

Кинетика и катализ, 2021, T. 62, № 2, стр. 224-232
Повышенная активность нового катализатора V2O5/HZSM-5 в гидроксилировании бензола
C. Li a, G. Li a, *, P. Dong a, **, H. Li a, W. Meng a, D. Zhang a
a College of Petrochemical Engineering, Lanzhou University of Technology
730050 Lanzhou, P.R. China
* E-mail: lgxwyf@163.com
** E-mail: dongpeng06190318@126.com
Поступила в редакцию 04.05.2020
После доработки 17.09.2020
Принята к публикации 05.10.2020
Аннотация
Приготовлен новый катализатор V2O5/HZSM-5, охарактеризованный методами рентгеновской дифракции, инфракрасной спектроскопии с преобразованием Фурье, сканирующей электронной микроскопии, трансмиссионной электронной микроскопии, энергодисперсионной рентгеновской спектроскопии, картирования, спектрометрии индуктивно связанной плазмы, физической десорбция азота и рентгеновской фотоэлектронной спектроскопии с целью выяснения взаимосвязи структуры и активности. Найдено, что в оптимальных условиях реакции конверсия бензола составляла 73.5%, селективность по фенолу превышала 99.0%, а также существенно улучшалась стабильность катализатора.
Фенол является важным сырьем для основного органического синтеза. Он широко используется в волокнах, пластмассах, фармацевтических препаратах, пестицидах, красителях, парфюмерии, покрытиях и при переработке [1, 2]. В настоящее время более 90% фенола в мире производится окислением кумола. Однако у этой промышленной технологии есть несколько ограничений, включая длительный технологический процесс, жесткие условия реакции, низкое атомное использование и образование эквимольного количества ацетона в качестве побочного продукта [3]. Следовательно, разработка экологически безопасного синтеза фенола представляет значительную ценность для промышленного применения и решения фундаментальных проблем. Оптимальным методом синтеза фенола является окисление бензола, что очень привлекательно, но сложно осуществимо. Проблема заключается в отсутствии катализаторов для эффективного гидроксилирования и подходящих окислителей.
В последние годы в качестве окислителей использовали N2O [4, 5] и O2 [6, 7] в газофазных реакциях, а в жидкофазных реакциях – H2O2 [8, 9]. Однако широкое использование N2O и O2 в газофазных реакциях было ограничено из-за низкой скорости превращения, высокой температуры реакции и быстрой дезактивации катализатора. Единственным же побочным продуктом окисления бензола до фенола с помощью H2O2 является вода, которая не загрязняет окружающую среду, и процесс протекает в мягких условиях [8, 10]. Таким образом, этот процесс был признан наиболее многообещающим для замены кумольного процесса. Кроме того, необходимо подчеркнуть, что выбор катализатора имеет решающее значение, поскольку он определяет не только выход фенола, но и экономическую выгоду всего процесса (например, доступность окислителя и сложность очистки продукта). Хорошо известно, что катализаторы, используемые в реакции гидроксилирования бензола в фенол, в основном представляют собой нанесенные катализаторы, такие как Fe/HZSM-5 [4, 5, 11, 12], Cu/Beta [13], Cu/APO-11 [14], V/g-C3N4 [2, 6, 15], V/SBA-15 [7], V/TiO2 [16] и V/NC-600 [8]. В последние годы исследования катализаторов на основе ванадия были особенно популярны, поскольку селективность таких катализаторов могла достигать более 90%. Активный компонент должен быть сильно диспергирован на носителе с высокой удельной площадью поверхности (на таком как HZSM-5), чтобы в полной мере использовать ванадийсодержащие активные центры.
Катализаторы на основе цеолита HZSM-5 имеют высокую удельную поверхность и кислотность, что способствует увеличению выхода фенола [4, 11]. Однако диффузия реагентов и продуктов серьезно затруднена из-за микропористой структуры HZSM-5, в результате чего потенциальная каталитическая активность цеолита не может быть эффективно использована, а конверсия и селективность значительно снижаются [12]. Поэтому для преодоления этих недостатков была использована иерархическая система пор [17]. Однако для получения катализаторов на основе ванадия ключевым фактором является метод их синтеза.
В данной работе была приготовлена серия катализаторов на основе ванадия. Структура катализаторов была охарактеризована с помощью методов рентгеновской дифракции (XRD), инфракрасной спектроскопии с преобразованием Фурье (FT-IR), сканирующей электронной микроскопии (SEM), трансмиссионной электронной микроскопии (TEM), энергодисперсионной рентгеновской спектрометрии (EDS), картирования, спектрометрии индуктивно связанной плазмы (ICP), физической адсорбции–десорбции N2 и рентгеновской фотоэлектронной спектроскопии (XPS). Активность и стабильность катализатора проверяли в реакции гидроксилирования бензола.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Химические реагенты
Бензол приобретен у “Yantai Shuangshuang Chemical Co., Ltd.”, ацетонитрил – у “Sinopharm Chemical Reagent Co., Ltd.” Пероксид водорода и лимонная кислота куплены в “Tianjin Damao Chemical Reagent Factory”. Иерархический цеолит HZSM-5 (SiO2/Al2O3 = 80) приобретен у “Zoran Environmental Technology Co., Ltd.” Все реагенты использовали без дополнительной очистки.
Приготовление катализатора
Катализатор V2O5/HZSM-5 готовили методом гидротермального синтеза. Сначала V2O5 диспергировали в деионизированной воде (60 мл). Некоторое количество цеолита HZSM-5 добавляли к этой суспензии при непрерывном перемешивании. Затем смесь переносили в автоклав из нержавеющей стали (емкостью 100 мл) и после перемешивания в течение 20 мин заполняли его деионизированной водой примерно до 70% вместимости. Автоклав герметизировали и выдерживали при 170°C в течение 20 ч. После охлаждения автоклава до комнатной температуры продукт отделяли центрифугированием, трижды промывали деионизированной водой, сушили при 80°C в течение 4 ч и прокаливали при 550°C в течение 6 ч. Продукты обозначали как X%V2O5/HZSM-5 (X показывает содержание ванадия).
Характеристики катализатора
Рентгенограммы катализатора снимали на рентгеновском дифрактометре D/MAX-2400 (“Rigaku”, Япония), излучение CuKα (λ = 154.2 Å), рабочее напряжение 40 кВ, ток 50 мА, диапазон сканирования образца 2θ = 5°~60°, скорость сканирования 10°/мин.
Фурье-ИК-спектрометр Nicolet Nexus 6700 (“Thermo Fisher Scientific”, США) использовали для измерения инфракрасного спектра. Предварительно катализатор измельчали и смешивали с соответствующим количеством спектрально чистого KBr, а затем проводили тестирование таблеток. Разрешение устройства составляло 4 см–1, а диапазон сканирования – 380~4000 см–1.
Морфологию и структуру катализатора определяли с помощью сканирующего электронного микроскопа Thermo Scientific FEI-Apreo (“Thermo Fisher Scientific”). Образец катализатора анализировали после ультразвукового диспергирования в растворе этанола в течение нескольких минут.
ТЕМ-изображения катализатора получали на просвечивающем электронном микроскопе высокого разрешения серии FEI-Talos F200X/S (“Thermo Fisher Scientific”).
Спектрометр с индуктивно связанной плазмой (“Thermo Fisher Scientific”) использовали для определения фактического содержания металла в катализаторе. Образец (0.005 г) растворяли в 4 мл царской водки, отбирали 1 мл раствора и для тестирования разбавляли в 10 раз после полного разложения.
Удельную площадь поверхности и распределение пор по размерам измеряли на приборе Quantachrome, Autosorb-iQ (“Quantachrome Instruments”, США) в экспериментах по определению физической адсорбции–десорбции азота при –196°C. Перед измерениями катализатор обязательно дегазировали в вакууме при 300°C в течение 10 ч.
Рентгеновский фотоэлектронный спектрометр PHI-5702 (“ULVAC-PHI Inс.”, США) использовали для анализа валентного состояния элементов в образце. Источником возбуждающего излучения служила мишень из алюминия, мощность 250 Вт, напряжение 14 кВ.
Реакция гидроксилирования бензола
Гидроксилирование бензола проводили в 100-мл трехгорлой колбе, в которую последовательно добавляли ацетонитрил (15 мл), бензол (4 мл), катализатор (0.2 г) и лимонную кислоту (0.075 г). Затем по каплям добавляли 30% H2O2 (12.5 мл) при нагревании до 70°C на водяной бане, снабженной механической мешалкой. Продолжительность реакции составляла 3 ч. Полученный продукт остужали и центрифугировали. Небольшую аликвоту супернатанта анализировали на газовом хроматографе GC-7890II (“Agilent Technologies Inc.”, США) в токе азота высокой чистоты в качестве газа-носителя на капиллярной колонке SE-30 (50 м × × 0.53 мм × 1 мкм) в режиме ПИД. Температуру поддерживали на уровне 60°C в течение 3 мин, повышали до 200°C со скоростью 15°C/мин и поддерживали на уровне 200°C в течение 1 мин. Температуру шприца и детектора устанавливали величиной 240°C. Объем инъекции микрошприца составлял 0.2 мкл. Для расчета конверсии бензола и селективности фенола использовали метод нормализации площадей.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Характеристики катализатора
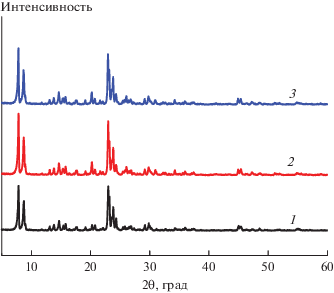
Рентгеновские дифрактограммы HZSM-5 и катализатора 6% V2O5/HZSM-5 до и после реакции показаны на рис. 1. В спектрах всех образцов видны пять отчетливых дифракционных пиков при значениях 2θ, равных 7.84°, 8.74°, 22.92°, 23.86° и 24.39°, что соответствует стандартным положениям пиков HZSM-5 и указывает на то, что кристаллическая структура образцов не изменяется после реакции [3, 9]. Характерные дифракционные пики оксида ванадия не обнаружены на дифрактограмме катализатора V2O5/HZSM-5, что позволяет предположить, что большая часть оксида ванадия равномерно диспергирована на поверхности HZSM-5.
Рис. 1.
Рентгенограммы катализаторов: 1 – HZSM-5, 2 – 6%V2O5/HZSM-5 (до реакции), 3 – 6%V2O5/HZSM-5 (после реакции).
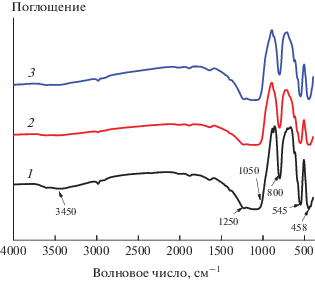
Фурье-ИК-спектры катализаторов HZSM-5 и 6%V2O5/HZSM-5 до и после реакции показаны на рис. 2. Характерная инфракрасная спектральная область колебаний каркаса находится в диапазоне 400~1600 см–1 для всех образцов. Пики ИК-поглощения для молекулярного сита HZSM-5 наблюдали вблизи 458, 545, 800, 1050 и 1250 см–1 [18], причем пик поглощения при 458 см–1 был отнесен к деформационному колебанию связи Si–O [19]. Пик поглощения около 545 см–1 характерен для сдвоенного кольца. Пик поглощения вблизи 800 см–1 обусловлен симметричным валентным колебанием связи Si–O–Si. Полоса поглощения при 1050~1250 см–1 является внешним или внутренним антисимметричным пиком валентных колебаний [20]. В инфракрасном спектре катализаторов отсутствуют пики поглощения, принадлежащие оксиду ванадия, что указывает на то, что оксид ванадия не входит в каркас катализатора. В то же время интенсивность валентного колебания ОН-групп при 3450 см–1 для катализатора 6%V2O5/HZSM-5 уменьшилась вследствие введения ванадия.
Рис. 2.
Фурье-ИК-спектры катализаторов: 1 – HZSM-5, 2 – 6%V2O5/HZSM-5 (до реакции), 3 – 6%V2O5/HZSM-5 (после реакции).
SEМ- и TEМ-изображения иерархического HZSM-5 и катализатора 6%V2O5/HZSM-5 показаны на рис. 3. Очевидно, что частицы иерархического цеолита HZSM-5 имеют форму лепестка с относительно шероховатой поверхностью. Отметим, что морфологии катализатора 6%V2O5/HZSM-5 и иерархического HZSM-5 очень похожи. Это свидетельствует о том, что введение оксида ванадия не приводит к нарушению морфологии цеолита при гидротермальном синтезе катализатора. Оксид ванадия не виден на внешней поверхности катализатора, что может быть связано с низким содержанием VOx или с высокой степенью дисперсности VOx на поверхности катализатора.

Элементные компоненты катализаторов были проанализированы с помощью метода TEM-EDS, чтобы определить, был ли оксид ванадия введен в катализатор. Результаты показаны на рис. 4. Очевидно, что катализатор содержит ванадий. Из картографической карты для катализатора 6%V2O5/HZSM-5 (рис. 5) можно видеть, что ванадий равномерно распределен по поверхности цеолита HZSM-5 [21, 22]. Это соответствует результатам характеризации методом XRD.

Адсорбцию–десорбцию N2 и данные ICP использовали для определения физической структуры катализатора и фактической загрузки ванадия с целью систематического анализа структуры и характеристик катализатора. Можно видеть, что кривые адсорбции–десорбции в областях низкого и среднего давления отклоняются вверх от изотерм адсорбции–десорбции N2 (рис. 6), что указывает на то, что в катализаторе имеется определенное количество микропор. Диаграмма распределения пор по размерам (вставка) показывает, что размер большей части микропор близок к 1.7 нм. Петля гистерезиса наблюдается между 0.45~0.99, что указывает на то, что в катализаторе все еще существует определенное количество мезопор (преимущественно 3.2 нм), которые могут быть причиной капиллярной конденсации. Для катализаторов характерны изотермы типа IV как до, так и после нанесения металла, а также наличие петель гистерезиса типа Н2 [8, 23]. Кривая адсорбции–десорбции и распределение пор катализатора по размерам сильно изменяются после реакции. Она соответствует изотерме типа IV с петлей гистерезиса типа H3. Кривая адсорбции–десорбции сильно отклоняется по оси y в зоне низкого давления, что в основном связано с резким уменьшением количества микропор. Накопление пор, вызванное серьезной агломерацией катализатора, приводило бы к сильному повышению адсорбционной способности в зоне высокого давления. Как видно из табл. 1, удельная поверхность, объем пор и размер пор значительно уменьшаются после нанесения оксида ванадия. В мезопористых каналах цеолита HZSM-5 присутствуют частицы различных оксидов ванадия (VOx) и частицы V2O5 в виде кластеров или кристаллических фаз. Возможно, они распределены на внутренней и внешней поверхностях катализатора, что может быть причиной блокировки каталитических каналов [11, 17]. Удельная поверхность и объем пор катализатора сильно уменьшаются после реакции, что в основном вызвано попаданием органических веществ в поры катализатора и осаждением их на поверхности катализатора во время реакции. Однако удельная площадь поверхности и объем пор катализатора до некоторой степени увеличиваются после прокаливания, приводящего к удалению части органического вещества. Фактическое содержание ванадия в катализаторе 6%V2O5/HZSM-5 было 4.74%. К сожалению, после реакции содержание ванадия в катализаторе сильно изменяется, и удаление органических веществ с поверхности катализатора путем прокаливания не приводит к значительному увеличению содержания ванадия, что в основном связано с потерей активных центров во время реакции.
Рис. 6.
Изотермы адсорбции–десорбции N2 и распределение пор по размерам (вставка) для HZSM-5 (a), 6%V2O5/HZSM-5 (б), 6%V2O5/HZSM-5 (без прокаливания после реакции) (в) и 6%V2O5/HZSM-5 (прокаливание после реакции) (г).
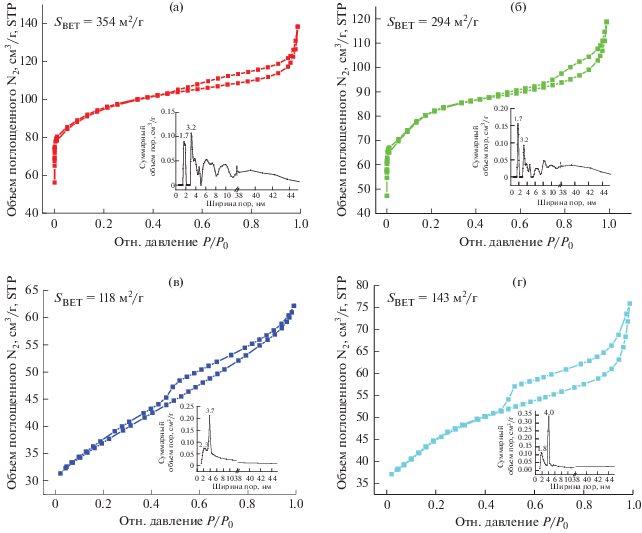
Таблица 1.
Физико-химические и химические свойства V-содержащих катализаторов
| Катализаторы | Содержание V (%) | Площадь поверхности (м2/г) | Объем пор (cм3/г) | ||||
|---|---|---|---|---|---|---|---|
| теорет. | реальное | БЭTe | микропорыf | внешняяf | микропорыf | общийg | |
| HZSM-5a | 0 | 0 | 354 | 285 | 69 | 0.12 | 2.14 |
| 6%V2O5/HZSM-5a | 6 | 4.74 | 294 | 240 | 54 | 0.10 | 1.83 |
| 6%V2O5/HZSM-5b, c | — | 3.75 | 118 | 60 | 58 | 0.03 | 0.09 |
| 6%V2O5/HZSM-5b, d | — | 3.97 | 143 | 92 | 51 | 0.05 | 0.10 |
На рис. 7 показана подгоночная кривая XPS ванадия для катализатора 6% V2O5/HZSM-5 до и после реакции. Область V2p катализатора содержит два пика при 521.43 и 517.43 эВ из-за связи орбитального и спинового моментов движения электронов, которые были приписаны соответственно V2p1/2 и V2p3/2 [24, 25]. Из рис. 7а следует, что ванадий в основном присутствует в форме V5+. Положение пика ванадия значительно изменяется после реакции (рис. 7б), т.е.: (1) на поверхности катализатора присутствует ванадий в двух валентных состояниях – V4+ (516.02 эВ в области V2p3/2) и V5+ (516.93 эВ в области V2p3/2 и 520.31 эВ в области V2p1/2); (2) пики с центрами при 522.75 и 524.15 эВ являются интерференционными пиками, генерируемыми другими элементами; (3) после реакции все пики смещаются в сторону низких энергий связи, что указывает на то, что часть V5+ восстанавливается до V4+ в ходе реакции [16, 26]. Кроме того, слабый темно-зеленый цвет раствора после реакции косвенно также подтверждает образование V4+. На основании многочисленных данных из литературных источников [23–25] можно предположить, что изменение цвета во время реакции соответствует первоначальному восстановлению V5+ до V4+ под действием H2O2, а затем бензол и H2O2 превращаются в фенол с участием восстановленного катализатора. В итоге V4+ окисляется до V5+ для завершения каталитического цикла. Цикл можно представить следующим образом [27]:
(1)
${{{\text{V}}}^{{5 + }}} + {{{\text{H}}}_{{\text{2}}}}{{{\text{O}}}_{2}} \to {{{\text{V}}}^{{4 + }}} + {{{\text{H}}}^{ + }} + {\text{HOO}},$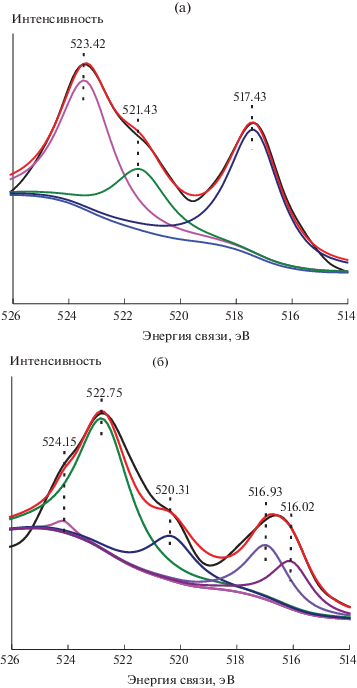
Каталитическая эффективность
Каталитическая активность катализаторов на основе ванадия для гидроксилирования бензола до фенола приведена в табл. 2. Как видно, катализатор HZSM-5 малоактивен, что обусловлено, главным образом, отсутствием активных центров в катализаторе. Кроме того, на активность катализатора также влияет источник ванадия. Структура пор цеолита также играет значительную роль в каталитической реакции, поскольку она ограничивает массоперенос реагентов в структуре микропор HZSM-5, тогда как наличие структуры мезопор способствует проникновению реагентов в поры. В то же время образующийся фенол может быстро десорбироваться с катализатора, чтобы не подвергаться дальнейшему окислению. Поскольку ванадий является активным центром каталитической системы, увеличение его содержания способствует росту выхода фенола. Однако при повышенных концентрациях ванадия каналы цеолита блокируются, что может легко вызвать агломерацию оксидов металлов на поверхности катализатора [9, 16].
Таблица 2.
Каталитическая активность катализаторов*
| Катализаторы | Конверсия бензола | Селективность фенола | Выход фенола |
|---|---|---|---|
| (%) | |||
| HZSM-5 | 0.3 | 49.7 | 0.15 |
| 6%V2O5/HZSM-5 | 73.5 | 99.0 | 72.77 |
Для достижения максимальной конверсии бензола и селективности фенола были найдены оптимальные условия реакции в этой каталитической системе. Конверсия бензола достигала 73.5%, а селективность фенола была более 99.0% в оптимальных условиях реакции: содержание WV – 6%, масса катализатора – 0.2 г, бензол – 4 мл, 30%-ный раствор пероксида водорода – 12.5 мл, ацетонитрил – 15 мл, лимонная кислота – 0.075 г, T = 70°C, время реакции – 3 ч. В результате более глубокого окисления фенола в реакции гидроксилирования бензола были также обнаружены небольшие количества побочных продуктов (гидрохинона и п‑бензохинона). Повышение степени конверсии бензола почти на 30% по сравнению с опубликованными литературными данными [1, 8, 10] является основным достижением данной работы. Высокая степень конверсии на катализаторе объясняется следующими основными причинами. Во-первых, присутствующие в цеолите мезопоры способствуют быстрой реакции реагентов с активным центром и увеличивают скорость десорбции фенола. Во-вторых, использование лимонной кислоты обеспечивает подходящую каталитическую среду для реакции гидроксилирования бензола, что благоприятно сказывается на повышении селективности по фенолу. Наконец, ключевым фактором является сам метод синтеза катализатора.
Повторное использование катализатора
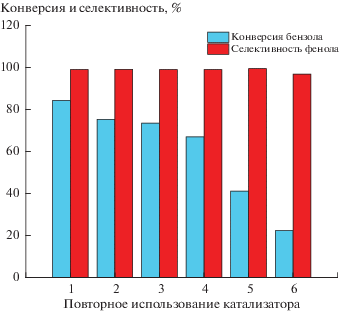
Помимо хороших каталитических характеристик, возможность выделения и повторного использования катализатора также являются важными показателями для оценки преимуществ катализаторов. Как показано на рис. 8, селективность по фенолу не изменяется после 5 циклов, снижается только выход фенола. Основной причиной снижения степени конверсии бензола является потеря активного компонента согласно данным ICP для катализатора 6%V2O5/HZSM-5 до и после реакции (табл. 3). Некоторые компоненты в реакционной системе адсорбируются на поверхности или в порах катализатора на его активных центрах, что приводит к уменьшению эффективной удельной поверхности, тем самым влияя на каталитическую активность катализатора. Следовательно, чтобы продлить срок службы использованного катализатора, его нужно соответствующим образом активировать. Так, использованный катализатор высушивают при 110°C в течение 12 ч и прокаливают при 550°C в течение 6 ч. Для следующего цикла каталитической реакции также необходимо добавить ~0.02 г свежего катализатора.
ВЫВОДЫ
Таким образом, катализатор V2O5/HZSM-5, приготовленный методом гидротермального синтеза, показал хорошую активность и селективность в реакции прямого гидроксилирования бензола до фенола с H2O2 в качестве окислителя, с ацетонитрилом в качестве растворителя и лимонной кислотой в качестве добавки. Это может быть обусловлено не только очень однородным распределением активных ванадиевых центров и различных форм ванадия, но и иерархической структурой пор цеолита HZSM-5, которая способствует быстрой диффузии реагентов и продуктов. Кроме того, присутствие лимонной кислоты может способствовать образованию ионов V4+ в реакции. Конверсия бензола составила 73.5%, а селективность по фенолу превышала 99.0% за счет оптимизации условий для катализатора 6%V2O5/HZSM-5. Стабильность катализатора также была улучшена.
Список литературы
Zhou Y., Ma Z.P., Tang J.J., Yan N., Du Y.H., Xi S.B., Wang K., Zhang W., Wen H.M., Wang J. // Nat. Commun. 2018. V. 9. № 1. P. 2931.
Xu J., Chen Y., Hong Y., Zheng H., Ma D., Xue B., Li Y.H. // Appl. Catal. A. Gen. 2018. V. 549. P. 31.
Shahid A., Lopez-Orozco S., Marthala V.R., Hartmann M., Schwieger W. // Micropor. Mesopor. Mater. 2017. V. 237. P. 151.
Zhang F.M., Chen X., Zhuang J., Xiao Q., Zhong Y.J., Zhu W.D. // Catal. Sci. Technol. 2011. V. 1. № 7. P. 1250.
Li L.L., Meng Q.L., Wen J.J., Wang J.G., Tu G.M., Xu C.H., Zhang F.M., Zhong Y.J., Zhu W.D., Xiao Q. // Micropor. Mesopor. Mater. 2016. V. 227. P. 252.
Wang C., Hu L.Y., Wang M.Y., Ren Y.H., Yue B., He H.Y. // Chinese J. Catal. 2016. V. 37. № 11. P. 2003.
Chen X., Zhao W.G., Wang F., Xu J. // J. Nat. Gas Chem. 2012. V. 21. № 5. P. 7.
Hu L.Y., Wang C., Yue B., Chen X.Y., He H.Y. // RSC Adv. 2016. V. 504. P. 6.
Xiao P.P., Wang Y., Kondo J.N., Yokoi T. // Appl. Catal. A. Gen. 2019. V. 579. P. 159.
Yang Y.W., Tang R.R. // Res. Chem. Intermediat. 2018. V. 44. № 10. P. 5911.
Cui O.Y., Li Y.X., Li J.W. // Catalysts. 2019. V. 9. № 1. P. 44.
Shahami M., Dooley K.M., Shantz D.F. // J. Catal. 2018. V. 368. P. 354.
Xiao P.P., Wang Y., Kondo J.N., Yokoi T. // Chem. Lett. 2018. V. 47. P. 1112.
Qi X.Y., Li J.Y., Ji T.H., Wang Y.J., Feng L.L., Zhu Y.L., Fan X.T., Zhang C. // Micropor. Mesopor. Mater. 2009. V. 122. P. 36.
Xu J., Jiang Q., Chen T., Wu F., Li Y.X. // Catal. Sci. Technol. 2015. V. 5. № 3. P. 1504.
Xu D., Jia L.H., Guo X.F. // Catal. Lett. 2012. V. 142. № 10. P. 1251.
Hu L.Y., Yue B., Chen X.Y., He Y.H. // Catal. Commun. 2014. V. 43. P. 179.
Phu N.H., Hoa T.T.K., Tan N.V., Thang H.V., Ha P.L. // Appl. Catal. B. Environ. 2001. V. 34. P. 267.
Deepanjan Sen, Hina Tasnim M., Jayasimha K.N., Suman Pawar, Nagaraju N. // Res. J. Chem. Environ. Sci. 2016. V. 4. P. 134.
Liu Y., Yu X.B., Qin L., Wang J.D., Yang Y.R. // Chine Pet. Process. PE. 2010. V. 1. P. 23.
Makgwane P.R., Ray S.S. // Appl. Catal. A. Gen. 2015. V. 492. P. 10.
Chen P., Chen L., Zeng Y., Ding F., Jiang X., Liu N., Au C.T., Yin S.F. // Appl. Catal. B. Environ. 2018. V. 234. P. 311.
Hu L.Y., Wang C., Yue B., Chen X.Y., He H.Y. // Mater. Today Commun. 2017. V. 11. P. 61.
Hu L.Y., Wang C., Ye L., Wu Y.N., Yue B., Chen X.Y., He H.Y. // Appl. Catal. A. Gen. 2015. V. 504. P. 440.
Xu J., Jiang Q., Chen T., Wu F., Li Y.X. // Catal. Sci. Technol. 2015. V. 5. № 3. P. 1504.
Xu J., Hong Y., Cheng M.J., Xue B., Li Y.X. // Micropor. Mesopor. Mater. 2019. V. 285. P. 223.
Ding G.D., Wang W.T., Jiang T., Han B.X., Fan H.L., Yang G.Y. // ChemCatChem. 2013. V. 5. P. 192.
Дополнительные материалы отсутствуют.
Инструменты
Кинетика и катализ