

Кинетика и катализ, 2021, T. 62, № 1, стр. 44-54
Активность катализаторов 5%СuО/Ce1 – xPrxOy в реакции окисления СО кислородом в избытке водорода
А. Н. Ильичев a, *, М. Я. Быховский a, З. Т. Фаттахова a, Д. П. Шашкин a, В. Н. Корчак a
a ФГБУН Институт химической физики им. Н.Н. Семенова РАН
119991 Москва, ул. Косыгина, 4, Россия
* E-mail: Ilichev-alix@yandex.ru
Поступила в редакцию 19.11.2019
После доработки 19.08.2020
Принята к публикации 29.08.2020
Аннотация
Катализаторы 5%CuO/Ce1 –xPrxOy синтезированы на основе окcидов СеО2, PrO2 и твердых растворов Ce1 – xPrxOy с х = 0.2, 0.5 и 0.8. Высокодисперсный оксид меди содержится в катализаторах 5%CuO/Ce1 –xPrxOy. При взаимодействии с носителями он образует активный кислород, который участвует в хемосорбции СО и низкотемпературной реакции окисления СО в присутствии водорода. Наибольшая величина конверсии СО в избытке Н2 (γmах(Т)), близкая к 100%, получена при температурах 120–160°С на катализаторе 5%CuO/CeO2. При модифицировании СеО2 катионами Pr, образец 5%Ce0.2Pr0.8Oy, она понижается до 65% при 220°С из-за увеличения прочности связи кислорода в медьсодержащих центрах. На образце 5%CuO/PrО2 максимальная конверсия СО (93%) зафиксирована при 200°С. При модифицировании PrO2 катионами Се активность катализаторов 5%CuO/Ce0.5Pr0.5Oy и 5%CuO/Ce0.2Pr0.8Oy не превышает таковую для 5%CuO/PrО2. С помощью метода ТПД изучены формы адсорбции СО и СО2 на образцах 5%CuO/Ce1 –xPrxOy. В области 170–500°С наблюдается десорбция кислорода носителей образцов 5%CuO/Ce0.5Pr0.5Oy и 5%CuO/PrOy. Обсуждаются особенности протекания реакции на катализаторах 5%CuO/Ce1 –xPrxOy. С учетом свойств комплексов СО, образующихся на медьсодержащих центрах окисления и адсорбции, рассмотрено их участие в реакции низкотемпературного окисления в водороде.
ВВЕДЕНИЕ
Топливные элементы превращают энергию химической реакции окисления водорода кислородом воздуха в электрическую и не загрязняют окружающую среду. В перспективе они могут найти широкое применение в автомобилестроении и теплоэнергетике. В промышленности водород производят из органического сырья. Он содержит около 2% оксида углерода, который понижает эффективность работы топливных элементов. Высокий коэффициент полезного действия генерации электроэнергии достигается в топливных элементах с платиновыми электродами при концентрации оксида углерода ниже 10 м. д., а для биметаллических систем PtRu – ниже 100 м. д. [1]. Водород может быть очищен от СО адсорбционным методом, с помощь мембран, а также в результате реакции СО + 3Н2 → СН4 + Н2О и селективного окисления 2СО + О2 → СО2 в избытке водорода. Селективное окисление, протекающее при низких температурах – наиболее перспективный метод удаления из водорода следов СО [2]. Выделяют три группы катализаторов для этой реакции: 1) системы с нанесенными благородными металлами, такими как Pt, Pd, Ir, Ru и Rh; 2) нанокатализаторы, содержащие Au; 3) оксидные системы с переходными металлами – Cо, Cr, Cu, Ni, Zn, нанесенными на оксиды с различными кислотными и оснóвными свойствами – MgO, La2O3, SiO2–Al2O3, CeO2, CeZrO2. Интерес к третьей группе обусловлен низкой стоимостью катализаторов по сравнению с таковой для первой и второй групп. Среди оксидных систем наибольшее внимание привлекает CuO–CeO2, не уступающий по активности катализаторам из 1 и 2 групп. Уникальность оксидной системы CuO–CeO2 связывают с ее способностью запасать кислород, быстрым восстановлением в парах Се4+–Се3+ и синергическим Сu–Се-взаимодействием. Эти свойства отвечают за высокую активность кислорода катализатора [3], хемосорбцию и окисление СО кислородом на катионах Сu+ при низких температурах [4, 5]. Однако все еще не решена проблема, связанная с относительно узким температурным окном для полной конверсии СО на CuO–CeO2.
Недавно в [6–10] было показано, что температурный интервал окисления СО в реакциях СО + O2 на M–СеО2 и СО + О2 + Н2 на CuО/M–CeO2 может быть расширен в низкотемпературную область при модифицировании оксида церия переходными и редкоземельными металлами (М). При изучении свойств твердых растворов, содержащих переходные металлы, в этих реакциях обнаружено, что наилучшими модификаторами являются оксиды Mn и Fe, которые повышают активность катализаторов в результате увеличения подвижности кислорода в образующихся кислородных вакансиях в CeO2 [6, 7, 10]. Другое поведение демонстрируют твердые растворы, в состав которых входят редкоземельные металлы. Среди них празеодим в Рr–CeO2 и Pr0.1Zr0.18Ce0.72O2 обеспечивает наиболее высокую активность катализаторов в реакции СО + О2 также за счет увеличения подвижности кислорода [8, 9]. Однако в процессе селективного окисления СО кислородом в присутствии водорода активность катализаторов CuO/хPrCeO2 (х = 5, 10 и 15) понижается при возрастании содержания Pr по сравнению с таковой для образца CuO/CeO2 [11]. Учитывая данные работ [3, 7], интерес представляют разбавленные твердые растворы с содержанием ионов празеодима х ≤ 1, при котором ожидаемо образование в CeO2 вакансий с активным кислородом, участвующим в низкотемпературном окислении СО.
Для проверки этого предположения в настоящей работе изучено влияние катионов празеодима в структуре носителя оксида церия на каталитические свойства медноцериевых катализаторов 5%CuO/CeO1 –хPrхОу с х = 0, 0.2, 0.5, 0.8 и 1.0 в реакции окисления СО кислородом в избытке водорода. Об активности образцов в реакции судили по зависимостям конверсии СО в СО2 от температуры при сопоставлении их с таковой для образца 5%СuO/CeO2, ранее показавшим наилучшие свойства в окислении CO кислородом катализатора при 20°С и в селективном окислении СО в ряду исследованных образцов (0.25–10)%СuO/CeO2 [5, 12].
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Носители CeO1 –хPrхОу с х = 0, 0.2, 0.5, 0.8 и 1 получали из гидроксидов, которые осаждали при добавлении растворов солей Ce(NO3)3, Pr(NO3)3 и их смесей в соответствующих соотношениях в раствор аммиака при рН 10. Осадки промывали в дистиллированной воде и высушивали при 150°С на воздухе. Далее гидроксиды разлагали при нагревании со скоростью 10°С/мин от 150 до 500°С и выдерживали при температуре 500°С в течение 30 мин. После термообработки носители пропитывали при 20°С раствором азотнокислой меди Сu(NO3)2 ⋅ 3H2O в такой концентрации, чтобы содержание СuO в готовом продукте составляло 5 мас. %. Затем образцы сушили, прогревали на воздухе, повышая температуру до 500°С со скоростью 10°С/мин, и выдерживали при 500°С в течение 1 ч.
Удельную поверхность образцов находили методом БЭТ по низкотемпературной адсорбции аргона. Дифрактограммы записывали на приборе ДРОН-3М (“Буревестник”, Россия) в диапазоне углов 2θ от 8° до 90°. Фазовый состав и параметры решетки определяли, сопоставляя полученные дифрактограммы с данными из международной картотеки JCPDS [13]. Средний размер кристаллитов оценивали по формуле Дебая–Шерера для линии с максимальной интенсивностью. По линиям дифрактограммы образца вычисляли параметры элементарной ячейки и рассчитывали их среднюю величину [14].
Реакцию окисления СО кислородом в избытке водорода проводили на установке проточного типа. Образец (смесь, содержащую 50 мг порошка катализатора и 70 мг кварца фракции 0.10−0.25 мм) помещали в кварцевый реактор (трубка с внутренним диаметром 3 мм) и прокаливали в токе кислорода при 500°С в течение 20 мин. Затем реактор охлаждали до 40°С и заменяли поток кислорода на реакционную смесь с объемным составом 98% Н2, 1% СО и 1% О2. Смесь подавали в реактор со скоростью 20 мл/мин. Активность катализатора оценивали по конверсии СО в СО2 при разных температурах. Температуру повышали ступенчато с шагом 20°С. Продукты реакции разделяли на колонках с молекулярными ситами 13Х и силикагелем и регистрировали с помощью детектора по теплопроводности на хроматографе Кристалл 2000 М (“Хроматэк”, Россия). Конверсии СО (γ, %) и О2 (β, %) определяли из отношений
Взаимодействие образцов с водородом изучили методом ТПВ-Н2 в потоке смеси 6% Н2 с Ar (30 мл/мин) при нагревании образца со скоростью 5°С/мин в области от 30 до 700°С. Навеску образца 100 мг помещали в U-образный реактор, прокаливали в потоке кислорода при 500°С в течение 10 мин, охлаждали до 30°С, заменяли поток кислорода на водородную смесь и регистрировали профиль ТПВ-Н2, используя детектор по теплопроводности. Количество поглощенного водорода определяли по площади пика ТПВ, сопоставляя ее с соответствующей величиной, полученной для стандарта (NiO). Образующуюся при восстановлении оксидов воду удаляли с помощь поглотителя Mg(Cl4)2. Ловушку с поглотителем устанавливали между реактором и детектором. Перед опытом поглотитель обезвоживали в вакууме при Р = 0.1 Па и Т = 150°С в течение 1 ч.
Адсорбцию и окисление СО исследовали методом ТПД СО в вакууме. Навеску образца 100 мг предварительно вакуумировали при 20°С, прогревали при температуре 500°С и остаточном давлении 10–4 Па в течение 1 ч, после чего напускали в реактор кислород до давления Р = 3 × 102 Па и выдерживали в течение 20 мин, а затем охлаждали до комнатной температуры и вакуумировали. Полученный катализатор далее называется окисленным. После этого на нем при 20°С в течение 10 мин адсорбировали СО при Р = 3.3 × 103 Па, откачивали газ в течение 20 мин и записывали профили ТПД СО в условиях постоянного вакуумирования при скорости нагрева образца 10°С/мин. Изменение давления в профиле ТПД отражает зависимость скорости десорбции (w) от температуры (Т). Давление регистрировали манометром Пирани с автоматической записью показаний [15]. Чтобы разделить на кривой ТПД пики десорбции СО и продукта окисления СО2, диоксид углерода вымораживали в U-образной ловушке, размещенной между образцом и манометром и охлаждаемой жидким азотом. Такая методика позволяла регистрировать профили совместной десорбции СО + + СО2 и, отдельно, десорбции СО, а вычитая второй профиль из первого, получать кривую десорбции СО2. Количество десорбированного СО2$\left( {N_{{{\text{С}}{{{\text{О}}}_{2}}}}^{{{\text{дес}}}}} \right)$ определяли по давлению СО2 после его размораживания, а количество десорбированного СО $\left( {N_{{{\text{СО}}}}^{{{\text{дес}}}}} \right)$ рассчитывали с помощью уравнения баланса: NдесСО = $N_{{{\text{СО + С}}{{{\text{О}}}_{2}}}}^{{{\text{дес}}}}$ – $N_{{{\text{С}}{{{\text{О}}}_{2}}}}^{{{\text{дес}}}}.$ При проведении расчета исходили из общего количества десорбированного газа $\left( {N_{{{\text{СО + С}}{{{\text{О}}}_{2}}}}^{{{\text{дес}}}}} \right),$ найденного в отдельном ТПД-опыте в замкнутом реакторе без вакуумирования. Точность определения $N_{{{\text{СО + С}}{{{\text{О}}}_{2}}}}^{{{\text{дес}}}}$ и $N_{{{\text{СО}}}}^{{{\text{адс}}}}$ соответствовала точности измерения давления газа и составляла 20%. Продукты десорбции также анализировали на масс-спектрометре МХ-7303 (СССР) методом отбора пробы через капилляр; СО, СО2 и О2 регистрировали по ионным пикам масс-спектров 28, 44 и 32 а.е. м. соответственно.
Для проведения адсорбционных исследований газы СО и О2 получали в вакуумных условиях по методикам, описанным в руководстве [16]. Для устранения неконтролируемых примесей газы СO и О2 вводили на образец через ловушку, охлаждаемую жидким азотом.
ПОЛУЧЕННЫЕ РЕЗУЛЬТАТЫ
Характеристики образцов
На рис. 1 представлены дифрактограммы синтезированных образцов Ce1 –xPrxOу c х = 0–1. Из дифрактограмм 1−5 видно, что образцы имеют кубическую модификацию (К) [13, 17]. При повышении содержания Pr в образцах рефлексы смещаются в область меньших значений углов 2θ из-за увеличения параметра решетки а (Å) элементарной ячейки куба. На рис. 2 приведена зависимость а от х. Величина а изменяется от 5.41 Å для кубической структуры CeO2 до 5.47 Å для кубической структуры Pr6O11. Рост значения а с повышением х свидетельствует об образовании твердых растворов, в которых присутствуют катионы Pr3+ и Pr4+.
Рис. 1.
Дифрактограммы образцов СeO2 (1), Сe0.8Pr0.2Oу (2), Сe0.5Pr0.5Oу (3), Сe0.2Pr0.8Oу (4) и PrOу (5).
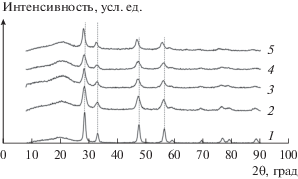
Рис. 2.
Изменение параметра решетки a (Å) от содержания празеодима x в образцах Ce1 – xPrxOу и 5%CuO/Ce1 – xPrxOу.
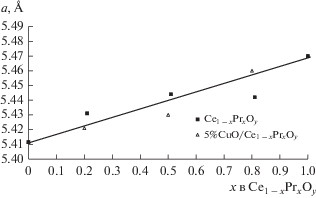
Из табл. 1 следует, что c возрастанием содержания Pr в Ce1 –xPrxOу от нуля до х = 0.8 удельная поверхность образцов (Sуд) сокращается от 120 до 13 м2/г, размер кристаллитов (L) увеличивается от 5 до 15 нм. Образец PrOу имеет поверхность 40 м2/г и L = 10 нм. Нанесение оксида меди уменьшает удельную поверхность твердых растворов на 5−20% и не влияет на размер кристаллитов. Дифрактограммы образцов 5%CuO/Ce1 –xPrxOу аналогичны таковым для носителей Ce1 –xPrxOу. Отсутствие в них рефлексов оксида CuO свидетельствует о его высокой дисперсности. Взаимодействие оксида меди с носителем не меняет зависимость параметров элементарной ячейки носителей от величины х (рис. 2).
Таблица 1.
Характеристики образцов Сe1 –xPrxOy и 5%СuO/Сe1 –xPrxOy
| Образец | Sуд, м2/г | Размер кристаллитов, нм | ${{N}_{{{{{\text{H}}}_{2}}}}}$ × 104 , моль/м2 | Np × 104, моль/м2 | ${{{{N}_{{{{{\text{H}}}_{2}}}}}} \mathord{\left/ {\vphantom {{{{N}_{{{{{\text{H}}}_{2}}}}}} {{{N}_{{\text{p}}}}}}} \right. \kern-0em} {{{N}_{{\text{p}}}}}}$ |
|---|---|---|---|---|---|
| CeO2 | 120 | 6 | 0.1 | − | − |
| Ce0.8Pr0.2Oу | 95 | 8 | 0.13 | − | − |
| Ce0.5Pr0.5Oу | 53 | 9 | 0.3 | − | − |
| Ce0.2Pr0.8Oу | 13 | 15 | 1.7 | − | − |
| PrOу | 40 | 10 | 0.42 | − | − |
| CuO/CeO2 | 103 | 6 | 0.18 | 0.06 | 3 |
| CuO/Ce0.8Pr0.2Oу | 86 | 8 | 0.19 | 0.07 | 2.7 |
| CuO/Ce0.5Pr0.5Oу | 56 | 9 | 0.4 | 0.1 | 4 |
| CuO/Ce0.2Pr0.8Oу | 9.4 | 15 | 2.9 | 0.65 | 3 |
Влияние содержания Pr на свойства кислорода в Ce1 –xPrxOу и 5%CuO/Ce1 –xPrxOу было исследовано методом ТПВ-Н2.
Термопрограммированное восстановление водородом образцов Ce1 – xPrxOу и 5%CuO/Ce1 – xPrxOу
На рис. 3 приведены профили ТПВ-Н2 предварительно окисленных образцов Ce1 –xPrxOу. Для оксида церия характерен широкий профиль поглощения водорода в области 300−600°С с максимумами при Тmах = 325, 400 и 530°С (профиль 1). Природа низкотемпературного пика при 325°С неясна. Вероятно, его следует отнести к поглощению водорода на дефектных структурах. Поглощение Н2 при 400°С связывают с восстановлением поверхности, а пик при 530°С соответствует образованию нестехиометрического оксида [18]. Суммарное количество поглощенного водорода на квадратном метре оксида $\left( {{{N}_{{{{{\text{H}}}_{2}}}}}} \right)$ определяли как отношение количества поглощенного водорода одним граммом катализатора к величине его удельной поверхности. Как видно из табл. 1, для СеО2 оно составляет 0.1 × 10–4 моль/м2. Так как окисление водорода кислородом из объема оксида СеО2 протекает при Т > 700°C, то эту величину следует отнести к окислению водорода кислородом поверхности [18, 19]. В профилях смешанных оксидов Ce0.8Pr0.2Oу, Ce0.5Pr0.5Oу и Ce0.2Pr0.8Oу, как и в профиле 5 оксида PrOу, пики поглощения Н2 также расположены в интервале от 300 до 600°С. Профиль 2 образца Ce0.8Pr0.2Oу содержит три пика при 353, 443 и 512°С. В профиле 3 образца Ce0.5Pr0.5Oу интенсивность этих пиков увеличивается. Основной пик имеет максимум при 432°С, а два дополнительных плохо разрешенных пика − в области температур 385 и 460°С. В профиле 4 образца Ce0.2Pr0.8Oу наблюдаются разделенные пики при 389 и 460°С и интенсивный пик при 503°С. Пики при 389 и 460°С могут соответствовать восстановлению частиц фазы оксида празеодима (рис. 3, профиль 5), которые, вероятно, присутствуют в образце вместе с частицами твердого раствора. С уменьшением содержания празеодима в катализаторах их интенсивность понижается. Частицы твердого раствора в образцах Ce0.8Pr0.2Oу и Ce0.5Pr0.5Oу восстанавливаются водородом при 430−440°С, а в случае образца Ce0.2Pr0.8Oу – при 503°С. Повышение температуры восстановления до 503°С свидетельствует об увеличение прочности связи кислорода в Ce0.2Pr0.8Oу, которое может быть связано с возрастанием размера кристаллитов (табл. 1).
Рис. 3.
Профили ТПВ-Н2 для образцов СeO2 (1), Сe0.8Pr0.2Oу (2), Сe0.5Pr0.5Oу (3), Сe0.2Pr0.8Oу (4) и PrOу (5).
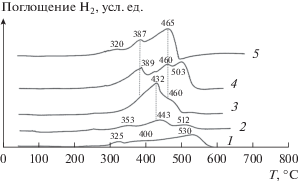
Поглощение водорода твердыми растворами (0.3 × 10–4−1.7 × 10–4 моль/м2) и оксидом PrOу (0.42 × 10–4 моль/м2) в разы превышает количество водорода, окисленного кислородом поверхности оксида СеО2 (0.1 × 10–4 моль/м2) (табл. 1). Такое различие позволяет полагать, что на твердых растворах и оксиде PrOу окисление водорода протекает, вероятно, с участием кислорода приповерхностного слоя.
Профили ТПВ-Н2 для 5%CuO/Ce1 –xPrxOу на рис. 4 отличаются от соответствующих профилей носителей Ce1 –xPrxOу по форме и расположению в температурных областях. В табл. 1 приведены экспериментально определенные количества поглощенного водорода ${{N}_{{{{{\text{H}}}_{2}}}}}$ катализаторами 5%CuO/Ce1 –xPrxOу и рассчитанные количества водорода Nр, необходимого для восстановления оксида меди в них по реакции CuО + + Н2 = Cu0 + Н2О в предположении, что все катионы меди в окисленных образцах находятся в состоянии Cu2+ согласно [5]. Из отношения ${{{{N}_{{{{{\text{H}}}_{2}}}}}} \mathord{\left/ {\vphantom {{{{N}_{{{{{\text{H}}}_{2}}}}}} {{{N}_{{\text{р}}}}}}} \right. \kern-0em} {{{N}_{{\text{р}}}}}}$ видно, что количества поглощенного водорода образцами с х = 0–1 больше того, что необходимо для восстановления в них оксида меди, в 2.7−5 раз. Избыточное поглощение Н2 указывает на восстановление оксида меди вместе с носителями.
Рис. 4.
Профили ТПВ-Н2 для образцов 5%CuO/СeO2 (1), 5%CuO/Сe0.8Pr0.2Oу (2), 5%CuO/Сe0.5Pr0.5Oу (3), 5%CuO/Сe0.2Pr0.8Oу (4), 5%CuO/PrOу (5) и CuO (6).
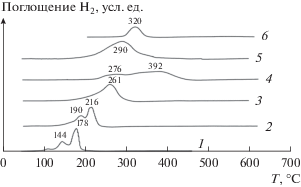
В профиле образца 5%CuO/СеO2 имеется два пика при Тmах = 144 и 178°С, что ниже температуры восстановления частиц моноклинной фазы СuO(М), составляющей 320°С (рис. 4, профили 1 и 6). Эти пики относят к восстановлению высокодисперсного оксида CuO, сильно взаимодействующего с носителем [20–25]. Авторы [20] полагают, что при 140°С восстанавливается только CuО, а при 180°С − CuO вместе с CеO2. Понижение температуры восстановления взаимодействующих оксидов СuO и CeO2 по сравнению с температурой восстановления их фаз (Тmах > 300) рассматривается как синергический эффект. Низкотемпературное восстановление носителя, неактивного в адсорбции Н2 при 180°C, протекает легче атомами водорода при их спилловере с меди, которая образуется при 140°С [21]. В профиле 2 образца 5%CuO/Ce0.8Pr0.2Oу пики поглощения водорода смещены в область бóльших температур относительно пиков в профиле 1. Две формы оксида меди восстанавливаются в водороде при Тmах = = 190 и 216°С. Согласно [9, 26] при 190°С восстанавливается высокодисперсный оксид CuO, сильно взаимодействующий с носителем, а при 216°С − крупные частицы оксида меди, у которых связь с носителем слабее, чем у высокодисперсного CuO. Для образца 5%CuO/Ce0.5Pr0.5Oу наблюдается один пик поглощения водорода при 260°С (профиль 3). На кривой ТПВ-Н2 катализатора 5%CuO/Ce0.8Pr0.2Oу можно выделить два широких пика при 276 и 382°С (профиль 4), а в профиле 5 образца 5%CuO/PrOy присутствует один пик при 290°С. Поглощение водорода в области температур 260−290°С связано с восстановлением небольших частиц фазы CuO на поверхности катализаторов, размер кристаллитов которых меньше, чем у частиц моноклинной фазы СuO(М) с L = 30 нм, восстанавливающихся при 320°С. Форма оксида меди, которая восстанавливается при 380°С, неясна. Отметим, что в этой температурной области может восстанавливаться носитель (рис. 3) и фаза оксида Cu2O [23].
Представленные данные показывают, что в области 300−600°С водород окисляется кислородом поверхности СеО2 и, вероятно, приповерхностного слоя образцов Ce1 –xPrxOу с х = 0.2–1. В образцах 5%CuO/Ce1 –xPrxOу наблюдается синергический эффект – взаимодействующие оксид меди и носители легче восстанавливаются водородом (100−290°С), чем их отдельные фазы (300−600°С). С возрастанием в смешанных оксидах содержания катионов Pr повышается температура совместного восстановления оксида меди и носителей вследствие увеличения прочности связи кислорода катализатора.
Окисление СО кислородом в избытке Н2 на 5%CuO/Ce1 – xPrxOу
На рис. 5 приведены зависимости конверсии СО в СО2 (γ) от температуры (Т) для катализаторов 5%CuO/Ce1 –xPrxOу (x = 0.2–0.8). Непосредственно для этих катализаторов дан ряд активности в водородсодержащей смеси СО + О2 + Н2. Видно, что для 5%CuO/CeO2 конверсия СО увеличивается до максимального значения γmах = 98–100% при изменении температуры от 40 до 160°С (кривая 1). При дальнейшем повышении Т конверсия уменьшается и при 220°С становится равной 70%. Подобные зависимости γ(Т) наблюдаются для других образцов (кривые 2−4). Из сравнения этих данных видно, что с возрастанием доли Pr в катализаторах от 0.2 до 0.8 температура реакции повышается с одновременным понижением конверсии γmах. Зависимость γmах(Т) от содержания Pr в катализаторах позволяет получить для них ряд активности: 5%CuO/CeO2 (~100%, 100−160°С) > > 5%CuO/Ce0.8Pr0.2Oу (98%, 160°C) > 5%Ce0.5Pr0.5Oу (91%, 180°С) > 5%CuO/Ce0.2Pr0.8Oу (65%, 220°C).
Рис. 5.
Зависимость конверсии СО в СО2 от температуры окисления СО кислородом в избытке водорода на образцах 5%CuO/СeO2 (1), 5%CuO/Сe0.8Pr0.2Oу (2), 5%CuO/Сe0.5Pr0.5Oу (3), 5%CuO/Сe0.2Pr0.8Oу (4), СeO2 (5), CuO (6) и PrOу (7).
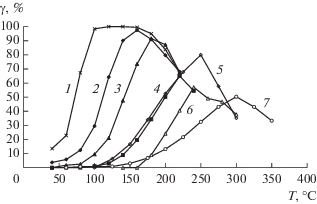
Для чистых оксидов зависимость γmах(Т) на рис. 5 дает следующий ряд активности: CeO2 (80%, 250°C) > CuO (57%, 240°С) > PrOу (50%, 300°С).
Высокая активность оксидных систем 5%CuO/ Ce1 –xPrxOу при низких температурах по сравнению с таковой для чистых оксидов свидетельствует о протекании реакции на оксиде меди, взаимодействующем с носителем.
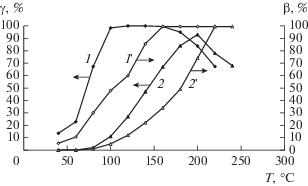
Одновременно с конверсией СО измеряли конверсию О2 (β). Зависимости γ(Т) и β(Т) для образцов 5%CuO/CeO2 и 5%CuO/PrOу приведены на рис. 6. Так, на образце 5%CuO/CeO2 реакция окисления СО начинается при 40°С. При повышении температуры до 110°С она протекает со 100% селективностью, так как при этих условиях отношение количеств расходуемых молекул СО и О2 соответствует их отношению, полученному из уравнения реакции СО + 0.5О2 → СО2 (кривые 1 и 1 '). В интервале 100−120°С поглощение кислорода замедляется с уменьшением содержания СО в потоке. При 110°С конверсия О2 составляет ~50% при конверсии СО близкой к 100%. С повышением температуры до 160°С конверсия О2 увеличивается из-за участия кислорода в окислении водорода. В соответствии с данными ТПВ-Н2 (рис. 4) в этой температурной области начинается поглощение водорода катализатором. Конверсия СО не изменяется и остается равной ~100%. При Т ≥ ≥ 160°С кислород из смеси полностью расходуется в двух реакциях. В условиях конкуренции за кислород конверсия СО понижается до 70% при 220°С из-за возрастающей скорости расходования кислорода в реакции с водородом $\left( {{{V}_{{{{{\text{H}}}_{2}}}}}} \right)$ относительно скорости его расходования в реакции с оксидом углерода (VCO). В случае образца 5%CuO/PrOу окисление СО начинается при 100°С. В интервале 100−160°C кислород расходуется только на окисление СО, а при Т > 160°C он участвует и в окислении Н2. Из-за протекания конкурирующих реакций, когда ${{V}_{{{{{\text{H}}}_{2}}}}}$ > VCO, конверсия СО достигает максимального значения 93% при 200°С. В этом случае в интервале температур 180−240°С селективность образования СО2 ниже 100%. Следует отметить, что в реакции окисления СО катализатор 5%CuO/PrOy (93%, 200°C) с высокой удельной поверхностью (Sуд= 37м2/г) может быть активнее, чем 5%CuO/Ce0.2Pr0.8Oу (65%, 220°C) с Sуд= 9.4 м2/г, из-за большего количества в нем активных центров оксида меди. Кроме того, согласно данным ТПВ-Н2 в образце 5%CuO/Ce0.2Pr0.8Oy кислород более прочно связан в медьсодержащих центрах.
Рис. 6.
Зависимости конверсии СО и О2 от температуры для образцов 5%CuO/СeO2 (1, 1') и 5%CuO/PrOy (2, 2').
Таким образом, активность оксидных систем 5%CuO/Ce1 –xPrxOу в реакции окисления СО кислородом в избытке водорода при низких температурах связана с оксидом меди, взаимодействующим с носителем. При увеличении содержания катионов Pr в CeO2 активность катализаторов падает: повышается температура начала реакции, уменьшается конверсия γmах(Т) со смещением ее в высокотемпературную область. Понижение γmах(Т) обусловлено конкуренцией за кислород в реакциях окисления СО и Н2.
Для образцов 5%CuO/Ce1 –xPrxOу разный вид зависимости конверсии СО от температуры на начальной стадии окисления СО может быть обусловлен влиянием температуры на превращения поверхностных интермедиатов, образующихся при адсорбции СО. С этой целью была исследована термостабильность адсорбционных комплексов на катализаторах методом ТПД СО.
Окисление СО кислородом катализаторов 5%CuO/Ce1 – xPrxOу
На рис. 7 приведены профили ТПД СО для окисленных образцов 5%CuO/Ce1 –хPrхOу после адсорбции на них СО при Р = 3.3 × 103 Па в течение 10 мин и вакуумирования в течение 20 мин при 20°С. В продуктах десорбции присутствуют СО и СО2. Они образуются при разложении адсорбционных комплексов, связанных с медьсодержащими центрами, так как после адсорбции СО на носителях Ce1 –хPrхOу количества десорбированного СО + СО2 (Nдес ≤ 1 × 1018 г–1) в температурной области 30−500°С значительно меньше таковых для катализаторов 5%CuO/Ce1 –хPrхOу (3 × 1020–0.7 × 1020 г–1).
Профиль 1 образца 5%CuO/CeO2 содержит пик десорбции СО при Тmах = 110°С и по крайней мере два сложных пика СО2 в области 150 и 350°С. Как было установлено в [5], десорбция СО обусловлена разложением линейных карбонильных комплексов Cu+−CO, а десорбция СО2 сопровождается разложением мостиковых, моно- и бидентатных карбонатных комплексов, которые образуются на центрах адсорбции и окисления, связанных с кластерами оксида меди.
Из сравнения профилей 1−4 на рис. 7 и соответствующих данных в табл. 2 видно, что при увеличении содержания Pr в образцах количество десорбированного СО в области 30−200°С уменьшается от 0.85 × 1018 до 0.04 × 1018 м–2, а количество десорбированного СО2 в области 50−500°С снижается от 2.4 × 1018 до 1.5 × 1018 м–2. Температура максимальной скорости десорбции СО2 (Тmах) повышается от 140 до 260°С. Способность центров хемосорбировать СО падает, так как η − отношение концентрации нанесенных катионов [Cu2+] к концентрациям десорбированных молекул [СО + + СО2] − возрастает от 1.3 для образца 5%CuOCeO2 до 6.3 для катализатора 5%CuO/PrOy (табл. 2).
Рис. 7.
Профили ТПД СО, полученные после адсорбции СО при 20°С на окисленных образцах 5%CuO/СeO2 (1), 5%CuO/Сe0.8Pr0.2Oу (2), 5%CuO/Сe0.5Pr0.5Oу (3) и 5%CuO/PrOу (4).
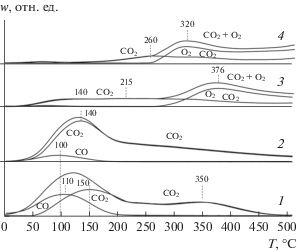
Таблица 2.
Десорбция СО и СО2 по данным ТПД СО образцов 5%CuO/Ce1 –xPrxOy
| Образец | Tmax, °C | [CO] × 10–18, м–2 | Tmax, °C | [CO2] × 10–18, м–2 | [Cu2+] × 10–18, м–2 | η* |
|---|---|---|---|---|---|---|
| 5%CuOCeO2 | 110 | 0.85 | 150 | 1.9 | 3.7 | 1.3 |
| 5%CuO/Ce0.8Pr0.2O2 | 100 | 0.12 | 140 | 2.4 | 4.4 | 1.8 |
| 5%CuO/Ce0.5Pr0.5O2 | 90 | 0.04 | 215 | 1.8 | 6.7 | 3.6 |
| 5%CuO/PrO2 | 74 | 0.08 | 260 | 1.5 | 10 | 6.3 |
В соответствии с [27] можно полагать, что на однотипных катализаторах 5%CuO/Ce1 –хPrхOу реакция окисления СО кислородом медьсодержащих центров протекает через формирование одинаковых поверхностных комплексов. Следовательно, по аналогии с 5%CuO/CeO2, на модифицированных образцах 5%CuO/Ce1 –хPrхOу при адсорбции СО на центрах адсорбции и окисления образуются карбонильные и карбонатные комплексы соответственно, которые разлагаются с десорбцией СО и СО2. Взаимодействие празеодима с центрами изменяет их способность к формированию и разложению адсорбционных комплексов. Для выяснения структуры этих комплексов необходимы дополнительные исследования.
Из рис. 7 следует, что профили 3 и 4 образцов 5%CuO/Ce0.5Pr0.5Oу и 5%CuO/PrOу содержат пики О2 при Тmах = 376 и 320°С. Десорбция кислорода не наблюдается в профилях 1 и 2 катализаторов 5%CuO/CeO2 и 5%CuO/Ce0.8Pr0.2Oу. На рис. 8 сопоставлены кривые десорбции кислорода, полученные в опытах ТПД СО, с кривыми ТПД О2 c окисленных образцов 5%CuO/Ce0.5Pr0.5Oу и 5%CuO/PrOу. Видно, что формы профилей 1, 3 и 2, 4 похожи, но профили 3 и 4 смещены относительно профилей 1 и 2 в высокотемпературную область на 30 и 50°С соответственно при сохранении их формы. Такой сдвиг может происходить из-за увеличения прочности связи кислорода в катализаторах в результате их восстановления в процессе десорбции СО2. Сравнение площадей под кривыми десорбции показывает, что количества десорбированного кислорода в опытах ТПД СО на ~10−20% меньше, чем в экспериментах ТПД О2. Причина таких изменений неясна. Возможно, это связано с участием небольшой части кислорода в окислении СО.
Рис. 8.
Кривые десорбции кислорода, полученные в опытах ТПД СО при вымораживании СО2 после адсорбции СО при 20°С на окисленных образцах 5%CuO/Сe0.5Pr0.5Oу (1), 5%CuO/PrOу (2) и в экспериментах ТПД О2 с окисленных образцов 5%CuO/Сe0.5Pr0.5Oу (3) и 5%CuO/PrOу (4).
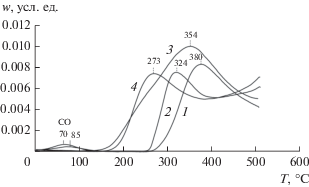
В профилях 3 и 4 на рис. 8 можно выделить перекрывающиеся пики в областях 160−300 и 300−500°С, которые соответствуют по крайней мере двум формам кислорода катализаторов с разной энергией связи. В образцах 5%CuO/PrOу и 5%CuO/Ce0.5Pr0.5Oу присутствует форма кислорода с низкой прочностью связи, которая разлагается при Тmах ≈ 270°С. В катализаторе 5%CuO/Ce0.5Pr0.5Oу вторая форма кислорода с более прочной связью характеризуется температурой разложения ~350°C. Скорость десорбции O2 с катализатора 5%CuO/PrOу в области 350−500°С монотонно возрастает и, вероятно, достигает максимального значения при Тmах ≥ 500°С. В этом случае энергия связи кислорода должна быть выше, чем в образце 5%CuO/Ce0.5Pr0.5Oу.
Профили ТПД О2 для носителей Ce0.5Pr0.5Oу и PrOу не отличаются от соответствующих профилей 3 и 4 катализаторов 5%CuO/Ce0.5Pr0.5Oу и 5%CuO/ PrOу на рис. 8. Следовательно, различие в прочности связи кислорода в образцах 5%CuO/Ce0.5Pr0.5Oу и 5%CuO/PrOу обусловлено Ce−Pr-взаимодействием. Следует также отметить, что Ce−Pr-взаимодействие облегчает окисление образца. Так, при выдерживании в кислороде при 20°С течение 20 мин образцов, предварительно восстановленных в опытах ТПД О2, запас кислорода в Ce0.5Pr0.5Oу восполняется на 70%, а в PrOу − только на 30% .
Таким образом, наблюдаемая десорбция кислорода с образцов 5%CuO/Ce0.5Pr0.5Oу и 5%CuO/PrOу генерируется носителями. Прочность связи кислорода в них зависит от Ce−Pr-взаимодействия, которое также способствует увеличению скорости реокисления кислородом образца 5%CuO/Ce0.5Pr0.5Oу по сравнению с таковой для 5%CuO/PrOу.
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Участие кислорода катализаторов 5%CuO/Ce1 – xPrxOу в реакциях окисления при низких температурах
С увеличением содержания Pr в носителях Ce1 –xPrxOу их удельная поверхность сокращается от 120 до 13 м2/г. Рост количества катионов Pr3+ в кристаллитах изменяет параметр элементарной ячейки куба а от 5.41 до 5.47 Å и способствует образованию анионных вакансий в носителях согласно [9, 28]. Наблюдаемая десорбция кислорода свидетельствует в пользу существования вакансий, так как Се−Pr-взаимодействие катионов понижает прочность связи кислорода в них. С другой стороны, диффузией атомов кислорода по вакансиям из объема к поверхности катализатора объясняется участие кислорода приповерхностного слоя в окислении водорода в ТПВ-Н2 на твердых растворах и оксиде PrOу.
В катализаторах 5%CuO/Ce1 –xPrxOу при сильном взаимодействии кластеров оксида меди с носителями образуется активный кислород, который участвует в окислении водорода при ТПВ-Н2, в хемосорбции СО при 20°С и в реакции окисления СО в смеси СО + О2 + Н2 при температурах ниже температур протекания реакции на отдельных фазах CuO, CeO2 и PrOy. В соответствии с [3, 29] температура гетерогенной реакции окисления зависит от энергии связи кислорода в катализаторе. Таким образом, повышение температуры окисления СО кислородом в смеси СО + О2 + Н2 на начальной стадии реакции с ростом содержания Рr в образцах 5%CuO/Ce1 –xPrxOу свидетельствует о возрастании прочности связи кислорода в катализаторах. Оно может быть связано как с взаимодействием катионов Pr с кислородсодержащими центрами кластеров, так и с увеличением размера кластеров оксида меди при уменьшении поверхности катализаторов, обусловленного повышением в них доли празеодима.
Из данных на рис. 6 видно, что кислород образца 5%CuO/CeO2 с Sуд = 100 м2/г участвует в окислении СО при 40−120°С. Его активность выше таковой для образца 5%CuO/PrOy с Sуд = 37м2/г, ведущего реакцию в области 100−200°С. Изменение активности не связано с изменением величины удельной поверхности образцов, так как ранее в [30] мы установили, что для катализатора 5%CuO/CeO2 с Sуд = 37 м2/г температура начала реакции повышается на 20°С, а температура достижения максимальной конверсии остается такой же, как и для образца 5%CuO/CeO2 с Sуд = = 100 м2/г − примерно 120°С. Это позволяет полагать, что при взаимодействии оксида меди с оксидом церия образуется кислород с менее прочной связью и более высокой активностью, чем в случае взаимодействия оксида меди с оксидом празеодима. Понижение активности образцов 5%CuO/Ce0.8Pr0.2Oy и 5%CuO/Ce0.5Pr0.5Oy с увеличением в них содержания Pr по сравнению с таковой для 5%CuO/CeO2 связано, как видно из рис. 5, с повышением энергии связи кислорода в результате взаимодействия катионов Pr с кислородсодержащими центрами, поскольку величины Sуд модифицированных образцов (86 и 56 м2/г соответственно) находятся в области значений 100−37 м2/г, в которой изменения активности от величины удельной поверхности катализатора 5%CuO/CeO2 невелики. Низкая активность образца 5%CuO/ Се0.2Pr0.8Oy с Sуд = 9.4 м2/г в области 200−220°С по сравнению с таковой для 5%CuO/PrOy, вероятно, обусловлено уменьшением количества кислородсодержащих центров при укрупнении кластеров оксида меди на поверхности образца.
Участие кислорода носителей в окисления СО в смеси СО + О2 + Н2 при низких температурах маловероятно, так как катализаторы 5%CuO/Ce1 –xPrxOу более активны в этой реакции, чем носители Ce1 –xPrxOу. Так, например, активность кислорода кластеров оксида меди в образце 5%CuO/PrO2 проявляется при 200°С. Она выше активности кислорода оксида PrO2, зафиксированной при 300°С, несмотря на то что десорбция кислорода с оксида празеодима наблюдается при Т ≥ 170°С и совпадает с началом реакции окисления СО (сравни рис. 5 и 8). То есть кислород в анионных вакансиях в оксиде PrO2 практически неактивен в окислении СО при ~200°С. Отметим, что разбавление оксида празеодима церием лишь незначительно повышает активность катализатора 5%CuO/Ce0.5Pr0.5Oy в реакции по сравнению с таковой для 5%CuO/PrO2, хотя прочность связи кислорода в носителях Ce0.5Pr0.5Oy и PrO2 различна, и первый реокисляется кислородом легче, чем второй (см. рис. 8). Это также может быть свидетельством в пользу низкой активности кислорода носителя в окислении СО при 200°С.
О механизме реакции окисления СО кислородом в присутствии Н2 на 5%CuO/Ce1 – xPrxOу
В ряду исследованных катализаторов 5%CuO/ Ce1 –xPrxOу в реакции окисления СО в смеси СО + + О2 + Н2 наибольшая конверсия СО при наименьшей температуре получена на образце 5%CuO/ CeO2 (~100%, 110°С). В работе [4] было показано, что активность катализаторов CuO/CeO2 линейно растет с увеличением в них количества катионов Cu+ в медьсодержащих центрах. Это означает, что лимитирующая стадия реакции протекает на катионах Cu+. В соответствии с [5] мы полагаем, что такой стадией может быть разложение мостиковых карбонатных комплексов с десорбцией СО2 в области 20−160°С. Предшествующие ей стадии, связанные с адсорбцией СO на кластерах CuO, такие как восстановление катионов Cu2+ до Сu+ при формировании карбонатных комплексов, формирование карбонильных комплексов Сu+−СО, а также окисления СО карбонильных комплексов до мостиковых карбонатов при адсорбции кислорода, происходящее вместе с окислением катализатора, являются быстрыми, так как протекают при 20°C.
Можно оценить минимальное количество катионов меди в кластерах СuO для образца 5%CuO/ CeO2 по количеству десорбированнного СО, полученного в опыте ТПД СО при разложении карбонильных комплексов. Если полагать, что кластеры оксида меди на поверхности имеют одинаковый химический и количественный состав и на каждой из них образуются один карбонильный комплекс, то количество катионов меди в кластере равно отношению [Cu2+]/[CO]. Так, с использованием данных табл. 2, согласно которым [CO] = 0.85 × 1018 м–2 и [Cu2+] = 3.7 × 1018 м–2, было установлено, что четыре катиона меди могут создать кластер с активным кислородом Сu4Oх. В соответствие с работой [31] в кластерах образцов CuOx/CeO2 катионы меди Cu+ находятся в линейной или треугольной координации с ионами кислорода, в то время как Cu2+ присутствует в тетрагональной и сильно искаженной октаэдрической координации. Отметим, что искаженная координация катионов Cu2+, расположенных внутри анионных вакансий или рядом с ними, может способствовать понижению прочности связи кислорода, локализованного на таких дефектах в оксиде CeO2 [3].
В реакции окисления СО кислородом в присутствии водорода при Т > 120°C на 5%CuO/CeO2 маршрут окисления СО карбонильных комплексов при адсорбции кислорода может быть не единственным. Так, в области 100−160°С, когда реакции окисления СО и Н2 конкурируют за кислород (рис. 6), высокое значение конверсии СО (~100%), возможно, поддерживается за счет дополнительной реакции разложения карбонатных комплексов на центрах окисления при этих температурах (рис. 7).
Согласно данным табл. 2, празеодим в образцах 5%CuO/Ce1 –xPrxOу ингибирует формирование карбонильных комплексов и снижает активность центров окисления в образовании карбонатных комплексов при адсорбции СО. Тогда уменьшение активности катализаторов в окислении СО кислородом в присутствии водорода при низких температурах обусловлено сокращением количества катионов Cu+, как это следует из зависимости активности образцов от содержания в них катионов Cu+. В этом случае реакция окисления СО может проходить на центрах окисления при образовании карбонатов с последующим их разложением с десорбцией СО2 в температурной области, соответствующей области протекания реакции (рис. 5 и 7).
ЗАКЛЮЧЕНИЕ
Катализаторы 5%CuO/Ce1 –xPrxOy синтезировали на основе оксидов СеО2, PrO2 и твердых растворов Ce1 –xPrxOy с х = 0.2–0.8, полученных при пиролизе гидроксидов.
Совместное восстановление водородом высокодисперсного оксида меди и носителей в катализаторах протекает значительно эффективнее (100−270°С), чем восстановление отдельных фаз CuO и Ce1 –xPrxOy (300−600°С) вследствие их сильного взаимодействия. Этим взаимодействием обусловлено и образование активного кислорода, который участвует в окислении водорода, хемосорбции СО и низкотемпературной реакции окисления СО в смеси СО + О2 + Н2.
В реакции окисления СО в избытке водорода активность образцов 5%CuO/Ce1 –xPrxOy с х = 0–0.8 понижается при возрастании содержания Pr. Повышение температуры начала реакции и уменьшение величины конверсии γmах(Т) со смещением ее в высокотемпературную область происходят в результате увеличения прочности связи кислорода в медьсодержащих центрах, взаимодействующих с катионами Pr.
Кислород анионных вакансий оксида PrOy менее активен, чем кислород медьсодержащих центров в 5%CuO/PrOy. Разбавление оксида празеодима церием лишь незначительно повышает активность образца 5%CuO/Ce0.5Pr0.5Oy по сравнению с таковой для 5%CuO/PrOy, хотя формa кислородa в носителях Ce0.5Pr0.5Oy и PrO2 различается по прочности связи, и первый реокисляется кислородом легче, чем второй.
На основании данных по термостабильности комплексов СО, образующихся на медьсодержащих центрах окисления и адсорбции образцов 5%CuO/Ce1 –xPrxOy, рассмотрено их участие в реакции низкотемпературного окисления в водороде.
Список литературы
Ола Д., Гепперт А., Пракаш С. Метанол и энергетика будущего. Когда закончатся нефть и газ. Москва: Бином, 2009. 416 с. (Olah G.A, Goeppert A., Prakash S. Beyond Oil and Gas: The Methanol Economy. Originally published by Wiley-VCH Verlag GmbH &Co, KGaA Boschstraße 12, D-69469 Weinheim, Federal Republic of Gemany.)
Mishra A., Prasad R. // Bull. Chem. React. Eng. Catal. 2011. V. 6. № 1. P. 1.
Yu K., Lou L.-L., Liu S., Zhou W. // Adv. Sci. 2020. V. 7. P. 1.
Martinez-Arias A., Gamarra D., Hungria A.B., Fernandez-Garcia M., Munuera G., Hornes A., Bera P., Conesa J.C., Camara A.L. // Catalysts. 2013. V. 3. P. 378.
Ильичев А.Н., Матышак В.А., Корчак В.Н. // Кинетика и катализ. 2015. Т. 56. № 1. С. 125. (Il’ichev A.N, Matyshak V.A., Korchak V.N. // Kinet. Catal. 2015. V. 56. № 1. P. 115.)
Venkataswany P., Jampaiah D., Aniz C.U., Reddy B.M. // J. Chem. Sci. 2015. V. 127. № 8. P. 1347.
Kim H.J., Jang M.G., Shin D., Han J.W. // ChemCatChem. 2020. V. 12. P. 11.
Singhania A. // Ind. Eng. Chem. Res. 2017. V. 56. № 46. P. 13 594.
Малютин А.В., Либерман Е.Ю., Михайличенко А.И., Аветисов И.Х., Кошкин А.Г., Конькова Т.В. // Катализ в промышленности. 2013. № 3.С. 54.
GuoX., QiuZ., MaoZ.Q., Zhou R. // Phys. Chem. Chem. Phys. 2018. V. 20. № 40. P. 25 983.
Zhao Z., Wang R., Zhao Q., Wang E., Su H., Zeng S. // Adv. Mater. Res. 2013. V. 773. P. 601.
Ильичев А.Н., Фирсова А.А., Корчак В.Н. // Кинетика и катализ. 2006. Т. 47. № 4. С. 602. (Il’ichev A.N., Firsova A.A., Korchak V.N. // Kinet. Catal. 2006. V. 47. № 4. P. 585.)
Powder Diffraction Fale. Alphabetical Indexes. Inorganic phases, JCPDS, International Center for Diffraction Data, Pennsylvania, USA, 1983.
Миркин Л.И. Справочник по рентгеноструктурному анализу поликристаллов. М: Государственное издательство физико-математической литературы, 1961. 864 с.
Третьяков И.И., Шуб Б.Р., Скляров А.В. // Журн. физ. химии. 1970. Т. 44. С. 2112.
Руководство по неорганическому синтезу / Под ред. Брауэра Г. Москва: Мир, 1985. Т. 2−3.
Narula C.K., Haack L.P., Chun W., Jen H.-W., Graham G.W. // J. Phys. Chem. B. 1999. V. 103. P. 3634.
Фирсова А.А., Ильичев А.Н., Хоменко Т.И., Горобинский Л.В., Максимов Ю.В., Суздалев И.П., Корчак В.Н. // Кинетика и катализ. 2007. Т. 48. № 2. С. 298. (Firsova A.A., Ilichev A.N., Khomenko T.I., Gorobinskii L.V., Maksimov Yu. V., Suzdalev I.P., and Korchak V.N. // Kinet. Catal. 2007. V. 48. № 2. P. 282.)
Fornasiero P., Balducci G., Monte R.D., Kaspar J., Sergo V., Gubitosa G., Ferrero A., Graziani M. // J. Catal. 1996. V. 164. P. 173.
Manzoli M., Monte R.D., Boccuzzi F., Coluccia S., Kaspar J. // Appl. Catal. B: Environ. 2005. V. 61. P. 192.
Luo M.F., Ma J.-M., Lu J.-Q., Song Y.-P., Wang Y.-J. // J. Catal. 2007. V. 246. P. 52
Gomez-Cortes A., Marquez Y., Arenas-Alatorre J., Diaz G. // Catal. Today. 2008. V. 133−135. P. 743.
Polster C.S., Naier H., Baertsch C.D. // J. Catal. 2009. V. 266. P. 308.
Moretti E., Storaro L., Talon A., Lenarda M., Riello P., Frattini R., Yuso M.V.M., Jimenez-Lopez A., Rodriguez-Gastellon E., Ternero F., Caballero A., Holgado J.P. // Appl. Catal. B: Enveron. 2011. V. 102. P. 627.
Arango-Diaz A., Cecilia J.A., Moretti E., Talon A., Nunez P., Morrero-Jerez J., Jimenez-Jimenez J., Jimenez-Lopez A., Rodriguez-Gastellon E. // Int. J. Hydrogen Energy. 2014. V. 39. P. 4102.
Wang S.-P., Zheng X.-C., Wang X.-Y., Wang S.-R., Zhang S.-M., Yu L.-H., Huang W.-P., Wu S.-H. // Catal. Lett. 2005. V. 105. № 3−4. P. 163.
Матышак В.А., Сильченкова О.Н. // Кинетика и катализ. 2019. Т. 60. № 5. С. 578. (Matyshak V.A., Sil’chenkova O.N. // Kinet. Catal. 2019, V. 60. № 5. P. 573.)
Иванова А.С. // Кинетика и катализ. 2009. Т. 50. № 6. С. 831.
Крылов О.В. Гетерогенный катализ. Москва Академкнига, 2004. 679 с.
Ильичев А.Н., Быховский М. Я., Фаттахова З.Т., Шашкин Д.П., Федорова Ю.Е., Матышак В.А., Корчак В.Н. // Кинетика и катализ. 2019. Т. 60. № 5. С. 654. (Il’ichev A.N., Bykhovskii M.Ya., Fattakhova Z.T., Shashkin D.P., Matyshak V.A., Korchak V.N. // Kinet. Catal. 2018. V. 59. № 2. P. 179.)
Skarman B., Grandjean D., Benfield R., Hinz A., Andersson A., Wallenberg L. R. // J. Catal. 2002. V. 211. P. 119.
Дополнительные материалы отсутствуют.
Инструменты
Кинетика и катализ