

Кинетика и катализ, 2021, T. 62, № 5, стр. 569-580
Синергетический эффект добавки оксида железа в состав носителя Pt/TiO2-катализатора в реакции окисления CO
А. А. Шутилов a, *, Г. А. Зенковец a
a ФГБУН ФИЦ Институт катализа им. Г.К. Борескова СО РАН
630090 Новосибирск, просп. Акад. Лаврентьева, 5, Россия
* E-mail: alshut@catalysis.ru
Поступила в редакцию 29.12.2020
После доработки 11.05.2021
Принята к публикации 19.05.2021
Аннотация
Исследовано влияние добавок оксида железа на формирование микроструктуры, электронное состояние платины и каталитические свойства нанесенных катализаторов 1 вес. % Pt/(Fe2О3–TiO2) в реакции окисления СО. При одинаковом содержании платины полученный катализатор характеризуется более высокой активностью по сравнению с катализатором 1 вес. % Pt/TiO2. Установлено, что при введении оксида железа в количестве до 10 вес. % формируется твердый раствор ионов железа в диоксиде титана со структурой анатаза. Нанесение платины приводит к уменьшению размера частиц и увеличению содержания платины в состоянии Ptδ+. Определен оптимальный химический состав катализатора, обеспечивающий наиболее высокую активность в исследуемой реакции.
Графический реферат
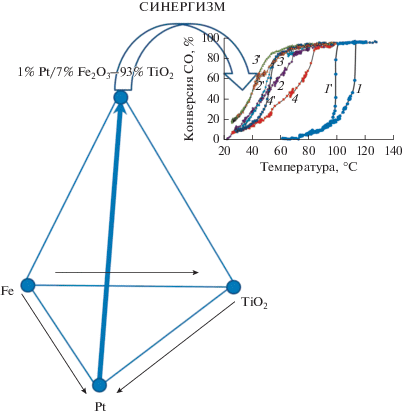
ВВЕДЕНИЕ
Катализаторы Pt/TiO2 проявляют достаточно высокую активность в реакции низкотемпературного окисления CO, для очистки воздуха, для очистки H2-содержащих смесей, использующихся в топливных элементах [1, 2] и фотокаталитического окисления вредных органических веществ [3–5].
Для увеличения активности Pt/TiO2-катализаторы модифицируют оксидами переходных элементов, среди которых оксид железа является одним из наиболее эффективных. Показано, что активность катализаторов в значительной степени зависит от количества введенного железа и метода его нанесения [6].
Известно, что каталитические свойства нанесенных платиновых катализаторов определяются размером частиц и электронным состоянием платины, распределением ее по поверхности [7–11]. В работе [6] PtFe/TiO2-катализаторы, содержащие 1 вес. % Pt при мольном соотношении Fe : Pt = = 0.2–1, были приготовлены пропиткой TiO2 по влагоемкости растворами ацетилацетонатов платины и железа. Показано, что при добавлении железа до соотношения Fe : Pt = 0.2 активность катализатора в реакции окисления СO повышается, а при дальнейшем возрастании концентрации железа снижается.
Как было продемонстрировано в [12], в катализаторах PtFe/TiO2, содержащих 1 вес. % Pt и 0.03–0.55 вес. % Fe и полученных пропиткой TiO2 водными растворами азотнокислой платины и железа, активность сначала увеличивается с ростом содержания железа до 0.28 вес. %, а затем при дальнейшем его повышении до 0.55 вес. % Fe снижается.
В реакции CO-PROX, наибольшей активностью характеризуются катализаторы Pt/(MOх)/ TiO2, где (M = Fe, Co), в которых Pt наносится на диоксид титана, предварительно модифицированный Fe или Co [2]. Было показано, что во всех PtFe/TiO2-катализаторах между атомами Pt и Fe (или Со) не происходит образования химической связи и формирования биметаллических частиц Pt–Fe. Авторы полагают, что в данном случае имеет место синергетический эффект: частицы оксида железа в высокодисперсном состоянии локализуются между частицами Pt и TiO2, создавая места для адсорбции и активации молекулярного O2, и промотируют реокисление Pt.
В работах [6, 13, 14] было обнаружено, что химическая природа носителя оказывает большое влияния на формирование активного компонента катализатора. Методом EXAFS установлено образование биметаллических Pt–Fe-частиц в нанесенных катализаторах PtFe/SiO2, в то время как в нанесенных PtFe/Al2O3 и PtFe/TiO2 они не образуются.
Целью настоящей работы является исследование влияния модифицирования нанесенных Pt/TiO2-катализаторов добавками оксида железа на формирование их структуры и каталитические свойства в реакции окисления CO.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
При получении катализаторов 1 вес. % Pt/TiO2 и 1 вес. % Pt/(1–10 вес. % Fe2O3–99–90 вес. % TiO2) сначала были приготовлены образцы носителей TiO2 и TiO2, допированного оксидом железа, в количестве 1–10 вес. %. В качестве исходного соединения титана использовали ксерогель TiO2 (анатаз), синтезированный по промышленной сернокислотной технологии. Как было показано ранее [5, 15], ксерогель состоит из крупных агрегатов (80–100 нм) рыхло упакованных высокодисперсных частиц с размером 3–6 нм. Диоксид титана в виде фракции 0.25–0.5 мм пропитывали по влагоемкости водным раствором азотнокислого железа с последующей сушкой на воздухе и прокаливанием в муфельной печи при температуре 500°С в течение 4 ч. Затем полученные носители пропитывали по влагоемкости водным раствором азотнокислой платины, сушили на воздухе и снова прокаливали в муфельной печи при 500°С 2 ч.
Рентгенографические исследования образцов проводили на дифрактометре D8 (“Bruker”, Германия) с монохроматизированным CuKα-излучением. Съемки рентгенограмм осуществляли в режиме сканирования в области 2θ = 15°–90° с шагом 0.05° и временем накопления в каждой точке 5 с. Размер кристаллических частиц анатаза оценивали по размеру областей когерентного рассеяния (Dокр), рассчитанных по формуле Селякова−Шерера, с использованием дифракционного пика 2.0.0. [16]. Параметры кристаллической решетки TiO2 (анатаза) уточняли с помощью программы “Поликристалл” по положениям максимумов 3−7 дифракционных пиков [17].
Состояние железа в образцах диоксида титана исследовали методом мессбауэровской спектроскопии. Спектры получали на спектрометре NZ-640 (“Tata Hűtőtechnika”, Венгрия), работающем в режиме постоянных ускорений с реверсом скорости, в режиме движущегося источника излучения Co в матрице металлического родия. Химические сдвиги приведены относительно α-Fe. Спектры регистрировали при комнатной температуре.
Изучение образцов методом просвечивающей электронной микроскопии высокого разрешения (ПЭМ-ВР) выполняли на микроскопе JEM 2010 (“Jeol”, Япония), при ускоряющем напряжении 200 кВ и разрешении по решетке 1.4 Å.
Удельную поверхность образцов (Sуд) определяли методом БЭТ [18] по термодесорбции аргона с применением прибора СОРБИ-М (“MЕТА”, Россия).
Термический анализ образцов проводили на анализаторе STA 449C Jupiter (“NETZSCH”, Germany). Образец нагревали на воздухе со скоростью 10°C/мин.
Рентгеновские фотоэлектронные спектры (РФЭС) записывали на спектрометре SPECS (“SPECS Gmbh”, Германия) с использованием излучения AlKα (hν = 1486.6 эВ, 205 Вт). Шкала энергий связи (Есв) была откалибрована по пикам Au4f7/2 (84.0 эВ) и Cu2p3/2 (932.67 эВ). Эффект подзарядки, возникающий в процессе фотоэмиссии электронов, учитывали с помощью метода внутреннего стандарта, которым служила линия C1s (Eсв = 284.8 эВ), соответствующая поверхностным углеводородным соединениям. Все спектры регистрировали при энергии пропускания анализатора 20 эВ с шагом 0.1 эВ. Разложение фотоэлектронных пиков Pt4f на отдельные спектральные компоненты проводили с помощью программы XPS Peak 4.1 [19] с вычитанием фона по методу Ширли [20]. Контуры пиков аппроксимировали функциями Гаусса и Лоренца, при этом константа спин-орбитального расщепления составляла 3.35 эВ, соотношение интенсивностей пиков Pt4f5/2/Pt4f7/2 принимали как 3/4, ширина линий на половине высоты была равна 1.9–2.1 эВ.
Исследование каталитических свойств образцов в реакции окисления оксида углерода осуществляли на проточно-циркуляционной установке BI-CATr(oxy) (Россия) [21] при постепенном повышении и последующем снижении температуры реакционной смеси. Основными частями блока реактора являются вертикально расположенный трубчатый металлический реактор и циркуляционный насос, соединенные в проточно-циркуляционный контур. Реактор состоит из двух частей – корпуса и съемного стакана. Образец катализатора в виде фракции 0.25–0.5 мм фиксировали на сборке, включающей металлическую решетку и фильтр. Температуру реакции измеряли термопарой, подведенной снизу к слою катализатора. Сигнал с термопары передавался на автоматическую цифровую плату, интегрированную в блок управления установки. Температуру реакции меняли в ручном режиме с шагом 1.7°С (погрешность не более 0.2°С).
Испытания проводили при следующем составе реакционной смеси: 1 об. % СО, 10 об. % О2, остальное − азот. Скорость подачи реакционной смеси составляла 4.46 × 10–3 моль/мин, навеска катализатора − 0.375 г, скорость циркуляции реакционной смеси − 1000 л/ч, кратность циркуляции − 167, что достаточно для установления в реакторе режима идеального смешения, т.е. достижения термической и концентрационной безградиентности в слое катализатора [22].
Состав реакционной смеси до и после реакции анализировали на встроенном в установку хроматографе MSR (ООО “СИБМИКРОРЕАКТОР”, Россия), оснащенном разделительной колонкой (300 × 3 × 2 мм), заполненной углем марки СКТ, и термокаталитическим детектором. Концентрацию CO2 определяли с помощью газоанализатора ТЕСТ-1 ( ООО “БОНЕР”, Россия).
Активность катализаторов характеризовали по конверсии СО в зависимости от температуры и скорости его превращения (W, мольСО${\text{г}}_{{{\text{кат}}}}^{{ - {\text{1}}}}$ мин–1). Скорость реакции рассчитывали при температуре 35°С, при которой конверсия CO не превышает 30%, по формуле:
где V − скорость подачи реакционной смеси (моль/мин), m – навеска катализатора (г), С0 – концентрация СО в исходной реакционной смеси (об. доли), С – концентрация СО в реакционной смеси при 35°С (об. доли).РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
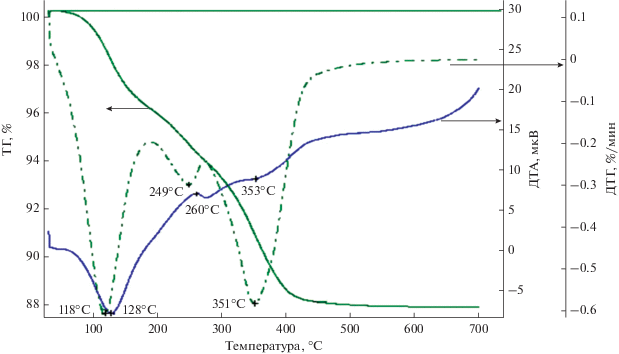
На рис. 1 приведены данные термического анализа образца 7 вес. % Fe2O3–93 вес. % TiO2, из которых можно видеть его формирование при термообработке. На кривой ДТА в области температур 118–128°С регистрируется эндоэффект, сопровождающийся потерей веса на кривой ТГ, обусловленный дегидратацией. В области температур 250–350°C зафиксированы эндоэффекты, сопровождающиеся потерей веса и обусловленные, по-видимому, разложением железа азотнокислого и его взаимодействием с диоксидом титана. В области температур 420–700°С никаких термоэффектов не обнаружено и изменения массы образца не происходит. Можно полагать, что при вышеуказанном способе приготовления формирование фазового состава образца завершается при 420°C, и при дальнейшем повышении температуры вплоть до 700°С его фазовый состав не меняется.
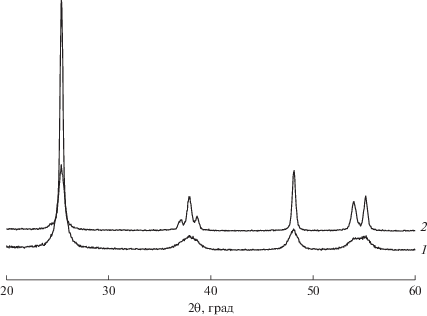
На рентгенограммах образцов носителей (1–10 вес. % Fe2O3–99–90 вес. % TiO2), прокаленных при 500°С, регистрируется только фаза анатаза; никаких примесей других окристаллизованных фаз не наблюдается (рис. 2). При этом в указанных образцах размер Dокр анатаза уменьшается по сравнению с немодифицированным TiO2 от 20 до 10–11 нм, а параметры кристаллической решетки анатаза (а и с) несколько возрастают при введении достаточно большого количества оксида железа (табл. 1). На основании полученных данных можно полагать, что при введении оксида железа формируется твердый раствор ионов железа, стабилизированных в решетке анатаза. Сравнительно небольшое изменение параметров решетки обусловлено тем, что радиусы ионов Fe3+ и Ti4+ близки и составляют соответственно 0.67 и 0.64 Å. Отметим, что при прокаливании при температуре 700°С во всех образцах, допированных оксидом железа, также регистрируется анатаз (рис. 3), что свидетельствует о высокой термостабильности образцов и согласуется с данными термического анализа. Использование в качестве исходного соединения титана ксерогеля высокодисперсных частиц анатаза с дефектной структурой, объединенных в крупные пористые агрегаты [23–25], позволяет получать твердые растворы ионов железа в TiO2 (анатаз) с высоким содержанием оксида железа. В ходе синтеза пористые агрегаты равномерно заполняются водным раствором железа азотнокислого, и на последующих стадиях сушки и термообработки происходит спекание кристаллитов анатаза с одновременной стабилизацией в их структуре ионов железа.
Рис. 2.
Рентгенограммы образцов носителей, прокаленных при температуре 500ºС: 1 – 3 вес. % Fe2О3–97 вес. % TiO2; 2 – 5 вес. % Fe2О3–95 вес. % TiO2; 3 – 7 вес. % Fe2О3–93 вес. % TiO2; 4 – 10 вес. % Fe2О3–90 вес. % TiO2.
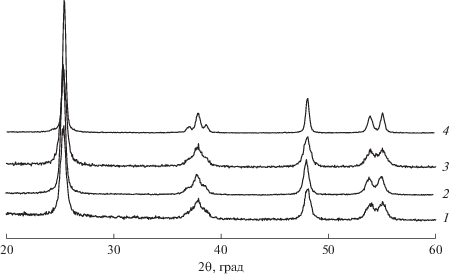
Таблица 1.
Структурные параметры образцов TiO2 (анатаза), модифицированного оксидом железа, прокаленных при температуре 500°С
| Измеряемый параметр | Химический состав, вес. % | ||
|---|---|---|---|
| TiO2 | 7% Fe2O3–93% TiO2 | 10% Fe2O3–90% TiO2 | |
| Dокр анатаза, нм | 16 | 11 | 10 |
| Параметры решетки фазы анатаза, Å | а = b = 3.786 c = 9.518 |
а = b = 3.795 c = 9.530 |
а = b = 3.794 c = 9.524 |
Рис. 3.
Рентгенограммы образца носителя 7 вес. % Fe2О3–93 вес. % TiO2, прокаленного при температуре: 500 (1) и 700°С (2).
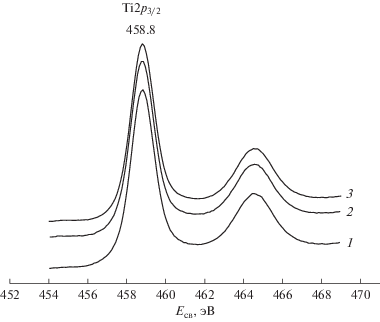
На рис. 4 представлен РФЭ-спектр TiO2 и спектры железо-титановых образцов 5 вес. % Fe2O3–95 вес. % TiO2 и 10 вес. % Fe2O3–90 вес. % TiO2, прокаленных при 500°C. Видно, что значения энергии связи пиков Ti2p3/2 в этих образцах одинаковы (458.8 эВ), и соответствуют состоянию Ti4+ в TiO2 [19].
Рис. 4.
РФЭ-спектры области Ti2p3/2 для образцов, прокаленных при 500°C: 1 – TiO2; 2 – 5 вес. % Fe2О3–95 вес. % TiO2; 3 – 10 вес. % Fe2О3–90 вес. % TiO2.
Мессбауэровские спектры образцов 5 вес. % Fe2O3–95 вес. % TiO2 и 10 вес. % Fe2O3–90 вес. % TiO2, прокаленных при 500°C, показаны на рис. 5. Спектры разложены на два симметричных дублета (D1 и D2), параметры которых приведены в (табл. 2). Полученные значения величин изомерных сдвигов всех дублетов соответствуют высокоспиновому состоянию Fe3+. При этом состояний, относящихся к Fe2+, и состояний с магнитным расщеплением, которые могли бы быть отнесены к магнитоупорядоченным оксидам железа, в спектрах не наблюдается [26].
Рис. 5.
Мессбауэровские спектры образцoв 5 вес. % Fe2О3–95 вес. % TiO2 (а) и 10 вес. % Fe2О3–90 вес. % TiO2 (б), прокаленных при 500°C.
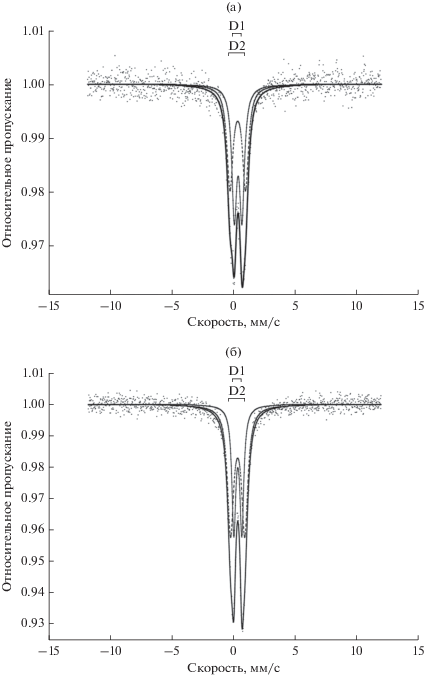
Таблица 2.
Параметры Мессбауэровских спектров образцов 5 вес. % Fe2О3–95 вес. % TiO2 и 10 вес. % Fe2О3–90 вес. % TiO2
| Образец, вес. % | Is, мм/с | Qs, мм/с | G, мм/с | Состояние железа | S, отн. % |
|---|---|---|---|---|---|
| 5% Fe2O3–95% TiO2 | 0.394 | 0.62 | 0.47 | [Fe3+ O6], D1 | 50 |
| 0.363 | 1.25 | 0.59 | [Fe3+ O5Vo], D2 | 50 | |
| 10% Fe2O3–90% TiO2 | 0.369 | 0.65 | 0.38 | [Fe3+ O6], D1 | 38 |
| 0.354 | 1.15 | 0.60 | [Fe3+ O5Vo], D2 | 62 |
Согласно данным [27] можно предположить, что дублет D1, характеризующийся меньшими величинами квадрупольного расщепления Qs = = 0.65 и Qs = 0.62 мм/с и изомерными сдвигами Is = 0.394 и Is = 0.369 мм/c, для образцов, содержащих 5 и 10 вес. % Fe2O3 соответственно, может быть связан с образованием твердого раствора замещения, в котором ионы Fe3+ замещают ионы Ti4+, находящиеся в несколько искаженном октаэдрическом кислородном окружении [Fe3+ O6].
Второй дублет D2 характеризуется большими величинами квадрупольного расщепления Qs = = 1.25 и Qs = 1.15 мм/с и изомерными сдвигами Is = 0.363 и Is = 0.354 мм/c соответственно для образцов, содержащих 5 и 10 вес. % Fe2O3. Этот дублет может быть также отнесен к ионам Fe3+, которые замещает ионы Ti4+ в структуре анатаза, но имеют кислородную вакансию в ближайшем своем окружении [Fe3+O5Vo]. С увеличением содержания оксида железа в TiO2 от 5 до 10 вес. % доля данного состояния возрастает.
Следовательно, при допировании диоксида титана оксидом железа формируется твердый раствор со структурой анатаза, в котором ионы титана находятся в состоянии Ti4+ в узельных позициях решетки анатаза в несколько искаженном октаэдрическом кислородном окружении. Ионы железа в состоянии Fe3+ частично замещают ионы Ti4+ в октаэдрическом и пятикоординированном кислородном окружении.
По данным РФА в катализаторах 1 вес. % Pt/(1–10 вес. % Fe2O3–99–10 вес. % TiO2) после прокаливания при температуре 500°С регистрируется анатаз с размером Dокр 10–11 нм. На электронно-микроскопическом снимке (рис. 6) наблюдаются однородные по размеру частицы платины (1.8–2.0 нм), распределенные достаточно равномерно по поверхности носителя. Оценка параметров кристаллической решетки частиц платины не выявила явного их изменения относительно стандартных значений металлической платины, в то время как в катализаторе 1 вес. % Pt/TiO2 этот показатель равен 1.5–5 нм [12]. Отметим, что после термообработки катализаторов при 500°С их удельная поверхность составляет около 100 м2/г.
Рис. 6.
Электронно-микроскопический снимок катализатора 1 вес. % Pt/(7 вес. % Fe2О3–93 вес. % TiO2), прокаленного при 500°C.

На рис. 7 для катализатора 1 вес. % Pt/(5 вес. % Fe2О3–95 вес. % TiO2) исходного и после работы в реакционной смеси приведено разложение РФЭ-спектров в области Pt4f на индивидуальные дублеты с параметрами спин-орбитального расщепления и интенсивностями компонент. Аналогичные разложения также были выполнены и для остальных катализаторов. В спектрах можно четко обозначить два основных состояния платины с Есв(Pt4f): дублет в области Есв(Pt4f7/2) = 71.2 эВ, соответствующий Pt0, и дублет в области Есв(Pt4f7/2) = 72.2 эВ, соответствующий Ptδ+. В табл. 3 дана количественная oценка состояний платины с Есв(Pt4f7/2) в исследованных образцах, из которой следует, что при одинаковом содержании платины ее электронное состояние в катализаторе изменяется в зависимости от количества введенного оксида железа. Так, в катализаторе Pt/TiO2 основным является состояние Pt0. При повышении содержания оксида железа до 7 вес. % регистрируется постепенное увеличение доли платины Ptδ+. Однако дальнейшее введение оксида железа до 10 вес. % приводит к снижению количества Ptδ+. После работы катализатора в реакционной смеси доля Ptδ+ возрастает по сравнению с исходным катализатором.
Рис. 7.
РФЭ-спектры Pt4f с их разложением на индивидуальные дублеты для катализатора 1 вес. % Pt/(5 вес. % Fe2О3–95 вес. % TiO2) исходного (1) и после реакции (2).
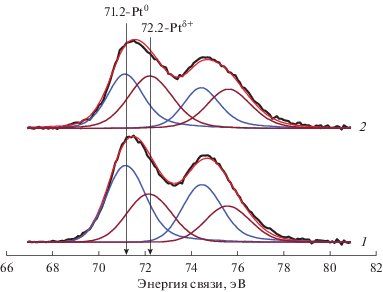
Таблица 3.
Состояния платины с Есв(Pt4f7/2) и их содержание в катализаторах, прокаленных при температуре 500°С, по данным РФЭС
| № | Катализатор, вес. % | Eсв(Pt4f7/2), эВ | Содержание, % | Ptδ+/Pt0 |
|---|---|---|---|---|
| 1 | 1% Pt/TiO2, исходный | 71.1 | 69.6 | 0.44 |
| 72.1 | 30.4 | |||
| 2 | 1% Pt/(5% Fe2О3–95% TiO2), исходный | 71.2 | 62.5 | 0.60 |
| 72.2 | 37.5 | |||
| 3 | 1% Pt/(5% Fe2О3–95% TiO2), после реакции | 71.2 | 47.6 | 1.1 |
| 72.2 | 52.4 | |||
| 3 | 1% Pt/(7% Fe2О3–95% TiO2), исходный | 71.2 | 42.5 | 1.35 |
| 72.2 | 57.5 | |||
| 4 | 1% Pt/(10% Fe2О3–90% TiO2), исходный | 71.1 | 50.5 | 0.98 |
| 72.1 | 49.5 |
В табл. 4 приведены результаты исследования каталитических свойств образцов в реакции окислении СO. Катализаторы, в состав которых входит оксид железа, демонстрируют более высокую активность по сравнению с катализатором 1 вес. % Pt/TiO2. Заметим, что для всех исследуемых образцов регистрируется явление гистерезиса. В сопоставимых условиях реакционной среды для катализатора 1 вес. % Pt/TiO2 температура 50% конверсии CO составляет 127°С при измерении по кривой нагрева и 109°С – по кривой охлаждения. В катализаторе 1 вес. % Pt/(7 вес. % Fe2О3–93 вес. % TiO2) температура 50% конверсии CO снижается до 38°C по кривой нагрева и до 37°C по кривой охлаждения. При дальнейшем повышении концентрации оксида железа до 10 вес. % наблюдается снижение активности катализатора (рис. 8) – температура 50% конверсии CO составляет 70°C по кривой нагрева и 53°C по кривой охлаждения. Оценка скорости реакции при температуре 35°C (когда конверсия CO не превышает 30%) для всех катализаторов показывает рост этого показателя при увеличении концентрации оксида железа в катализаторе до оптимального содержания 7 вес. %, а при дальнейшем его увеличении до 10 вес. % скорость реакции снижается.
Таблица 4.
Каталитические свойства образцов Pt/TiO2 и Pt/(Fe2О3–TiO2) с различным содержанием железа в реакции окисления СО
| Состав катализатора, вес. % | $T_{{x = 50\% }}^{1},$ °С | $T_{{x = 50\% }}^{2},$ °С | W × 103, мольСО${\text{г}}_{{{\text{кат}}}}^{{ - 1}}$ мин–1 |
|---|---|---|---|
| 1% Pt/TiO2 | 127 | 109 | 0 |
| 1% Pt/(1% Fe2О3–99% TiO2) | 57 | 46 | 1.1 |
| 1% Pt/(3.5% Fe2О3–96.5% TiO2) | 49 | 47 | 1.78 |
| 1% Pt/(5% Fe2О3–95% TiO2) | 47 | 46 | 2.07 |
| 1% Pt/(7% Fe2О3–93% TiO2) | 38 | 37 | 3.57 |
| 1% Pt/(10% Fe2О3–90% TiO2) | 70 | 53 | 1.19 |
Рис. 8.
Зависимость конверсии CO от температуры в реакции окисления CO при нагреве (1, 2, 3, 4) и охлаждении реакционной смеси (1', 2', 3', 4') для катализаторов: 1, 1' – 1 вес. % Pt/TiO2; 2, 2' – 1 вес. % Pt/(5 вес. % Fe2О3–95 вес. % TiO2); 3, 3' – 1 вес. % Pt/(7 вес. % Fe2О3–93 вес. % TiO2), 4, 4' – 1 вес. % Pt/(10 вес. % Fe2О3–90 вес. % TiO2).
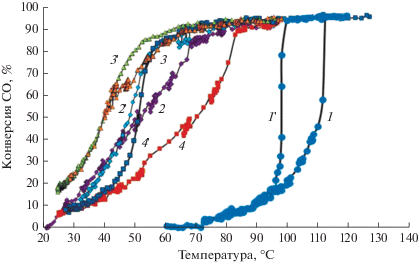
Как видно из данных табл. 4 и рис. 8, при возрастании содержания оксида железа в катализаторе от 1 до 7 вес. % ширина петли гистерезиса уменьшается, а при дальнейшем повышении до 10 вес. % снова увеличивается. На основании данных исследования катализаторов методом мессбауэровской спектроскопии можно предположить, что снижение активности катализатора с ростом количества оксида железа больше оптимального может быть обусловлено сокращением содержания кислорода в поверхностном слое за счет повышения концентрации кислородных вакансий. Уменьшение концентрации поверхностного кислорода при введении в катализатор оксида железа (более оптимального) может приводить к возрастанию прочности связи поверхностного кислорода и снижению подвижности решеточного кислорода, в результате каталитическая активность будет падать. Однако в настоящее время о природе активного состояния данного типа катализаторов в литературе нет однозначного мнения. Например, понижение активности диоксида титана наблюдалось в реакции фотокаталитического разложения метилоранжа при увеличении содержания оксида железа [27, 28]. В другом случае, например [29], наличие кислородных вакансий в катализаторе, представляющим собой диоксид титана, допированный оксидом цинка, приводило к увеличению активности в реакции фотокаталитического окисления CO. Поэтому эти предположения требуют дальнейшего исследования.
В литературе существуют две основные точки зрения, объясняющие явление гистерезиса. Считается, что под действием реакционной среды происходит изменение состояния катализатора, например, увеличение концентрации Ptδ+, которое затем длительное время сохраняется и при его охлаждении, обеспечивая высокую активность при понижении температуры [30]. По другой версии, температурный гистерезис имеет скорее физическую природу [31] и обязан своим происхождением локальному перегреву активных центров катализатора в результате выделения избыточного количества тепла в экзотермической реакции, что в нашем случае при использовании низкоконцентрированной смеси (1% CO) представляется маловероятным.
ЗАКЛЮЧЕНИЕ
Таким образом, в настоящей работе синтезированы нанесенные катализаторы 1 вес. % Pt/(Fe2O3–TiO2), которые характеризуются более высокой активностью в реакции окисления СО по сравнению с образцом 1 вес. % Pt/TiO2. Катализаторы приготовлены нанесением раствора азотнокислой платины на специально приготовленный носитель TiO2, допированный оксидом железа в количестве 1–10 вес. %, с последующей сушкой и прокаливанием на воздухе при температуре 500°С. Носитель представляет собой твердый раствор ионов Fe3+ в TiO2 со структурой анатаза, в котором ионы Fe3+, частично замещая ионы Ti4+, стабилизируются как в октаэдрическом, так и пятикоординированном кислородном окружении с образованием кислородной вакансии в его ближайшем окружении. С увеличением содержания оксида железа в носителе от 1 до 7 вес. % каталитическая активность возрастает. Дальнейшее повышение до 10 вес. % (больше оптимального) приводит к снижению активности, что, предположительно, может быть обусловлено значительным уменьшением содержания кислорода в поверхностном слое вследствие роста концентрации кислородных вакансий.
При нанесении платины в катализаторе Pt/(Fe2O3–TiO2) формируются частицы размером 1.8–2 нм, которые равномерно распределяются по поверхности носителя, в то время как в образце 1 вес. % Pt/TiO2 частицы платины имеют больший размер – 1.5–5 нм. Следовательно, при введении добавок оксида железа, как можно полагать, происходит изменение взаимодействия платины с носителем, что приводит к изменению электронного состояния платины (увеличение доли Ptδ+) и росту каталитической активности. При этом между атомами Pt и Fe не наблюдается образования интерметаллической связи. По-видимому, синергетический эффект добавки оксида железа в оптимальных количествах оказывает основное влияние на физико-химические взаимодействия платины с носителем, что способствует повышению каталитической активности катализаторов Pt/(Fe2O3–TiO2) в реакции окисления CO.
Список литературы
Bamwenda G.R., Tsubota S., Nakamura T., Haruta M. // Catal. Lett. 1997. V. 44. P. 88.
Navas-Cardenas C., Benito N., Wolf E.E., Gracia F. // Appl. Catal. A: Gen. 2019. V. 576. P. 11.
Yang J., Li D., Zhang Z., Li Q., Wang H. // J. Photochem. Photobiol. A: Chem. 2000. V. 137. P. 197.
Blake N.R., Griffin G.L. // J. Phys. Chem. 1988. V. 92. P. 5697.
Kryukova G.N., Zenkovets G.A., Shutilov A.A., Wilde M., Gunter K., Fassler D., Richter K. // Appl. Catal. B: Env. 2007. V. 71. № 3–4. P. 169.
Einaga H., Urahama N., Tou A., Neraoka Y. // Catal. Lett. 2014. V. 144. P. 1653.
Harris P.J.F. // Int. Mater. Rev. 1995. V. 40. P. 97.
Abbet S., Heiz U., Yakkimen H., Landman U. // Phys. Rev. Lett. 2001. V. 86. P. 5950.
Heiz U., Sanches A., Abbet S., Schneider W.-D. // J. Amer. Chem. Soc. 1999. V. 121. P. 3214.
Бухтияров В.И. // Успехи химии. 2007. Т. 76. № 6. С. 596.
Dulub O., Hebenstreit W., Diebold U. // Phys. Rev. Lett. 2000. V. 84. № 16. P. 3646.
Шутилов А.А., Зенковец Г.А., Олейник А.А., Пахаруков И.Ю., Просвирин И.П. // Кинетика и катализ. 2015. Т. 56. № 6. С. 800.
Woo H.S., Fleisch T.H., Foley H.C., Uchiyama S., Delgass W.N. // Catal. Lett. 1990. V. 4. P. 93.
Lin L-W., Kou V., Zou M., Yan Z. // Phys. Chem. Chem. Phys. 2001. V. 3. P. 1789.
Зенковец Г.А., Гаврилов В.Ю., Шутилов А.А., Цыбуля С.В. // Кинетика и катализ. 2009. Т. 50. № 5. С. 790.
Guinier A. Theorie et Technique de la Radiocristallographie. Paris: Dunot, 1956. 604 p.
Цыбуля С.В., Черепанова С.B., Соловьева Л.П. // ЖСХ. 1996. Т. 37. № 2. С. 379.
Brunauer S., Emmett P.H., Teller E. // J. Amer. Chem. Soc. 1938. V. 60. P. 309.
Handbook of X-ray Photoelectron Spectroscopy / Eds. Moulder J.F., Stickle W.F., Sobol P.E. and Bomben K.D. Eden Prairie, Minnesota: Perkin-Elmer Corp. 1992.
Shirley D.A. // Phys. Rev. B. 1972. V. 5. P. 4709.
Пахаруков И.Ю. Исследование особенностей кинетики гетерогенных каталитических реакций проточно-циркуляционным методом с усовершенствованной постановкой эксперимента. Дис. … к. х. н. Новосибирск, 2009.
Боресков Г.К. Катализ. Изд. ”НАУКА”, Новосибирск. 1971. 266 с.
Шутилов А.А., Зенковец Г.А., Цыбуля С.В., Гаврилов В.Ю., Крюкова Г.Н. // Кинетика и катализ. 2012. Т. 53. № 3. С. 424.
Гаврилов В.Ю., Зенковец Г.А. // Кинетика и катализ. 1993. Т. 34. № 2. С. 357.
Зенковец Г.А., Цыбуля С.В., Бургина Е.Б., Крюкова Г.Н. // Кинетика и катализ. Т. 40. № 4. С. 623.
Ahouari H., Rousse G., Rodriguez-Carvajal J., Sougrati M.-T., Saubanere M., Courty M., Recham N., Taarscon J.-M. // Chem. Mater. 2015. V. 27. № 5. P. 1631.
Korolenko M.V., Fabritchnyi P.B., Afanasov M.I. // Mendeleev Commun. 2020. V. 30. P. 383.
Korolenko M.V., Fabritchnyi P.B., Afanasov M.I., Labrugère C. // Mendeleev Commun. 2019. V. 29. P. 718.
Барсуков Д.В., Субботина И.Р. // Изв. Акад. наук. Серия химич. 2018. № 2. Р. 0243.
Гудков Б.С., Субботин А.Н., Якерсон В.И. // Природа. 2001. № 6. С. 24.
Субботин А.Н., Гудков Б.С., Воробьева М.П., Голосман Е.З., Якерсон В.И., Кустов Л.М. // Катализ в промышленности. 2005. № 3. С. 52.
Дополнительные материалы отсутствуют.
Инструменты
Кинетика и катализ