 | 2.7 | 30.4 | 1.4 | |
| Ni/ГПК/Al2O3(E) | 330 | 1 | 99 | 0 | 100 | 2.9 | 63.1 | 3.5 | 29.4 | 1.1 |
| 450 | 19 | 81 | 1 | 99 | 1.5 | 63.5 | 2.5 | 31.2 | 1.3 | |
После обработки водородом при 450°С доля Ni0 существенно возрастает. В немодифицированных образцах Ni/Al2O3 содержание Ni0 составляет 8–9%, для модифицированных образцов приближается к 20%. Таким образом, модификация носителя ГПК облегчает восстановление никеля.
Спектры РФЭС W4f модифицированных ГПК образцов вследствие спин-орбитального расщепления содержат две линии: W4f5/2 и W4f7/2. Весь вольфрам в образцах, обработанных водородом при 330°С, находится в состоянии W6+, поскольку компонента W4f7/2 имеет энергию связи 36.0–36.2 эВ. Это несколько выше, чем величина 35.0 эВ для W6+ в составе промышленных катализаторов NiW/γ-Al2O3, приведенная в работе [81], где калибровку проводили также по линиям Al2p. В литературе приводятся также значения 35.7 эВ для WO3 и 36.1 эВ для гидроксида WO3(OH2)n [82], а также 35.8 эВ [83] и 36.3 эВ [84] для вольфрама в составе гетерополикислот. В результате обработки при 450°С в образцах, содержащих ГПК, появляется около 1% W0 (табл. 3). Значительный сдвиг линии W4f7/2 в W0 (31.2–31.6 эВ) относительно линий W6+ (36.0–36.2 эВ) позволяет уверенно говорить о его присутствии. Линии W4+ с энергией связи 32.7 эВ [81] и W5+ с энергией связи 35.4 эВ [85] в спектрах наших образцов не наблюдаются. Согласно [62], W4+ восстанавливается до W0 в одну стадию, что обусловлено нестабильностью промежуточных оксидов.
Отметим также, что при повышении температуры восстановления образцов отмечается уменьшение содержания никеля, а для модифицированных образцов – увеличение содержания вольфрама на поверхности (табл. 3), обусловленное миграцией этих элементов. Особенно заметно данный эффект проявляется для Ni/ГПК/Al2O3(E). Снижение доли углерода в образцах, восстановленных при 450°С, согласуется с высказанным ранее предположением о возможности гидрирования углеродсодержащих функциональных групп в условиях ТПВ, однако этот процесс скорее влияет на поглощение водорода в высокотемпературной области.
Таким образом, методом РФЭС in situ подтверждено повышение способности никеля к восстановлению при модификации носителя ГПК. По данным РФЭС никель в составе катализатора Ni/Al2O3(A) обладает несколько большей способностью к восстановлению при относительно низкой температуре (330°С). Согласно результатам ТПВ добавление ГПК в сравнении с Ni/Al2O3 (E) приводит к существенному изменению степени связывания нитрата никеля с поверхностью носителя и набора форм окисленного никеля, присутствующих в термообработанных на воздухе при 250°С прекурсорах катализаторов, а также их способности к восстановлению. При повышении температуры восстановления до 450°С содержание Ni0 возрастает. Данные ТПВ и РФЭС косвенно свидетельствуют об образовании на поверхности модифицированных ГПК систем вольфрамата никеля, о каталитическом действии никеля на восстановление вольфрама из оксида, а также подтверждают различия в степени связывания никеля с поверхностью двух использованных в работе типов Al2O3.
2.5. Исследование методом ИК ДО с адсорбцией СО
Восстановленные in situ Н2 при 330°С катализаторы исследовали методом ИК ДО-спектроскопии с адсорбцией СО (рис. 7).
Рис. 7.
Спектры ИК ДО с адсорбцией CO после восстановления при 330°С прекурсоров катализаторов Ni/Al2O3(Е) (а), Ni/Al2O3(А) (б), Ni/ГПК/Al2O3(Е) (в) и Ni/ГПК/Al2O3(A) (г), полученные при давлении CO равном 5 (1), 20 (2) и 50 (3) мм рт. ст.
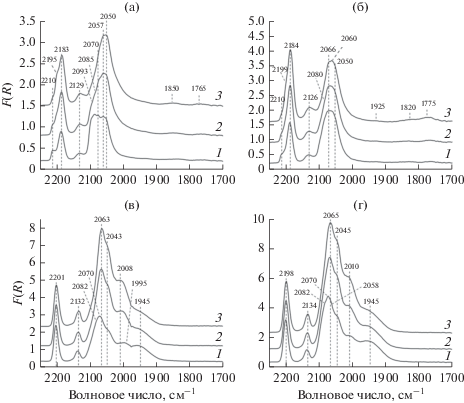
ИК ДО-спектры исходных носителей изучены ранее в работе [41]. В результате адсорбции СО на льюисовских кислотных центрах (ЛКЦ) в спектре Al2O3(A) наблюдается интенсивная полоса поглощения (п. п.) при 2200 см–1, в спектре Al2O3(E) – две п. п. при 2200 и 2236 см–1.
Адсорбция СО на поверхности Ni/Al2O3(E) и Ni/Al2O3(A) (рис. 7а и 7б) сопровождается появлением трех групп перекрывающихся п. п. В высокочастотной области спектра отмечается суперпозиция п. п. 2183–2185, 2198 и 2210 см–1. Полосу поглощения при 2183–2185 см–1 относят к карбонильным комплексам СО с катионами Ni2+ [86, 87]. Частоты п. п. 2198 и 2210 см–1 совпадают с характерными для комплексов СО с ЛКЦ Al2O3, однако в спектре отсутствуют другие присущие этим комплексам полосы при 2234–2240 и 2216–2219 см–1 [41]. Поэтому более правильно отнести наблюдаемые п. п. к комплексам с координационно ненасыщенными катионами Ni2+, входящими в структуру Ni2+–O–Ni2+ [87]. Такое отнесение хорошо согласуется с полученными ранее данными о типах ЛКЦ использованных в настоящей работе носителей, для которых характерна высокая интенсивность полосы наиболее координационно ненасыщенных катионов Al3+ (на вершине ступеньки или изломе ее линии) [41]. В этом случае возможно формирование заметной концентрации мостиковых структур типа Al3+(на вершине ступеньки)–Ni2+–O–Ni2+–Al3+(у основания ступеньки). Также никель может быть локализован на месте отсутствующего катиона Al3+ с образованием структуры типа Ni2+(на вершине ступеньки)–O–Ni2+(у основания ступеньки).
Поглощение при 2126–2130 см–1 относится к карбонильным комплексам с катионами Ni+. В области частот 2040–2100 см–1 наблюдается суперпозиция нескольких перекрывающихся п. п. Полосы при 2065–2073 и 2059–2060 см–1 соответствуют комплексам СО с атомами Ni0 в различном координационном окружении [87]. К линейным комплексам СО с Ni0 на поверхности микрочастиц или кластеров металлического никеля можно отнести п. п. при 2050–2053 см–1 [88].
Интересной особенностью спектра образца Ni/Al2O3(Е) является появление после адсорбции СО при 5 мм рт. ст. полос поглощения при 2085 и 2095 см–1, интенсивность которых падает при повышении давления, т.е. степени заполнения поверхности молекулами СО. Авторы [89] относят эти п. п. к дикарбонилам Ni+(CO)2, однако в этом случае снижение интенсивности указанных полос при росте давления СО должно сопровождаться уменьшением интенсивности п. п. 2130 см–1, чего мы не наблюдаем. Поэтому мы считаем, что эти полосы характеризуют комплексы СО с катионами Niδ+ (δ < 1) [90], которые возникают в результате взаимодействия расположенных рядом атома Ni0 и катиона Ni+ [91]. Понижение интенсивности п. п. 2085 и 2095 см–1 при увеличении степени заполнения поверхности молекулами СО вызвано изменением состояния такого адсорбционного центра при взаимодействии катионов никеля с молекулами СО.
В спектре Ni/Al2O3(A) интенсивность п. п. комплексов СО с Ni0 ниже по сравнению с таковой для комплексов с Ni2+, тогда как для образца Ni/Al2O3(E) наблюдается обратная картина. Следовательно, доля Ni0, образовавшегося на поверхности в ходе обработки водородом при 330°С, заметно больше в образце Ni/Al2O3(E) по сравнению с Ni/Al2O3(A). Эти данные не согласуются с полученными методом РФЭС результатами.
В спектрах обоих образцов при 1820–1850 и 1765–1775 см–1 наблюдаются п. п. мостиковых форм адсорбции СО. Первую из них можно отнести к комплексам СО, координированного атомом углерода с тремя или четырьмя атомами никеля, вторая соответствует мостиковой форме адсорбции с участием обоих атомов в молекуле СО [89].
Модифицирование поверхности Al2O3 ГПК приводит к значительным изменениям в спектре адсорбированного СО (рис. 7в и 7г), которые указывают на нивелирование структурных различий в распределении никеля на поверхности, вызванных использованием двух типов Al2O3. В высокочастотной области спектра для обоих модифицированных образцов наблюдается только одна симметричная п. п. при 2198–2200 см–1. Отсутствие п. п. 2210 см–1 может быть связано с координацией ГПК по ЛКЦ оксида алюминия. Максимум п. п. комплексов СО с катионами Ni2+ сдвигается в высокочастотную область на 14–17 см–1, а с катионами Ni+ – на 3–8 см–1. Следовательно, электроноакцепторная способность ЛКЦ в результате модифицирования ГПК повышается. При этом интенсивность п. п., соответствующих комплексам СО с Ni2+ (2200 см–1), возрастает в 1.5 раза по сравнению с немодифицированными системами, а интенсивность суперпозиции п. п. комплексов СО с атомами Ni0 (с максимумом при 2063 см–1) увеличивается более чем в 3 раза. Это указывает на бóльшую доступность никеля для СО в модифицированных ГПК системах. В спектрах модифицированных катализаторов вклад п. п. при 2050 см–1, которую мы относим к адсорбции СО на Ni0 в окружении других атомов никеля, ниже, чем для немодифицированных, что может свидетельствовать об уменьшении размера частиц никеля.
В области частот ниже 2050 см–1 присутствуют новые п. п. при 1945–1955 и 1995–2010 см–1, которых не было в спектрах систем без ГПК, но исчезают п. п. с частотами ниже 1900 см–1. Полосу поглощения 1945–1955 см–1 можно отнести к мостиковой адсорбции СО атомом углерода на двух расположенных рядом атомах Ni0 [92]. Природа п. п. 1995–2010 см–1 неясна, ранее такую п. п. не наблюдали в спектрах никельсодержащих систем. Возможно, она относится к мостиковой адсорбции СО на атомах никеля и вольфрама. К сожалению, комплексы СО с катионами W6+ и W4+ на Al2O3 выделить не удается, поскольку их п. п. (2200 см–1 в случае W6+, 2176 и 2154 см–1 в случае W4+ [93]) перекрываются с п. п. комплексов СО с катионами никеля.
Снижение вклада в спектр комплексов СО с п. п. 2050 см–1, наличие мостиковых форм адсорбции СО на двух атомах никеля и отсутствие комплексов СО с тремя/четырьмя атомами никеля (п. п. при 1820–1850 и 1765–1775 см–1) косвенно свидетельствуют о более высокой дисперсности никеля в модифицированных ГПК образцах.
Таким образом, данные ИК ДО-спектроскопии указывают на присутствие Ni0 в заметной концентрации на поверхности всех восстановленных при 330°С катализаторов, в то время как по данным РФЭС Ni0 имеется не во всех образцах, а в тех, где он есть, его содержание очень мало (менее 2%).
Такое различие в данных ИК ДО и РФЭС может быть связано с тем, что в процессе восстановления частицы металлического никеля мигрируют в объем образца. В результате поверхностная концентрация никеля, определенная методом РФЭС, снижается. Глубина анализа методом ИК ДО выше, и он регистрирует мигрировавшие внутрь образца частицы Ni0, которые не обнаруживаются с помощью РФЭС.
Из представленных результатов отчетливо видны различия в составе и структуре адсорбционных центров в никелевых катализаторах на разных типах Al2O3, а также влияние модификации ГПК на эти параметры.
2.6. Тестирование каталитических систем в реакции ГДХ ХБ
Все катализаторы после восстановления при 330°С проявили существенную активность в парофазном ГДХ ХБ с образованием бензола и циклогексана. Методом ГХ/МС в составе продуктов на модифицированных катализаторах обнаружено также присутствие следовых количеств продуктов скелетной изомеризации циклогексана (метилциклопентан).
На рис. 8а приведены кривые зависимости конверсии ХБ от температуры в ходе повышения и снижения температуры реакции. Каждая точка на этих кривых получена как среднее значение конверсии ХБ в трех последовательных пробах после достижения стабильных величин этого параметра в изотермическом режиме. В присутствии обоих немодифицированных катализаторов ХБ начинает восстанавливаться уже при относительно низких температурах (150–200°C). Так, конверсия ХБ на Ni/Al2O3(E) составляет 38% уже при 150°С (рис. 8а). При 200°С она достигает 99% и не меняется при дальнейшем повышении температуры. Катализатор Ni/Al2O3(А) начинает эффективно работать при более высоких температурах. При 200°С конверсия ХБ (45%) на этом катализаторе вдвое ниже, чем в присутствии Ni/Al2O3(E), и приближается к 100% лишь при 250°С.
Рис. 8.
Зависимость конверсии ХБ (а) и селективности образования циклогексана (б) от температуры при ступенчатом нагреве и охлаждении.
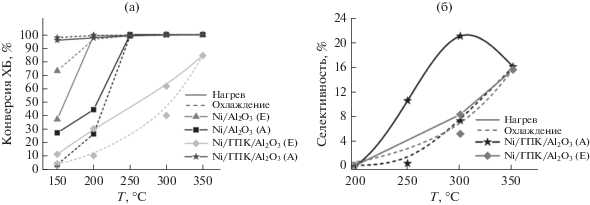
Модификация ГПК по-разному влияет на каталитические свойства образцов на носителях Al2O3(А) и Al2O3(Е). Катализатор Ni/ГПК/Al2O3(Е) во всем исследованном интервале температур обеспечивает меньшую конверсию ХБ по сравнению с обоими немодифицированными образцами: при 150°С конверсия ХБ составляет лишь 12%, при повышении температуры почти линейно увеличивается до максимального значения 85% при 350°С (рис. 8а). Напротив, модификация ГПК образца Ni/Al2O3(А) приводит к значительному улучшению каталитических свойств. Уже при 150°С конверсия ХБ составляет 96%, при повышении температуры возрастает до 98–100% (рис. 8а).
Преобладающим продуктом реакции на немодифицированных катализаторах является бензол, т.е. они активируют связь C–Cl и H2, однако активации С–С-связей в бензольном кольце не происходит, и бензол десорбируется с поверхности сразу после образования. Циклогексан обнаружен лишь в количествах менее 1%. В присутствии модифицированных катализаторов в интервале температур 200–350°С наблюдается заметное образование циклогексана (ЦГ) (рис. 8б): его максимальное содержание в смеси составляет 21% при 300°С на образце Ni/ГПК/Al2O3(А) и 16% при 350°С на катализаторе Ni/ГПК/Al2O3(E).
В присутствии Ni/ГПК/Al2O3(А) значения конверсии ХБ, достигнутые при определенных температурах в ходе повышения и снижения температуры реакции, практически совпадают. Три других катализатора по-разному ведут себя при нагревании и охлаждении. Конверсия ХБ на Ni/Al2O3(E) при 150°С выше при охлаждении (73%), чем при нагревании (38%). Напротив, образец Ni/Al2O3(А) в ходе снижения температуры работает хуже: при 150°С конверсия составляет 28% в начале опыта и только 4% в конце. Падение конверсии в ходе понижения температуры реакции по сравнению с наблюдаемой в ходе ее повышения отмечается и для модифицированного Ni/ГПК/Al2O3(E).
Образец Ni/Al2O3(А) после каталитических испытаний исследовали методом ТПВ (рис. 5). Видно, что профиль ТПВ этого образца сильно меняется: он не содержит пиков в низкотемпературной области, высокотемпературный пик имеет меньшую интенсивность и ширину и сдвинут в сторону бóльших температур по сравнению с профилем ТПВ исходного образца.
3. ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Каталитические свойства Ni/Al2O3 зависят от типа Al2O3, использованного в качестве носителя. В интервале 150–200°С более эффективен катализатор на основе Al2O3(Е), хотя в его составе методом ААС обнаружено меньшее содержание никеля (5.26 вместо 6.45 мас. %), а методом низкотемпературной адсорбции азота – более низкая удельная поверхность, радиус и объем пор по сравнению с Ni/Al2O3(А). По данным РФЭС содержание никеля на поверхности этих катализаторов практически не различается, а степень восстановления никеля на поверхности после обработки водородом при 330°С не превышает 1%. Судя по отсутствию рефлексов никельсодержащих фаз на дифрактограммах, дисперсность никелевых частиц во всех катализаторах высокая. Однако текстурные факторы могут способствовать образованию несколько более крупных частиц никеля при восстановлении образца Ni/Al2O3(E), что может играть важную роль для структурно-чувствительной реакции ГДХ. Это предположение находит подтверждение в данных ИК ДО с адсорбцией СО, т.к. в спектре образца Ni/Al2O3(E) выше интенсивность полос, обусловленных координацией СО на нескольких частицах никеля. В спектрах ИК ДО обработанных водородом при 330°С катализаторов Ni/Al2O3(А) и Ni/Al2O3(Е) присутствуют п. п., характерные для комплексов СО с никелем в трех степенях окисления (0, 1+ и 2+). Однако в составе Ni/Al2O3(E) преобладает Ni0, а в составе Ni/Al2O3(А) – окисленные формы никеля.
Соотношения разных типов сильных ЛКЦ на поверхности носителей Al2O3 (А) и (Е) различаются [41]. На поверхности Al2O3(Е) часть ЛКЦ относится к типу L1 по классификации Мортерры [94, 95], и они преимущественно заполняются при низких давлениях СО, но имеются также ЛКЦ типов L3 и L2. На поверхности же Al2O3(А) типы L3 и L2 становятся преобладающими. Следовательно, координация Ni(NO3)2 в ходе нанесения на носитель из раствора по-разному происходит на поверхности этих двух типов носителя. Присутствие на поверхности Al2O3(E) более сильных ЛКЦ по сравнению с Al2O3(А) в ходе приготовления катализаторов может приводить к усилению взаимодействия между Ni2+ и Al2O3 [96], и это согласуется с результатами ТПВ. Действительно, в профиле Ni/Al2O3(E) (рис. 5 и табл. 2) относительное содержание химически связанного NiO заметно выше.
Бóльшая каталитическая эффективность Ni/Al2O3(E) при 150°С, вероятно, связана с повышенным содержанием сильных ЛКЦ. Хлорбензол способен адсорбироваться и активироваться как на металле, так и на носителе. При использовании Al2O3 адсорбция ХБ с высокой вероятностью протекает на ЛКЦ носителя, далее реагент гидрируется активированным водородом, поступившим к носителю с поверхности металла по механизму спилловера. За счет особенностей кислотности можно ожидать более сильной адсорбции ХБ на носителе Al2O3(Е).
Существенные различия в составе двух немодифицированных катализаторов найдены методом ТПВ (рис. 5): низкотемпературный пик, обусловленный процессами разложения и восстановления нитрата и гидроксинитратов никеля, в профиле ТПВ прекурсора Ni/Al2O3(Е) имеет меньшую относительную площадь по сравнению с высокотемпературным пиком и с аналогичным пиком в профиле прекурсора Ni/Al2O3(А). Следовательно, особенности структуры носителя Ni/Al2O3(E) способствуют более полному разложению нитрата никеля в ходе термической обработки при низкой температуре (250°С). Кроме того, в профиле ТПВ Ni/Al2O3(E) наблюдается небольшой пик при 186°С, отсутствующий на профиле ТПВ Ni/Al2O3(А), который может свидетельствовать о присутствии легко восстанавливаемой формы никеля. По-видимому, структурные особенности Ni/Al2O3(E) обуславливают более высокую эффективность образующихся в ходе восстановления прекурсоров частиц никеля в ГДХ хлорбензола при относительно низких температурах.
Наблюдаемые различия в активности катализаторов при низких температурах реакции коррелируют с относительно большим содержанием Ni0 в Ni/Al2O3(E), обнаруженным методом ИК ДО спектроскопии. При Т ≥ 250°С оба катализатора проявляют одинаково высокую эффективность в связи с общим возрастанием скорости реакции и достижением температур, при которых может осуществляться дополнительное восстановление никеля водородом из реакционной среды.
Для системы Ni/Al2O3(E) наблюдалось увеличение конверсии ХБ ходе снижения температуры реакции от 350 до 150°С по сравнению с величинами, достигнутыми в ходе ее повышения. По данным ИК ДО спектроскопии никель в этом образце легче подвергается восстановлению при относительно низкой температуре (330°С), так что при длительном пребывании в реакционной среде, содержащей H2, может происходить восстановление дополнительных количеств никеля, обеспечивающее повышение степени превращения ХБ при “возвращении” к температуре 150°С.
Модификация ГПК приводит к существенным изменениям физико-химических и каталитических свойств. Так, в профилях ТПВ модифицированных катализаторов повышается относительная интенсивность высокотемпературного пика и происходит его сдвиг в сторону бóльших температур. Это может быть обусловлено изменением силы связывания нитрата никеля с поверхностью Al2O3, облегчающим разложение нитрата и гидроксинитратов при относительно низкой температуре термообработки (250°С), а также сильным взаимодействием между никелем и WO3 в составе ГПК. По данным ТПВ, в составе Ni/ГПК/Al2O3(E) связь никеля с носителем существенно сильнее, чем в Ni/ГПК/Al2O3(А). В литературе отмечено формирование связей Ni–O–W в материалах, содержащих соли никеля и гетерополисоединения на основе вольфрама [71, 72].
При взаимодействии никеля с модифицированным ГПК носителем может образоваться как поверхностный алюминат, так и NiWО4 в ходе координации с WO3 в составе ГПК. По данным [62] вольфрамат восстанавливается водородом при 500°С, а алюминат никеля при еще более высоких температурах, так что в восстановленных при 330°С системах возможно присутствие обоих типов поверхностных соединений. Ранее отмечалось, что взаимодействие NiO c WO3 способствует возрастанию активности в диссоциативной адсорбции водорода, а также активации связей С–С [70, 97].
По данным ИК ДО спектроскопии, в результате модификации ГПК существенно повышается льюисовская кислотность каталитических систем, что согласуется с данными о сильном взаимодействии никеля с вольфрамсодержащим модификатором, полученными методом ТПВ. После нанесения ГПК сдвигается в высокочастотную область пик, соответствующий поверхностным комплексам CO с Ni2+. Следовательно, добавление ГПК повышает электроноакцепторную способность никеля.
Заметно возрастает интенсивность широкой неоднородной п. п. в интервале 2000–2060 см–1, характеризующей адсорбцию CO на Ni0, то есть модификация облегчает восстановление никеля. Неоднородность указанной п. п. свидетельствует о присутствии в образцах Ni/ГПК/Al2O3(Е) и Ni/ГПК/Al2O3(А) нескольких форм Ni0, обеспечивающих возможность различных вариантов мостиковой адсорбции СО [86, 92, 98]. В работе [92] показано, что доля мостиковой адсорбции Ni0(CO)2 с п. п. при 1940 см–1 высока на образце Ni/SiO2, модифицированном этиленгликолем, за счет большей дисперсности частиц металлического никеля и лучшей восстановительной способности по сравнению с немодифицированным аналогом. Ранее [41] было обнаружено, что введение ГПК в каталитическую систему Pd/Al2O3 способствует уменьшению размера частиц Pd. Вполне вероятно, что в случае никелевых катализаторов на аналогичных носителях также происходит повышение дисперсности активного компонента, что находит подтверждение в данных ИК ДО спектроскопии с адсорбцией СО. Изменения в способности NiO к восстановлению могут быть связаны с сильным взаимодействием никеля с W или Si с образованием структур Ni–O–W или Ni–O–Si. Методом ИК ДО спектроскопии не найдено существенных различий в структуре поверхности и способности к восстановлению между модифицированными ГПК катализаторами на разных типах Al2O3.
Повышение способности никеля к восстановлению в результате модификации Al2O3 нанесением ГПК находит подтверждение в данных РФЭС in situ. Содержание Ni0 в обоих модифицированных катализаторах после восстановления как при 330°С, так и при более высокой температуре значимо увеличивается по сравнению с немодифицированными образцами. Также этим методом обнаружено обеднение поверхности никелем в модифицированных ГПК системах, особенно заметное для Ni/ГПК/Al2O3(E).
Изменения кислотности поверхности и электронного состояния никеля, его сильное взаимодействие с вольфрамсодержащим модификатором носителя приводят к иным каталитическим свойствам модифицированных катализаторов. Особенно интересно существенное увеличение эффективности катализатора Ni/ГПК/Al2O3(А) по сравнению с немодифицированным аналогом. Наиболее вероятной причиной является повышение степени восстановления никеля в результате модификации. Кроме того, рост активности может быть вызван дополнительной активацией водорода на вольфрамсодержащих компонентах катализатора – ГПК [35] и поверхностных структурах типа вольфрамата никеля. В ходе частичной деградации ГПК могут образовываться анионные вакансии, на которых протекает гетеролитическая диссоциация водорода [9]. Хлорорганические соединения адсорбируются на анионных вакансиях, титруются водородом и десорбируются с поверхности. Протоны ГПК участвуют в образовании стабильного гидрирующего слоя, который перетекает с металла и ГПК на носитель по механизму спилловера. Кроме того, изменения в каталитическом действии могут быть вызваны более прочной хемосорбцией хлорбензола на ЛКЦ модифицированной подложки.
Особенностями структуры, по-видимому, обусловлена повышенная способность Ni/ГПК/Al2O3(А) к образованию циклогексана (рис. 8б). Оно происходит в том случае, когда на поверхности присутствуют сильные центры адсорбции ХБ. Сильная адсорбция может обеспечивать гидрирование ХБ до циклогексана без десорбции промежуточного продукта – бензола. Отрицательные значения энергии Гиббса, характерные для реакции гидрирования бензола до циклогексана при относительно низких температурах, при нагревании реакционной системы выше 300°С сменяются положительными, что объясняет экстремальную зависимость селективности образования циклогексана от температуры реакции в ходе ее повышения (рис. 8б). Падение селективности по циклогексану, наблюдаемое в ходе ступенчатого снижения температуры реакции, может быть связано со структурными и химическими изменениями, происходящими в реакционной среде при 350°С (эта температура выше, чем температура восстановления катализатора), приводящими к уменьшению силы адсорбции ХБ на поверхности.
В статье [99] обсуждались электронные взаимодействия между оксидами NiO и WO3, приводящие к понижению электронной плотности на никеле и переносу ее к вольфраму. Это облегчает диссоциативную адсорбцию водорода. Поверхностные центры Ni–W, например, в составе NiWO4, могут служить прекурсорами активных центров. Однако их появление более вероятно при более высоких температурах термообработки на воздухе, чем использованные в нашей работе (250°С) [74]. Также в подобных системах возможно формирование вольфрамовых бронз вида HxWO3. Атомы водорода, активированного на Ni, по механизму спилловера поступают к терминальному кислороду оксида вольфрама и встраиваются в его решетку с образованием бронз, проявляющих высокую активность в реакциях гидрирования [42].
Таким образом, увеличение конверсии ХБ на Ni/ГПК/Al2O3(А) по сравнению с Ni/Al2O3(А) может быть обусловлено ростом содержания Ni0 в восстановленном модифицированном образце и формированием новых активных центров в результате взаимодействия между никелем и вольфрамом в составе ГПК или продуктов ее частичного разложения.
Сложнее объяснить снижение эффективности катализатора на носителе Al2O3(Е) после модификации ГПК. По данным ААС и адсорбционных измерений содержание никеля в этих образцах и текстурные свойства существенно не различаются. Однако на профиле ТПВ прекурсора Ni/ГПК/Al2O3(E) наблюдается минимальная относительная интенсивность пика восстановления при низких температурах, хотя интенсивность высокотемпературного пика сравнима с таковой для Ni/ГПК/Al2O3(А), что может свидетельствовать о меньшем содержании способных к легкому восстановлению форм никеля. Кроме того, методом РФЭС обнаружена наибольшая степень миграции никеля в объем образца Ni/ГПК/Al2O3(Е) в ходе обработки водородом. Возможно, падение активности связано с дополнительной миграцией части никеля под воздействием водорода реакционной смеси при проведении реакции при температурах 300–350°С.
Уменьшение значений конверсии ХБ в ходе снижения температуры реакции от 350 до 150°С по сравнению с величинами, полученными в ходе повышения температуры, которое наблюдали для катализаторов Ni/Al2O3(А) и Ni/ГПК/Al2O3(E), может быть вызвано постепенной дезактивацией катализаторов под действием реакционной среды. Наиболее вероятные ее причины – взаимодействие с адсорбированным на поверхности хлором, приводящее к окислению никеля, спекание частиц никеля и изменение степени их взаимодействия с носителем. Образование NiCl2 может приводить к потере активных центров и блокировке оставшихся [100, 101]. В литературе обсуждается также агрегация никелевых частиц под действием реакционной среды: так, в работе [102] наблюдали снижение дисперсности 5% Ni/SiO2 с 37 до 18% после проведения ГДХ 1,2-дихлорпропана.
Профиль ТПВ образца Ni/Al2O3(A) после использования в реакции ГДХ хлорбензола (рис. 5) содержит один пик с максимумом при 500°С, а в области относительно низких температур наблюдается лишь незначительное поглощение водорода. В работе [103] показано, что допирование хлором Ni/CeO2 приводит к высокотемпературному смещению пика восстановления в профиле ТПВ от 335 до 400°С, однако оно существенно меньше, чем наблюдаемое в нашей работе. По-видимому, сдвиг пика восстановления никеля в область высоких температур после проведения каталитической реакции вызван комплексом причин, включающих хлорирование активного компонента, изменение размера частиц никеля, а также усиление взаимодействия с подложкой.
Хотя отрицательный гистерезис свидетельствует о дезактивации образцов, значительного падения активности в ходе каталитических испытаний не наблюдали. Продолжительность работы каждого катализатора в проточном режиме задавалась необходимостью достижения стабильных показателей конверсии ХБ в трех последовательных анализах методом ГЖХ, каждый длился около 30 мин, и не всегда это были первые три анализа. Таким образом, каждый катализатор в ходе испытаний в режиме нагрев–охлаждение в большинстве опытов работал не менее 20 ч. Расчет показывает, что за время каталитического эксперимента при повышении и последующем снижении температуры в присутствии Ni/ГПК/Al2O3(А) переработано в продукты (преимущественно бензол) 373 г ХБ/г Ni в катализаторе, и признаков дезактивации не отмечалось. Это позволяет говорить о высокой стабильности катализатора Ni/ГПК/Al2O3(А), для которого гистерезис не наблюдается. Возможно, его удалось бы определить при использовании меньшей навески этого высокоактивного катализатора. В работе [104] образование структур типа Ni2+–O3W–O–WO3 в ходе сильного взаимодействия никеля с вольфрамсодержащими соединениями способствовало большей устойчивости катализатора к сернистым соединениям. В нашем случае формирование таких соединений способно повышать устойчивость катализаторов к воздействию хлора.
Безусловно, представляет интерес существенное увеличение доли Ni0 в образцах, восстановленных при 450°С. Такая обработка может обеспечить повышение эффективности катализаторов, но она не исключает разложения ГПК, поэтому эти системы будут изучены и подробно описаны в будущих публикациях.
ЗАКЛЮЧЕНИЕ
В представленной работе проведено исследование физико-химических и каталитических свойств Al2O3, различающихся морфологией и текстурой, немодифицированных и модифицированных нанесением ГПК на основе вольфрама и кремния. Чтобы предотвратить разложение ГПК и обеспечить возможности для корректного сравнения, термообработку на воздухе и восстановление модифицированных и не содержащих ГПК материалов проводили при относительно низких температурах (250 и 330°С соответственно). Методом ТПВ показано, что воздушная термообработка при 250°С не обеспечивает полного превращения исходного нитрата никеля в оксид, в связи с чем на поверхности прекурсоров катализаторов присутствует значительное количество нитратов и гидроксинитратов, дающих на профиле ТПВ суперпозицию пиков при температуре ниже 350°С. Методами РФЭС in situ, ИК ДО спектроскопии с адсорбцией СО и ТПВ обнаружены различия в составе и силе ЛКЦ на поверхности двух типов Al2O3, которые приводят к неодинаковой координации нитрата и разной способности никеля к восстановлению. Так, на поверхности Ni/Al2O3(E) никель несколько легче восстанавливается и, возможно, образует более крупные наночастицы. Эти факторы оказывают гораздо более существенное влияние на показатели конверсии ХБ при парофазном гидродехлорировании, чем различия в текстуре и даже в содержании никеля в катализаторе.
Модификация Ni/Al2O3 гетерополикислотой состава H8Si(W2O7)6 ⋅ nH2O изменяет кислотные свойства поверхности и способ координации нитрата никеля в ходе пропитки, облегчает разложение нитрата и повышает способность никеля к восстановлению. В результате сильного взаимодействия никеля и вольфрама в составе ГПК увеличивается соотношение Ni0 и Ni2+ на поверхности восстановленных систем (по данным РФЭС in situ и ИК ДО с адсорбцией СО), а также адсорбционная способность в отношении хлорбензола, появляются новые центры диссоциативной адсорбции водорода и, возможно, меняется размер частиц активного компонента. В зависимости от природы носителя модификация ГПК приводит к улучшению (Ni/ГПК/Al2O3(А)) или ухудшению (Ni/ГПК/Al2O3(E)) каталитической эффективности. При высоких температурах в системах с модификатором селективность по бензолу снижается вследствие гидрирования бензола до циклогексана.
Эффективность катализаторов возрастает в ряду Ni/ГПК/Al2O3(E) < Ni/Al2O3(А) < Ni/Al2O3(E) < < Ni/ГПК/Al2O3(А). Наиболее активный катализатор Ni/ГПК/Al2O3(А) является также самым стабильным и не дезактивируется в ходе каталитических испытаний.
Список литературы
Lokteva E.S., Golubina E.V., Likholobov V.A., Lunin V.V. / Disposal of chlorine-containing wastes. In: Chemistry beyond chlorine. Springer, Netherlands. 2016. P. 559.
Wei G.L., Liang X.L., Li D.Q., Zhuo M.N., Zhang S.Y., Huang Q.X., Yuan Z.J. // Environ. Int. 2016. V. 92. P. 373.
van Mourik L.M., Gaus C., Leonards P.E.G., de Boer J. // Chemosphere. 2016. V. 155. P. 415.
Huang B., Lei C., Wei C., Zeng G. // Environ. Int. 2014. V. 71. P. 118.
Keane M.A. // ChemCatChem. 2011. V. 3. P. 800.
Amorim C., Keane M.A. // J. Hazard. Mater. 2012. V. 211. P. 208.
Hashimoto Y., Uemichi Y., Ayame A. // J. Jpn. Pet. Inst. 2005. V. 48. P. 127.
Локтева Е.С., Голубина Е.В., Антонова М.В., Клоков С.В., Маслаков К.И., Егоров А.В., Лихолобов В.А. // Кинетика и Катализ. 2015. Т. 56. № 6. С. 753.
Навалихина М.Д., Кавалерская Н.Е., Локтева Е.С., Перистый А.А., Голубина Е.В., Лунин В.В. // Журн. физ. химии. 2012. Т. 86. С. 1792.
Li F., Liu Y., Wang L., Li X., Ma T., Gong G. / Application of Heterogeneous Catalysts in Dechlorination of chlorophenols. In: Organochlorine by Aurel Nuro, IntechOpen, 2018. P. 346.
Meshesha B.T., Chimenta R.J., Medina F., Sueiras J.E., Cesteros Y., Salagre P., Figueras F. // Appl. Catal. B: Environ. 2009. V. 87. P. 70.
Coq B., Ferrat G., Figueras F. // J. Catal. 1986. V. 101. P. 434.
Benitez J.L., Del Angel G. // React. Kinet. Catal. Lett. 2000. V. 70. P. 67.
Генцлер А.Г., Симагина В.И., Нецкина О.В., Комова О.В., Цыбуля С.В., Абросимов О.Г. // Кинетика и Катализ. 2007. Т. 48. С. 60.
Klokov S.V., Lokteva E.S., Golubina E.V., Chernavskii P.A., Maslakov K.I., Egorova T.B., Chernyak S.A., Minin A.S., Konev A.S. // Appl. Surf. Sci. 2019. V. 463. P. 395.
de Jong V., Louw R. // Appl. Catal. A: Gen. 2004. V. 271. P. 153.
Srikanth C.S., Kumar V.P., Viswanadham B., Chary K.V.R. // Catal. Commun. 2011. V. 13. P. 69.
Diaz E., Mohedano A.F., Casas J.A., Shalaby C., Eser S., Rodriguez J.J. // Appl. Catal. B: Environ. 2016. V. 186. P. 151.
Comandella D., Woszidlo S., Georgi A., Kopinke F.-D., Mackenzie K. // Appl. Catal. B: Environ. 2016. V. 186. P. 204.
Amorim C., Wang X., Keane M.A. // Chin. J. Catal. 2011. V. 32. P. 746.
Ruiz-García C., Heras F., Gilarranz M.Á., Aranda P., Ruiz-Hitzky E. // Appl. Clay Sci. 2018. V. 161. P. 132.
Arevalo-Bastante A., Álvarez-Montero M.A., Bedia J., Gómez-Sainero L.M., Rodriguez J.J. // Appl. Catal. B: Environ. 2015. V. 179. P. 551.
Ordóñez S., Sastre H., Díez. F.V. // Appl. Catal. B: Environ. 2000. V. 25. P. 49.
Babu N.S., Lingaiah N., Pasha N., Kumar J.V., Prasad P.S.S. // Catal. Today. 2009. V. 141. P. 120.
Lingaiah N., Prasad P.S.S., Rao P.K., Berry F.J., Smart L.E. // Catal. Commun. 2002. V. 3. P. 391.
Jujjuri S., Ding E., Shore S.G., Keane M.A. // Appl. Organomet. Chem. 2003. V. 17. P. 493.
Shao Y., Xu Z., Wan H., Chen H., Liu F., Li L., Zheng S. // J. Hazard. Mater. 2010. V. 179. P. 135.
Babu N.S., Lingaiah N., Prasad P.S.S. // Appl. Catal. B: Environ. 2012. V. 111. P. 309.
Trueba M., Trasatti S.P. // Eur. J. Inorg. Chem. 2005. V. 17. P. 3393.
Kim P., Kim Y., Kim H., Song I.K., Yi J. // J. Mol. Catal. A: Chem. 2004. V. 219. P. 87.
Bonarowska M., Kaszkur Z., Kępiński L., Karpiński Z. // Appl. Catal. B: Environ. 2010. V. 99. P. 248.
Babu N.S., Lingaiah N., Kumar J.V., Prasad P. S.S. // Appl. Catal. A: Gen. 2009. V. 367. P. 70.
Díaz E., Faba L., Ordóñez S. // Appl. Catal. B: Environ. 2011. V. 104. P. 415.
Gómez-Sainero L.M., Seoane X.L., Fierro J.L.G., Arcoya A. // J. Catal. 2002. V. 209. P. 279.
Навалихина М.Д., Крылов О.В. // Кинетика и Катализ. 2001. Т. 42. С. 264.
Eswaramoorthi I., Geetha Bhavani A., Lingappan N. // Appl. Catal. A: Gen. 2003. V. 253. P. 469.
Puello-Polo E., Diaz Y., Brito J.L. // Catal. Commun. 2017. V. 99. P. 89.
Lee K.-Y., Misono M. / Heteropoly Compounds. In: Handbook of Heterogeneous Catalysis. 2nd. ed., Wiley-VCH, Weinheim, 2008. P. 318.
Okuhara T., Mizuno N., Misono M. / Catalytic Chemistry of Heteropoly Compounds. In: Advances in Catalysis, Academic Press, 1996. V. 41. P. 113.
Alcañiz-Monge J., El Bakkali B., Trautwein G., Reinoso S. // Appl. Catal. B: Environ. 2018. V. 224. P. 194.
Голубина Е.В., Локтева Е.С., Гурбанова У.Д., Харланов А.Н., Егорова Т.Б., Липатова И.А., Власкин М.С., Школьников Е.И. // Кинетика и Катализ. 2019. Т. 60. № 3. С. 315.
Xi Y., Chen Z., Gan Wei Kiat V., Huang L., Cheng H. // Phys. Chem. Chem. Phys. 2015. V. 17. P. 9698.
Tarlani A., Abedini M., Khabaz M., Amini M.M. // J. Colloid Interf. Sci. 2006. V. 292. P. 486.
Green S. V., Kuzmin A., Purans J., Granqvist C.G., Niklasson G.A. // Thin Solid Films. 2011. V. 519. P. 2062.
Liang Y., Zhao M., Wang J., Sun M., Li S., Huang Y., Zhong L., Gong M., Chen Y. // J. Ind. Eng. Chem. 2017. V. 54. P. 359.
Newman A.D., Brown D.R., Siril P., Lee A.F., Wilson K. // Phys. Chem. Chem. Phys. 2006. V. 8. P. 2893.
Liu L., Wang B., Du Y., Borgna A. // Appl. Catal. A: Gen. 2015. V. 489. P. 32.
Школьников Е.И., Волков В.В. // Доклады РАН. Физ. Химия. 2001. Т. 378. № 4. С. 507.
Shkolnikov E.I., Shaitura N.S., Vlaskin M.S. // J. Supercrit. Fluids. 2013. V. 73. P. 10.
Brown G.M., Noe-Spirlet M.R., Busing W.R., Levy H.A. // Acta Crystallogr. Sect. B: Struct. Sci. 1977. V. 33. P. 1038.
Hernández-Cortez J.G., Manríques M., Lartundo-Rojas L., López-Salinas E. // Catal. Today. 2014. V. 220–222. P. 32.
Rao P.M., Wolfson A., Kababya S., Vega S., Landau M.V. // J. Catal. 2005. V. 232. P. 210.
de Mattos F.C.G., de Carvalho E.N.C.B., de Freitas E.F., Paiva M.F., Ghesti G.F., de Macedo J.L., Dias S.C.L., Dias J.A. // J. Braz. Chem. Soc. 2017. V. 28 P. 336.
Jin H., Yi X., Sun X., Qiu B., Fang W., Weng W., Wan H. // Fuel. 2010. V. 89. P. 1953.
Shen H., Li Y., Huang S., Cai K., Cheng Z., Lv J., Ma X. // Catal. Today. 2019. V. 330. P. 117.
Atia H., Armbruster U., Martin A. // J. Catal. 2008. V. 258. P. 71.
Li C., Chen Y.W. // Thermochim. Acta. 1995. V. 256. P. 457.
Chary K.V.R., Ramana Rao P.V., Venkat Rao V. // Catal. Commun. 2008. V. 9. P. 886.
Małecka B., Łącz A., Drożdż E., Małecki A. // J. Therm. Anal. Calorim. 2015. V. 119. P. 1053.
Elmasry M.A.A., Gaber A., Khater E.M.H. // J. Therm. Anal. Calorim. 1998. V. 52. P. 489.
Ho S.C., Chou T.C. // Ind. Eng. Chem. Res. 1995. V. 34. P. 2279.
Scheffer B., Molhoek P., Moulijn J.A. // Appl. Catal. 1989. V. 46. P. 11.
Bartholomew C.H., Farrauto R.J. // J. Catal. 1976. V. 45 P. 41.
Weigel D., Imelik B., Laffitte P. // Bull. Soc. Chim. Fr. 1962. P. 345.
Shimoda N., Koide N., Kasahara M., Mukoyama T., Satokawa S. // Fuel. 2018. V. 232. P. 485.
Molina R., Poncelet G. // J. Catal. 1998. V. 173. P. 257.
Lamber R., Schulz-Ekloff G. // Surf. Sci. 1991. V. 258. P. 107.
Rynkowski J.M., Paryjczak T., Lenik M. // Appl. Catal. A: Gen. 1993. V. 106. P. 73.
Chen D., Christensen K.O., Ochoa-Fernández E., Yu Z., Tøtdal, B., Latorre, N., Monzón, A., Holmen A. // J. Catal. 2005. V. 229. P. 82.
Cao Y., Wang J., Li Q., Yin N., Liu Z., Kang M., Zhu Y. // J. Fuel Chem. Technol. 2013. V. 41. P. 943.
Палчева Р., Спожакина А., Тюлиев Г., Иратова К., Петров Л. // Кинетика и Катализ. 2007. Т. 48. № 6. С. 905.
Palcheva R., Dimitrov L., Tyuliev G., Spojakina A., Jiratova K. // Appl. Surf. Sci. 2013. V. 265. P. 309.
Mangnus P.J., Bos A., Moulijn J.A. // J. Catal. 1994. V. 146. P. 437.
Southmayd D.W., Contescu C., Schwarz J.A. // J. Chem. Soc. Faraday Trans. 1993. V. 89. P. 2075.
Yan Y., Dai Y., He H., Yu Y., Yang Y. // Appl. Catal. B: Environ. 2016. V. 196. P. 108.
Golubina E.V., Peristyy A.A., Lokteva E.S., Maslakov K.I., Egorov A.V. // React. Kinet. Mech. Cat. 2020.
Velon A., Olefjord I. // Oxid. Met. 2001. V. 56. P. 415.
Legrand D.L., Nesbitt H.W., Bancroft G.M. // Am. Mineral. 1998. V. 83. P. 1256.
Mansour A.N. // Surf. Sci. Spectra. 1994. V. 3. P. 211.
Salagre P., Fierro J.L.G., Medina F., Sueiras J.E. // J. Mol. Catal. A: Chem. 1996. V. 106. P. 125.
Ng K.T., Hercules D.M. // J. Phys. Chem. 1976. V. 80. P. 2094.
Shpak A.P., Korduban A.M., Medvedskij M.M., Kandyba V.O. // J. Electron Spectrosc. 2007. V. 156. P. 172.
Jalil P.A., Faiz M., Tabet N., Hamdan N.M., Hussain Z. // J. Catal. 2003. V. 217. P. 292.
Winoto H.P., Fikri Z.A., Ha J.M., Park Y.K., Lee H., Suh D.J., Jae J. // Appl. Catal. B: Environ. 2019. V. 241. P. 588.
Watmanee S., Suriye K., Praserthdam P., Panpranot J. // Top. Catal. 2018. V. 61. P. 1615.
Garbarino G., Campodonico S., Perez A.R., Carnasciali M.M., Riani P., Finocchio E., Busca G. // Appl. Catal. A: Gen. 2013. V. 452. P. 163.
Давыдов A.A. ИК-спектроскопия в химии поверхности окислов. Новосибирск: Наука, 1984. 245 с.
Zarfl J., Ferri D., Schildhauer T.J., Wambach J., Wokaun A. // Appl. Catal. A: Gen. 2015. V. 495. P. 104.
Mihaylov M., Lagunov O., Ivanova E., Hadjiivanov K. // Top. Catal. 2011. V. 54 P. 308.
Peri J.B. // J. Catal. 1984. V. 86. P. 84.
Zaki M.I. // Stud. Surf. Sci. Catal. 1995. V. 100. P. 569.
Liu Y., Sheng W., Hou Z., Zhang Y. // RSC Adv. 2018. V. 8. P. 2123.
Hadjiivanov K.I., Vayssilov G.N. / Characterization of oxide surfaces and zeolites by carbon monoxide as an IR probe molecule. In: Advances in Catalysis, Academic Press, 2002. P. 307.
Morterra C., Magnacca G. // Catal. Today. 1996. V. 27. P. 497.
Morterra C., Bolis V., Magnacca G. // Langmuir. 1994. V. 10. P. 1812.
Li H., Xu Y., Gao C., Zhao Y. // Catal. Today. 2010. V. 158. P. 475.
Kang H., Jeong Y.-K., Oh S.-T. // Int. J. Refract. Met. H. 2019. V. 80. P. 69.
Sarkar A., Seth D., Jiang M., Ng F.T.T., Rempel G.L. // Top. Catal. 2014. V. 57. P. 730.
Cao Y., Wang J., Kang M., Zhu Y. // J. Mol. Catal. A: Chem. 2014. V. 381. P. 46.
Keane M.A., Park C., Menini C. // Catal. Lett. 2003. V. 88. P. 89.
Park C., Menini C., Valverde J.L., Keane M.A. // J. Catal. 2002. V. 211. P. 451.
Choi Y.H., Lee W.Y. // Catal. Lett. 2000. V. 67. P. 155.
Gao Z., Zhang S., Ma H., Li Z. // J. Rare Earth. 2017. V. 35. P. 977.
Mogica-Betancourt J.C., López-Benítez A., Montiel-López J.R., Massin L., Aouine M., Vrinat M., Berhault G., Guevara-Lara A. // J. Catal. 2014. V. 313. P. 9.
Дополнительные материалы отсутствуют.
Инструменты
Кинетика и катализ