

Кинетика и катализ, 2021, T. 62, № 2, стр. 175-186
Определяющая роль законов цепных реакций в процессах горения, взрыва и детонации газов
В. В. Азатян *
Объединенный институт высоких температур РАН
125412 Москва, ул. Ижорская, 13, стр. 2, Россия
* E-mail: vylenazatyan@yandex.ru
Поступила в редакцию 28.10.2020
После доработки 12.11.2020
Принята к публикации 12.11.2020
Аннотация
Показано, что открытые Н.Н. Семеновым явление разветвления реакционных цепей и особенности механизма цепного горения при низких давлениях (в отсутствие саморазогрева) играют решающую роль также при высоких давлениях, в распространении пламени, во взрыве и детонации. Выяснен физико-химический механизм, обеспечивающий большие скорости и ускорения реакций, расходование реагентов за стотысячные доли секунды, переход горения во взрыв и детонацию. Получили объяснение основные закономерности всех режимов горения, необъяснимые ранее. Разработаны химические методы управления горением, взрывом и детонацией, основанные на законах цепных реакций.
Имя всемирно известного ученого, лауреата Нобелевской премии Николая Николаевича Семенова неразрывно связано с наукой о химической кинетике, о процессах горения и взрыва. Нашей общественности Н.Н. Семенов хорошо известен как один из основных создателей ядерного щита нашей страны и как руководитель последующих испытаний созданного атомного оружия. Известно, что руководимый Н.Н. Семеновым Институт химической физики тоже участвовал в разработке водородной бомбы. Настоящая статья посвящена роли теории цепных реакций Н.Н. Семенова в развитии стратегически важной области науки о процессах распространения пламени, взрыва и детонации газов.
Открытие Н.Н. Семеновым разветвленно-цепных процессов (РЦП) [1–3] ознаменовало наступление качественно нового этапа в развитии науки о кинетике и механизме химических процессов и науки о процессах горения. Значимость открытия состоит не только в обнаружении не имеющих аналогов закономерностей процессов горения, возникающих и протекающих практически без повышения температуры, при давлениях в сотни раз ниже атмосферного. Еще более значимым оказалось выявление принципиального механизма горения, реализующегося несмотря на крайне низкие концентрации. Было установлено, что большие скорости этих процессов определяются участием в них высокоактивных частиц – свободных атомов и радикалов, которые в реакциях с исходными реагентами не только регенерируются, но и размножаются. В результате таких реакций реализуются разветвленные реакционные цепи, обеспечивающие прогрессирующее самоускорение процесса даже без повышения температуры. Таким образом, Н.Н. Семеновым был выявлен новый рациональный канал использования системой внутренней энергии исходных реагентов для самоускорения процесса, принципиально отличный от саморазогрева. Последующие исследования показали, что цепной путь использования энергетических ресурсов для горения значительно рациональнее не только при крайне низких давлениях, но также при всех более высоких давлениях. Поэтому теория разветвленно-цепных реакций во многом служит базой для учения о горении газов.
В настоящей работе рассматривается развитие представлений о физико-химических механизмах и факторах, определяющих закономерности горения, взрыва и детонации газов, учитывающих их цепную природу, а также роли теории цепных реакций Н.Н. Семенова в развитии науки об этих процессах.
Известно, что к воспламенению и горению способны привести два разных по своей природе фактора – цепная лавина и саморазогрев.
ТЕПЛОВОЕ ВОСПЛАМЕНЕНИЕ
Горение, вызванное прогрессивным ускорением реакции в результате саморазогрева, принято называть тепловым. Н.Н.Семеновым [3, 4] было показано, что тепловое воспламенение реализуется, если скорость тепловыделения (q+) больше, чем скорость теплоотвода (q–) и если при этом с повышением температуры тепловыделение ускоряется больше, чем теплоотвод:
(2)
${{{\text{d}}{{q}_{ + }}} \mathord{\left/ {\vphantom {{{\text{d}}{{q}_{ + }}} {{\text{d}}T}}} \right. \kern-0em} {{\text{d}}T}} \geqslant {{{\text{d}}{{q}_{--}}} \mathord{\left/ {\vphantom {{{\text{d}}{{q}_{--}}} {{\text{d}}T}}} \right. \kern-0em} {{\text{d}}T}}.$Соотношение (1) относится к саморазогреву. Соотношение же (2) соответствует режиму, в котором при повышении температуры скорость тепловыделения возрастает больше, чем скорость теплоотвода. Одновременное выполнение условий (1) и (2) приводит к прогрессирующему накоплению тепла в системе, к более интенсивному ускорению реакции, к воспламенению. Теорию горения, рассматривающую саморазогрев как единственный фактор самоускорения реакции, принято называть тепловой.
Вскоре после открытия РЦП Н.Н. Семеновым на примере горения паров фосфора такого же типа механизм был выявлен в горении водорода С. Хиншельвудом [5]. Длительное время цепное горение изучали лишь для выяснения закономерностей, вызванных цепной природой реакций. С этой целью реакции проводили при давлениях во много десятков и сотен раз ниже атмосферного, при которых горение практически не сопровождается саморазогревом. Поэтому теория цепных реакций рассматривала только их изотермические режимы. При более высоких давлениях саморазогрев становится значительным, и такие процессы считались тепловым горением без заметной роли реакционных цепей. В настоящей работе рассматривается роль цепного механизма в реакциях горения, взрыва и детонации газов.
ЦЕПНОЕ ВОСПЛАМЕНЕНИЕ
Разветвленно-цепным процессам соответствует следующая обобщенная схема:
где x и y – свободные атомы и радикалы, A и B – исходные молекулы, Р – конечный продукт.Например, в модельном разветвленно-цепном процессе окисления водорода атомы и радикалы – носители цепей (НЦ), образуются, регенерируются и размножаются в следующих реакциях:
Поскольку НЦ, появившиеся в стадиях развития цепи (I)–(III), участвуют в развитии реакционных цепей, образуя новые ветви, то цепь разветвляется. Методом частичных квазистационарных концентраций [3] система кинетических уравнений атомов и радикалов приводится к одному уравнению, относящемуся к той частице, реакции которой лимитируют разветвление цепей. Из этой схемы следует, что скорость расходования О2 равна
(4)
$~W = - \frac{{{\text{d}}\left[ {\text{B}} \right]}}{{{\text{d}}t}} = {{\omega }_{0}} + {{k}_{1}}\left[ {\text{B}} \right]n,$НЦ вступают также в реакции образования малоактивных продуктов, неспособных участвовать в цепном горении. В этих реакциях цепь обрывается. В процессе окисления водорода такими стадиями являются адсорбция НЦ и реакция
если радикал HO2 затем адсорбируется или вступает, например, в реакцию а также реакция с ингибитором (In) Изменению величины n соответствует следующее уравнение [2, 3]:(5)
$\frac{{{\text{d}}n}}{{{\text{d}}t}} = {{\omega }_{0}} + \left( {f - g} \right)n = {{\omega }_{0}} + \varphi n,$(6)
$f = 2{{k}_{1}}\left[ {\text{B}} \right] = 2k_{1}^{0}{{e}^{{--~\frac{{{{E}_{1}}}}{{RT}}}}}\left[ {\text{B}} \right],\,\,\,\,g = {{k}_{{{\text{эф}}}}},$В условиях, при которых g > f, устанавливается низкая концентрация НЦ, определяемая соотношением скоростей их образования и гибели. В условиях же, когда происходит лавинное размножение НЦ, лавинообразно ускоряется также расходование исходных реагентов. Таким образом, условием цепного воспламенения является неравенство
Долгое время было принято считать, что в силу экспоненциальной зависимости скорости реакции от температуры саморазогрев ускоряет реакцию намного сильнее, чем цепная лавина. Поэтому в условиях саморазогрева воспламенение считалось результатом только тепловыделения. До последнего времени в теории горения, взрыва и детонации газов, а также в работах, использующих основные положения этой теории, во всех фундаментальных монографиях и энциклопедиях роль реакционных цепей в горении с саморазогревом игнорировали, а в ряде случаев даже отрицали [5–12]. В теории теплового горения химический процесс представляют гипотетической одностадийной реакцией между исходными молекулами. Такой реакции приписывают эффективную константу скорости, определенную из кинетических данных брутто-процесса при допущении о его первом или редко втором кинетическом порядке. Согласованность расчета с той или иной экспериментальной величиной получается в результате того, что используются уравнение гипотетической одностадийной реакции и входные параметры, рассчитанные из данных по изучению горения при том же допущении об одностадийности реакции (например, [11]).
Температурной зависимости скорости реакции приписывают закон Аррениуса, вопреки тому, что этот закон относится не к скорости реакции, а к константе скорости. Публикации, игнорирующие цепной характер горения, встречаются и в настоящее время [13–15].
При численном моделировании горения преследуется цель лишь описать (но не объяснить) ту или иную частную закономерность на базе принятых реакционных схем. Достоверность результатов невелика в силу ограниченности информации о механизмах реакций и константах скоростей. Утверждения о якобы известном детальном механизме горения углеводородов неправильные, даже если не считать неопределенности, вызванные образованием диспергированной сажи и ее реакциями. Формальный характер моделирования проявляется, например, в том, что в некоторых работах [16, 17] говорится о тепловом, а не цепном характере горения водорода. Между тем известно, что межмолекулярные реакции чрезвычайно медленны и не способны поддержать горение даже после сильного инициирования (см. ниже). Без учета цепной природы горения остаются нерешенными фундаментальные проблемы, относящиеся к существу явления горения во всех его режимах. Ниже приводятся примеры.
НЕВОЗМОЖНОСТЬ ОБЪЯСНЕНИЯ ГОРЕНИЯ, ВЗРЫВА И ДЕТОНАЦИИ ГАЗОВ БЕЗ УЧЕТА ЦЕПНОЙ ПРИРОДЫ РЕАКЦИИ
1. Теория горения, не учитывающая цепную природу реакций горения, взрыва и детонации газов, не способна объяснить даже факт протекания этих процессов. Действительно, энергии активации реакции, например, Н2 и СН4 непосредственно с О2 более 220 кДж/моль [18, 19]. Предэкспоненциальные же множители констант скорости, естественно, не превышают известную частоту двойных столкновений, близкую 10–10 в расчете на одну частицу в 1 см3. Поэтому характеристические времена таких реакций в области атмосферного давления при температурах горения в тысячи раз больше характеристического времени теплоотвода, и межмолекулярные реакции даже не сопровождаются саморазогревом. Между тем, в указанных условиях стехиометрические смеси водорода и метана с воздухом воспламеняются и выгорают за доли миллисекунды, показывая, что реальная реакция протекает не по межмолекулярному пути.
2. Теория теплового горения не может объяснить важное для практики сильное влияние малых примесей на горение и взрыв газов. Эта теория не объясняет также зависимость характеристик горения от химических свойств контактирующих твердых поверхностей.
3. Учет реакции только валентно-насыщенных соединений не позволяет объяснить наблюдаемое сильное ускорение реакции при повышении температуры. Например, при нагревании смеси Н2 с О2 при 1 атм от 823 до 853 К константа скорости межмолекулярной реакции (0), характеризующейся энергией активации 225 кДж/моль, увеличится лишь на ~5%. На такую же малую величину возрастет скорость межмолекулярной реакции. Между тем, эксперимент показывает, что при таком нагреве смесь воспламеняется и выгорает за малые доли миллисекунды, т.е. скорость от температуры зависит несравненно сильнее, чем экспоненциальная функция [12].
Анализ таких противоречий поставил перед нами кардинальный вопрос: каковы особенности физико-химического механизма реакций, обеспечивающие столь быстрое протекание горения, взрыва и детонации газов? Изучение закономерностей этих процессов привело нас к выводу, что в области атмосферного и повышенных давлений эти реакции, вопреки общепринятым представлениям, протекают при решающем участии свободных атомов и радикалов, реакции которых представляют собой неизотермические цепные процессы.
ДОКАЗАТЕЛЬСТВО ЦЕПНОЙ ПРИРОДЫ ГОРЕНИЯ ГАЗОВ ПРИ АТМОСФЕРНОМ ДАВЛЕНИИ
Цепной характер всех режимов горения газов при атмосферном и повышенных давлениях был доказан экспериментами, показавшими, что при исключении цепной лавины воспламенение и взрыв не происходят, несмотря на неизменность всех остальных характеристик реакционной смеси, при которых эти процессы протекают при наличии реакционных цепей [20–25]. Для исключения цепного пути реакций использовали явление ингибирования, при котором малые присадки ингибиторов перехватывают свободные атомы и радикалы, обрывают реакционные цепи и блокируют горение.
Реакции проводили в стальных реакторах и в ударной трубе. Горение заранее составленной смеси инициировали искрой, взрывом раскаленной проволоки или падающей ударной волной. Сигналы датчиков хемилюминесценции и давления поступали в многоканальный осциллограф, связанный с компьютером. Воспламенение регистрировали по появлению пиков на осциллограммах хемилюминесценции и давления, а также по падению давления смеси после завершения горения вследствие уменьшения числа молей реагентов. На рис. 1 видно, что смеси, содержащие менее 3% водорода и 2% изобутена (область 0 вблизи центра координат), воспламенить не удается, а 2.5% ингибитора предотвращают воспламенение всех смесей, содержащих более 20% Н2. После запуска искры скачки давления и хемилюминесценция, присущие воспламенению, не регистрируются. Хроматографический анализ показывает, что после опытов с ингибитором концентрация О2 в смеси остается неизменной с точностью 0.03%. Отсутствие хемилюминесценции и скачка давления, неизменность концентраций исходных реагентов после искры однозначно показывают, что присадкой ингибитора воспламенение предотвращено. Обрыв цепей происходит по реакции:
(VII)
${\text{Н}} + i{\text{ - }}{{{\text{С}}}_{4}}{{{\text{Н}}}_{8}} = {{{\text{С}}}_{3}}{{{\text{Н}}}_{7}}.$Рис. 1.
Области воспламенения смесей водорода с воздухом в присутствии: изобутена (1), пропилена (2) и азота (3). Смеси, содержащие менее 3% водорода и 2% изобутена (область 0 вблизи центра координат), воспламенить не удавалось.
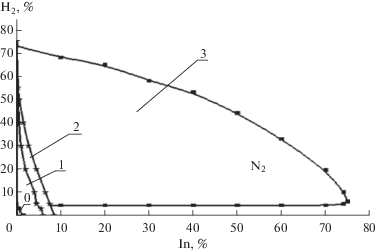
Неизменность концентрации О2 показывает также, что в смеси с ингибитором искра не способна вызвать реакцию непосредственно между Н2 и О2. Аналогичное влияние оказывают присадки других низших олефинов. На рис. 1 видно, что, в отличие от ингибиторов, при таких же добавках N2 смеси водорода с воздухом воспламеняются и горят так же, как и в отсутствие присадок.
Воспламенение имеет цепную природу также при высоких начальных температурах [22–25]. Разработанные в этих исследованиях методы позволяют повысить температуру газа c помощью ударной волны за доли микросекунды на тысячи градусов, инициировать горение и следить за влиянием ингибиторов на развитие горения при заданных начальных температурах, однородных по объему реактора. Установка была снабжена аппаратурой для измерения скорости ударной волны (УВ), регистрации давления, методиками эмиссионной и абсорбционной спектроскопии. Камеры высокого давления (КВД) и низкого давления (КНД) ударной трубы разделены диафрагмой. КВД заполняли толкающим газом. КНД служил реактором. Разогрев смеси до заданной температуры создавался падающей УВ, возникающей при разрыве диафрагмы. Параметры газа во фронте УВ рассчитывали с использованием известной программы по измеренной скорости УВ и начальным параметрам смеси. При сильном инициировании горение переходило в детонацию. Эксперименты показывают, что в присутствии ингибиторов воспламенение не происходит даже при высоких начальных температурах и при очень сильном инициировании [22–25]. Например, смесь, воспламеняющаяся в отсутствие ингибиторов при 930 К и 60 кПа, не воспламеняется даже при 1130 К и давлении 84 кПа в результате подавления цепной лавины 0.7% пропилена. Для воспламенения смеси с таким содержанием пропилена приходится увеличить скорость инициирующей УВ и тем самым повысить температуру и давление соответственно до 1140 К и 0.85 атм. При дальнейшем увеличении содержания ингибитора минимальная температура самовоспламенения становится еще выше. Повышаются и критические давления воспламенения. Таким образом, даже при очень высоких температурах молекулярные реагенты непосредственно между собой не реагируют и, значит, в согласии с нашим выводом, в отсутствие цепной лавины высокие температуры условия горения не обеспечивают. Более подробно методика и результаты описаны в публикациях [23, 25].
Эксперимент и расчеты показывают, что при атмосферном давлении саморазогрев и его влияние становятся заметными лишь в уже развивающемся цепном горении. Определяющую роль цепной лавины в уже развившемся горении показывают эксперименты по гашению пламени под воздействием ингибиторов, образованных в ходе горения [23].
Таким образом, реакции горения газов при атмосферном давлении целиком являются цепными и протекают по законам цепных процессов. Объяснение получили большие скорости реакций, обеспечивающие выгорание смеси за доли миллисекунды. Также получен ответ на фундаментальный вопрос о причинах и физико-химическом механизме, обеспечивающем протекание быстрых реакций воспламенения, горения и взрыва. Указанная же выше важная особенность горения – очень сильное самоускорение реакций, определяется специфическим характером температурной зависимости скорости цепных процессов.
ОСОБЕННОСТИ ТЕМПЕРАТУРНОЙ ЗАВИСИМОСТИ СКОРОСТИ РЕАКЦИИ В ГОРЕНИИ И ДЕТОНАЦИИ
Теория теплового горения и ее методы основаны на представлениях, по которым температурная зависимость скорости соответствует закону Аррениуса (например, [3–13]). Считается также, что зависимость скорости реакции и ее константы от температуры зависят тем сильнее, чем больше энергия активации (например, [7, 10, 11]). В работах [22–24], однако, было показано, что эти представления неправильные, противоречат экспериментам и не объясняют скорости реакций в горении и детонации. Было выяснено также, что экспоненциальный множитель, входящий в константу скорости, не описывает зависимость скорости реакции от температуры, присущую горению. Представления о соответствии температурной зависимости скорости реакции функции Аррениуса противоречат, прежде всего, закону действующих масс, согласно которому скорость равна произведению константы скорости и концентрации реагентов. Эти множители являются разными функциями от температуры и в ходе горения изменяются по разным законам. Даже в простейшей реакции первого кинетического порядка скорость равна
(8)
$--\frac{{{\text{d}}C}}{{{\text{d}}t}} = W\left( {T,t} \right) = k\left( {T,t} \right)C\left( {T,t} \right).$(9)
$\frac{{\partial W}}{{\partial T}} = k\left( {T,t} \right)\frac{{\partial C\left( {T,t} \right)}}{{\partial T}} + C\left( {T,t} \right)\frac{{\partial k\left( {T,t} \right)}}{{\partial T}}.$Зависимость концентрации от температуры незначительна только в начальный момент. В ходе же реакции функция C(T, t) изменяется по кинетическим законам, разным в разных реакциях и в разных условиях. Коренное различие температурных зависимостей скорости и константы скорости покажем сначала на примере реакции первого порядка. Интегрирование уравнения (11) приводит к следующим выражениям:
(10)
$С\left( {T,t} \right) = ~\,\,{{C}_{0}}\exp \left[ {--\int\limits_{{{t}_{0}}}^t {k\left( {T,t} \right)} } \right]{\text{d}}t,$(11)
$W\left( {T,t} \right) = k\left( {T,t} \right){{C}_{0}}{\kern 1pt} {\kern 1pt} {\text{exp}}\left[ {--\int\limits_{{{t}_{0}}}^t {k\left( {T,t} \right)} } \right]{\text{d}}t,$Неправильными оказались также представления, по которым более сильной температурной зависимости W(T) и k(T) соответствует большая энергия активации. Ошибка вызвана тем, что вместо абсолютных величин изменений W(T) и k(T) рассматриваются относительные величины, т.е. $\frac{{\Delta k}}{k}~$ и $\frac{{\Delta W}}{W}~{\text{,}}$ которые не определяют кинетику реакции. Влияния температуры на скорости реакции и тепловыделения определяются не относительными величинами изменений k(T) и W(T), а абсолютными величинами $~\Delta k~$ и $\Delta W{\text{.}}$ Именно абсолютная величина изменения скорости определяет величину саморазогрева, переходы во взрыв и в детонацию.
Из выражения константы скорости
следует, что(13)
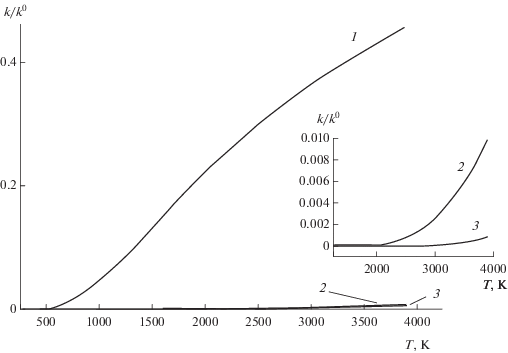
${\text{d}}k = \frac{{\partial k}}{{\partial T}}{\text{d}}T = {{k}^{0}}{{e}^{{--~\frac{E}{{RT}}}}}\frac{E}{{R{{T}^{2}}}} = k\frac{E}{{R{{T}^{2}}}}.$Поскольку константы скорости реакций атомов и радикалов намного больше, чем константы межмолекулярных реакций, то температурная зависимость констант скоростей реакций активных частиц так же, как и самих скоростей, намного сильнее. Это различие показано на графиках функции (12) на рис. 2, а также в формуле (13). Из этого рисунка следует, что межмолекулярные реакции не могут обеспечить горение не только вследствие очень малых величин констант скорости, но также вследствие того, что даже при сильном повышении температуры внешним источником рост абсолютной величины их констант скорости крайне мал.
Рис. 2.
Графики функции k/k0= e–E/RT при различных значениях энергии активации, кДж/моль: 1 – 25, 2 – 150, 3 – 225. На вставке показан участок высоких температур ([22]).
ЗАКОН ЭКСПОНЕНТЫ В ПОЛОЖИТЕЛЬНОЙ ЭКСПОНЕНТЕ
При реальных предэкспоненциальных множителях констант скорости бимолекулярных реакций функция ${{e}^{{--~\frac{E}{{RT}}}}}^{~}$ не описывает наблюдаемый резкий рост скорости реакции в развивающемся процессе независимо от значения Е и режима горения. Это указывает на то, что характер реальной температурной зависимости скорости коренным образом отличается от используемой в этих целях функции Аррениуса.
Чрезвычайно сильная температурная зависимость скорости определяется следующей особенностью кинетики изменения концентраций НЦ. В каждый момент времени и при каждых данных величинах f и g скорость изменения концентрации НЦ находится в обратной связи с концентрацией n, как это видно также из уравнения (6). При этом, если f > g, то обратная связь положительная, и, значит, величина n возрастает во времени экспоненциально, даже при постоянной температуре. В эту экспоненту в качестве множителя времени t входит разность f – g. В величину f входит константа скорости разветвления k1 со своим фактором Больцмана в соответствии с выражением (7). Таким образом, при f > g величины n и W зависят от температуры по закону экспоненты, находящейся в положительном показателе степени. Поскольку функциональная зависимость “экспоненты в положительной экспоненте” осуществляется при каждой данной температуре, то она выполняется в ходе горения. При наличии диффузионного члена в уравнениях указанная обратная связь сохраняется.
При временах t > t0 ≅ 3/φ, когда в уравнениях (5) и (6) уже можно пренебречь величиной w0. Интегрирование уравнения (6) с учетом выражения f по формуле (7) и температурной зависимости k1 приводит к следующей зависимости n от температуры и времени:
(14)
$n = \,\,~{{n}_{0}}{\kern 1pt} {\text{exp}}{\kern 1pt} \int\limits_{{{t}_{0}}}^t {\left[ {\exp \left( {--\frac{{{{E}_{{\text{p}}}}}}{{RT}}} \right) - g} \right]} {\text{d}}t.$Большие скорости и резкое ускорение процесса при повышении температуры вызваны, прежде всего, сильным ускорением реакций активных частиц Н и ОН с О2 и Н2 соответственно. Благодаря большим величинам констант скорости k1 и k2 этих реакций, велики также скорости роста их абсолютных величин с температурой (рис. 2). Ускоряющееся в этих реакциях размножение активных частиц по закону экспоненты в положительной экспоненте (14) вместе с возрастающей k1 обеспечивают те скорости процесса, которые присущи взрыву и детонации.
При подстановке n из выражения (14) в уравнение скорости (5) получается [21–24]
(15)
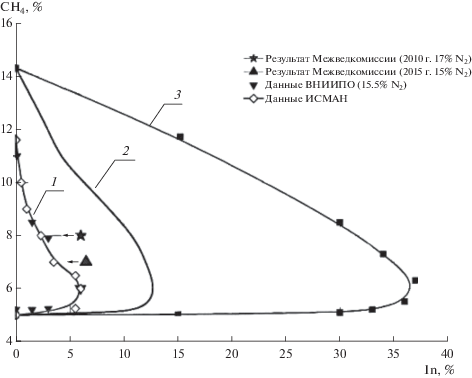
$\frac{W}{{\left[ {\text{B}} \right]}} = ~\,\,{{k}_{1}}{{n}_{0}}\exp \int\limits_{{{t}_{0}}}^t {\left[ {{{f}_{0}}\exp \left( {--{E \mathord{\left/ {\vphantom {E {RT}}} \right. \kern-0em} {RT}}} \right)--g} \right]} {\kern 1pt} {\text{d}}t.$В силу очень сильной зависимости от температуры концентраций активных частиц и скорости крайне велика роль градиентов этих величин. Градиенты во многом определяются гетерогенными реакциями. Из уравнения (15) следует также, что при разбавлении смеси инертным газом вследствие снижения концентрации окислителя В экспоненциально уменьшаются скорости реакции и тепловыделения. Таким образом, даже небольшие присадки инертных газов экспоненциально усиливают тормозящее воздействие ингибиторов. Этот эффект (синергизм инертным газом) был использован для усиленного ингибирования возгораний метано-воздушных смесей. На рис. 3 приведены результаты проведенных в различных организациях испытаний по предотвращению возгораний и взрывов метано-воздушных смесей с использованием (кривая 1) и без использования явления синергизма (кривая 2) [26]. Критические условия предотвращения возгорания, инициированного локальным источником, не зависят от объема реактора, поскольку ингибитор подавляет возгорание у самого очага инициирования. Этим объясняется количественное согласие результатов всех испытаний, проведенных в различных испытательных центрах в камерах разной формы и в объемах от 3.2 л до 43 м3.
Рис. 3.
Сужение концентрационных пределов возгорания метано-воздушных смесей ингибитором CF3H с использованием эффекта синергизма инертным газом (1) и без использования его (2) по данным различных организаций. Кривая (3) – влияние азота на концентрационные пределы воспламенения смесей метана с воздухом.
ОПРЕДЕЛЯЮЩАЯ РОЛЬ ЦЕПНОЙ ЛАВИНЫ В РАСПРОСТРАНЕНИИ ПЛАМЕНИ И ДЕТОНАЦИИ
Очевидно, что необходимой стадией детонации является повышение температуры свежей смеси, вызванное теплом, поступающим из прилегающего горящего слоя, и сжатием ударной волной. В теории теплового горения объяснение воспламенения газов при нагревании было основано на модели реакции валентно-насыщенных соединений [5–13]. Однако, как было показано выше (рис. 3) и в работах [23, 24], вследствие больших величин энергий активации таких реакций крайне малы не только их скорости, но и абсолютные величины роста скорости при нагревании даже до 4000 К. Таким образом, теория, учитывающая реакции только валентно-насыщенных соединений, при учете реальных расчетных параметров не может объяснить воспламенение, вызванное нагреванием и, значит, не объясняет также распространение пламени и детонацию. Прежняя теория описывала (но не объясняла) наблюдаемый процесс горения при допущении об одностадийности реакции и на основании гипотезы о ее температурной зависимости по закону Аррениуса. Поскольку, однако, как мы увидели, к скорости реакции этот закон не относится, а функция ${{e}^{{--~\frac{E}{{RT}}}}}$ не описывает сильную зависимость скорости от температуры, то описание было неубедительным.
Воспламенение газа, вызванное нагреванием при атмосферном давлении, получило объяснение в работе [27], в которой на основании экспериментальных данных было установлено, что крайне медленная реакция между Н2 и О2 вне области воспламенения является цепным процессом, скорость которого при каждой данной температуре, медленно возрастая, достигает стационарного значения. Даже непосредственно под третьим пределом (лишь на 0.2% ниже критической температуры) характеристическое время реакции и тепловыделения в 700 раз больше времени теплоотвода. Поэтому саморазогрев не регистрируется. Вместе с тем, измеренные скорости расходования О2 в десятки раз больше, чем скорость межмолекулярной реакции Н2 с О2. При нагревании скорость этой реакции, стационарная при каждой данной температуре, прогрессивно возрастает. Поскольку, в этих условиях g > f, то скорости реакции соответствует выражение:
(16)
${{W}_{{{\text{стац}}}}} = --\frac{{{\text{d}}\left[ {{{{\text{O}}}_{2}}} \right]}}{{{\text{d}}t}} = {{\omega }_{0}}\left\{ {1 + \frac{{{{k}_{1}}\left[ {{{{\text{O}}}_{2}}} \right]}}{{g~--~f}}\left[ {1--{{е}^{{--\left( {g~--~f} \right)t}}}} \right]} \right\}.$После того, как f становится больше g, характер зависимости НЦ и W от времени качественно изменяется: рост n становится прогрессирующим согласно выражению:
(17)
$n = {{n}_{0}}\left[ {{{e}^{{\left( {f~--~g} \right)\left( {t~--~{{t}_{0}}} \right)~}}}--~1} \right].$(18)
$W = --\frac{{{\text{d}}\left[ {{{{\text{O}}}_{2}}} \right]}}{{{\text{d}}t}} = {{\omega }_{0}}\left\{ {1 + \frac{{{{k}_{1}}\left[ {{{{\text{O}}}_{2}}} \right]}}{{f~--~g}}\left[ {{{е}^{{\left( {f~ - ~g} \right)t}}}--~1} \right]} \right\}.$При сильной цепной лавине выполняется условие тепловой лавины, т.е. соотношение (2). Вследствие ускорения реакции в соответствии с выражением (19) характеристическое время тепловыделения становится короче времени выравнивания давления и температуры на расстояниях, заметно превышающих размеры очага реакции. Это соответствует определению взрыва, согласно которому произведение характеристического времени тепловыделения (tp) на скорость звука (v) в реакторе значительно меньше, чем линейный размер (L) очага горения [29]
В очаге горения газ не успевает расширяться, возникает локальный скачок давления и температуры, т.е. цепно-тепловой взрыв. Границы скачка продвигаются со скоростью звука, соответствующей температуре зоны горения и, значит, со сверхзвуковой скоростью в еще не нагретом газе. Образуется ударная волна. В силу своей цепной природы взрыв легко ингибируется [22–26]. Ингибированием предотвращаются взрыв и детонация водорода в силовых установках [20, 26], а также метана в испытательном штреке и в шахтах [23].ЦЕПНАЯ ПРИРОДА РЕАКЦИЙ В ДЕТОНАЦИИ ГАЗОВ
Интенсивные исследования привели к разработке теории газодинамики взрыва и детонации. Однако фундаментальные проблемы химических и физико-химических аспектов теории этих процессов стали получать свое решение лишь в последние два–три десятилетия. До этого теория детонации газов основывалась на гипотетической модели одностадийной реакции только исходных молекул (например, [30, 32]) вопреки тому, что в силу больших энергий активации такие реакции чрезвычайно медленные и не поддерживают горение. Реакциям условно приписывали первый кинетический порядок. О наличии активных частиц в детонации говорили авторы работ [30, 33, 34], которые, однако, представляли процесс в виде одностадийной реакции между исходными реагентами. В некоторых более поздних работах детонацию по-прежнему описывают реакциями только валентно-насыщенных соединений с нереальными константами скорости [35, 36]. Возможность ингибирования детонации отрицалась [37–39], что равносильно отрицанию ее цепной природы. Вопрос о причинах, о механизме крайне больших скоростей реакций в детонации не ставился. Однако из скорости детонационной волны, например, в водородо-воздушной смеси и толщины слоя пламени следует, что реакция завершается за миллионные доли секунды. За такие же времена происходит ускорение реакции до столь больших скоростей. Такие большие скорости и ускорения указывают на неспособность прежних представлений объяснить детонацию и взрыв.
Вопреки этим представлениям в серии работ [20–25, 40] было показано, что реакции в газофазной детонации являются цепными и протекают по специфическим законам неизотермических цепных процессов. Необходимость цепной лавины для реализации детонации была доказана путем предотвращения и полного подавления детонации малыми присадками ингибиторов на примерах реакций окисления водорода и оксида углерода воздухом. Высокая эффективность олефинов в качестве ингибиторов определяется наличием в их молекулах двойной связи, быстро захватывающей свободные атомы и радикалы. Насыщенные углеводороды менее эффективны, поскольку при ингибировании с их участием происходит разрыв прочной связи С–Н.
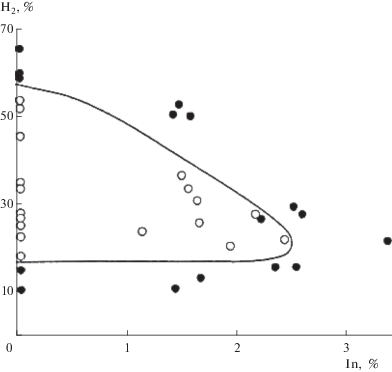
Эксперименты проводили на различных стендах, в том числе на модели испытательного стенда прямоточного воздушно-реактивного двигателя [22, 24, 40]. На рис. 4 видно, что по мере увеличения содержания ингибитора концентрационные пределы перехода горения в детонацию сужаются и при присадках более 2.6 об. % детонация предотвращается при любых соотношениях концентраций водорода и воздуха. При содержаниях ингибитора выше 8.5% подавляется также дефлаграционное горение. Методика описана в работах [23, 40, 41]. Очевидно, что предотвращение ингибиторами детонации является однозначным доказательством того, что детонация происходит только благодаря цепной природе реакции. Обрыв цепей протекает по реакции, аналогичной приведенному выше акту (VII).
Рис. 4.
Зависимость концентрационных пределов перехода горения водородо-воздушной смеси от содержания ингибитора в детонацию рабочей модели прямоточного воздушно-реактивного двигателя: ⚫ – горение без перехода в детонацию, ⚪ – детонация ([22]).
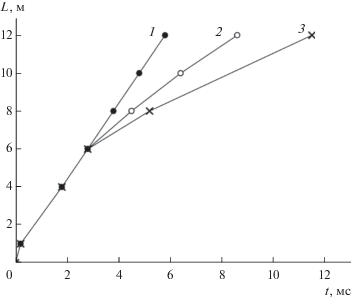
Путем разрушения стационарной волны детонации был доказан цепной характер реакции также в стационарной детонации. На рис. 5 видно, что детонационная волна после вхождения в реактор с водородо-воздушной смесью в отсутствие ингибитора и прохождения 0.9–1 м продвигается с постоянной скоростью (прямая 1). Из наложения точек (обозначенных крестиками и кружками) на прямой 1 (рис. 5) видно, что скачкообразный рост давления и свечение в каждом сечении трубы регистрируются одновременно, т.е. фронт горения и ударная волна продвигаются совместно, что присуще детонации. Скорость волны (1985 ± 5 м/с) находится в хорошем согласии с известными экспериментальными данными (например, [12]). В присутствии же 3% пропилена на расстоянии 6 м фронт горения начинает отставать от ударной волны. Детонация распадается на затухающую ударную волну (кривая 2) и все больше отстающую от нее затухающую волну горения (кривая 3). Подавление детонации тем эффективнее, чем активнее ингибитор и чем больше его содержание.
Рис. 5.
L–t-диаграммы ударной волны (-⚪-) и скачка хемилюминесценции (-×-) в смеси 33% Н2 с воздухом в отсутствие пропилена (1) и в присутствии 3% пропилена (2, 3) ([22]).
Очевидно, что в теории газодинамических характеристик детонации следует учесть цепную природу реакций и законы их развития, определяющие все основные закономерности процесса.
Необходимость цепного механизма в газофазных процессах взрыва и детонации определяется тем, что только благодаря большим константам скорости реакций НЦ, цепной лавине и рассмотренной выше специфической очень сильной температурной зависимости скорости по закону “экспоненты в положительной экспоненте” быстро создаются очень высокие концентрации активных частиц, в силу чего расходование исходных реагентов завершается за стотысячные доли секунды. Только большие скорости и ускорения цепной реакции обеспечивающие такое же ускорение саморазогрева, создают условия перехода горения в режим самоподдерживающейся взрывной волны–детонации.
Отрицание и игнорирование некоторыми исследователями цепного характера реакций в детонации и взрыве является следствием непонимания физико-химической сущности детонации и взрыва, а также неосознанности того, что без участия свободных атомов и радикалов горение в газовой фазе невозможно.
ВЫВОДЫ
Таким образом, работами, развитыми на базе открытия Н.Н. Семеновым разветвленно-цепных реакций при низких давлениях и созданной им теории, установлено, что цепной механизм является определяющим в горении газов также при любых более высоких давлениях, в режимах распространения пламени, взрыва и детонации. При этом получили объяснение все основные закономерности этих процессов, не находившие объяснения ранее, и возникло новое научно-техническое направление – развитие теории горения, взрыва и детонации газов, а также разработка химических методов управления их закономерностями.
Список литературы
Ceмeнoв H.H. // Z. Phys. 1927. V. 46. P. 109.
Семенов Н.Н. Цепные реакции. Л.: Госхимтехиздат, 1934. 555 с.
Семенов Н.Н. Избранные труды. М.: Наука, 2005. Т. 3. 499 с.
Семенов Н.Н. // Успехи физических наук. 1940. Т. 23. № 1. С. 251.
Льюис Б., Эльбе Г. Горение, пламя и взрывы в газах. М.: Мир, 1968, 592 с.)
Вильямс Ф.А. Теория горения. М.: Наука, 1971. 615 с.
Зельдович Я.Б., Баренблат Г.И., Либрович В.Б., Махвиладзе Г.М. Математическая теория горения. М.: Наука, 1980. 478 с.
Химическая энциклопедия. Горение. М.: Сов. энциклопедия, 1988. Т. 1. С. 594.
Физическая энциклопедия. Горение. М.: Сов. энциклопедия, 1988. Т. 1. С. 515.
Мержанов А.Г., Хайкин Б.И. Теория волн горения в гомогенных средах. Черноголовка: Изд-во Ин-та структурной макрокинетики РАН, 1992. 160 с.
Франк-Каменецкий Д.А. Основы макрокинетики, диффузия, теплопередача в химической кинетике. Долгопрудный: Интеллект, 2008. 407 с.
Lewis B., von Elbe G. Combustion, Flames and Explosions of Gases. N.Y.: Acad. Press, 1987. 739 p.
Девисилов В.А., Дроздова Т.И., Тимофеева С.С. Теория горения и взрыва. М.: Форум, 2012. 351 с.
Палесский Ф.С., Фурсенко Р.В., Минаев С.С. // Физика горения и взрыва. 2014. Т. 50. № 6. С. 3.
Сабденов К.О. // Хим. физика. 2017. Т. 36. № 11. С. 39. (Sabdenov, K.O., Russ. J. Phys. Chem. B, 2017, vol. 11, no. 6, p. 942.)
Варнатц Ю., Маас У., Диббл Р. Горение. Физические и химические аспекты, моделирование, эксперименты, образование загрязняющих веществ. М.: Физматлит, 2003. 351 с.
Frassoldati A., Couci A., Faravelli T., Niemann U., Ranzi E., Seirer R., Seshadri K. // Combust. Flame. 2010. V. 157. P. 2.
Baulch D.L., Bowman C.T., Cobos C.J., Cox R.A., Just T., Kerr J.A., Pilling M.J., Stoker D., Troe J., Tsang W., Walker R.W.,Warnatz J. // J. Phys. Chem. Ref. Data. 2005. V. 34. № 3. P. 757.
Srinivasan N.K., Michael J.V., Harding L.B., Klipperstain S.J. // Combust. Flame. 2007. V. 149. № 1–2. P. 104.
Азатян В.В., Бакулев В.И., Калканов В.А., Романенко Н.Т., Федосов Л.Н., Харламов А.В., Шавард А.А. Способ питания газового двигателя внутреннего сгорания. Пат. RUS 1835139, 1992.
Азатян В.В. // Успехи химии. 1999. Т. 68. № 12. С. 1122.
Азатян В.В. // Кинетика и катализ. 2020.Т. 61. № 3. С. 291.
Азатян В.В. Цепные реакции в процессах горения, взрыва и детонации газов. Черноголовка: Изд-во ИПХФ РАН, 2017. 448 с.
Азатян В.В. // Журн. физ. химии. 2015. Т. 89. № 11. С. 1731.
Азатян В.В., Павлов В.А., Шаталов О.П. // Кинетика и катализ. 2005. Т. 46. № 6. С. 835.
Азатян В.В., Ведешкин Г.К., Филатов Ю.М. // Вестник РАН. 2019. Т. 89. № 2. С. 279.
Азатян В.В., Прокопенко В.М. // Журн. физ. химии. 2018. Т. 92. № 12. С. 1925.
Прокопенко В.М., Азатян В.В. // Журн. физ. химии. 2018. Т. 92. № 1. С. 55.
Большая российская энциклопедия. Взрыв. 2006. Т. 5. С. 242.
Зельдович Я.Б., Компанеец А.С. Теория детонации. М.: Гостехиздат, 1955. 268 с.
Митрофанов В.В. Детонация гомогенных и гетерогенных систем. Новосибирск. Изд-во СО РАН, 2003. 199 с.
Progress in Astronautics and Aeronautics. Advances in Combustion Science: in Honour of Ya.B. Zeldovich, 1997. V. 173. P. 95.
von Neumann J. Theory of Detonation Waves. Progress Report to the National Defense Research Committee Div. B, OSRD report № 549, 1942. PB 31090.
Doring W. // Ann. Physik. 1943. P. 421.
Deflagrative and Detonanive Combustion / Под ред. Фролова С.М. Moscow: TORUS PRESS, 2010. 468 с.
Дубровский А.В., Иванов В.С., Зангиев А.Э., Фролов С.М. // Хим. физика, 2016. Т. 35. № 6. С. 49.
Соколик А.С. Самовоспламенение, пламя и детонация в газах. М.: Изд-во АН СССР, 1960. 428 с.
Зельдович Я.Б. Избранные труды. М.: Наука, 1984. Т. 1. 374 с.
Гельфанд Б.Е. // Физика горения и взрыва. 2002. Т. 38. № 5. С. 101.
Azatyan V.V., Wagner G.G., Vedeshkin G.K. Gaseous and Heterogeneous Detonations. M.: ENAS Publishers, 1999. P. 331.
Азатян В.В., Абрамов С.К., Прокопенко В.М., Ратников В.И., Туник Ю.В. // Кинетика и катализ. 2013. Т. 54. № 5. С. 553.
Дополнительные материалы отсутствуют.
Инструменты
Кинетика и катализ