

Кинетика и катализ, 2022, T. 63, № 1, стр. 61-76
Закоксование катализаторов: механизмы, модели, влияние
Н. М. Островский *
Euro Gas d.o.o.,
24000 Subotica, Senćanski put 75, Serbia
* E-mail: n-ostr@yandex.ru
Поступила в редакцию 21.07.2021
После доработки 15.09.2021
Принята к публикации 15.09.2021
- EDN: OLIGJK
- DOI: 10.31857/S0453881122010063
Аннотация
Настоящая работа имеет обзорный характер. Представлен механизм полислойного образования кокса на катализаторах различных типов и проанализированы возможные его варианты. Дан детальный вывод уравнений динамики накопления разных видов кокса и кинетики его дезактивирующего действия. Выведены обоснованные зависимости относительной активности катализатора от концентрации кокса. Представлены конкретные примеры таких зависимостей. Они подтверждают преимущества строгих моделей, основанных на механизме явления, по сравнению с формальным описанием экспериментов, не имеющим предсказательной силы.
ВВЕДЕНИЕ
Проблема дезактивации катализаторов коксом всегда была ключевой для процессов нефтепереработки и нефтехимии, являющихся самыми многотоннажными в химической технологии. Это подтверждает множество публикаций, появившихся начиная с 50-х гг. ХХ в. Среди них особое место занимает монография Р.А. Буянова “Закоксование катализаторов” [1], в которой он не просто суммировал наиболее важные на начало 80-х гг. результаты. Уже в самой структуре монографии были обозначены важнейшие проблемы коксообразования, такие как:
– механизмы образования углеродных структур на катализаторе;
– состав, типы и морфология коксовых отложений;
– характер дезактивации катализатора при его закоксовании;
– кинетика процесса образования кокса;
– методы борьбы с дезактивацией катализаторов коксом.
Р.А. Буянов сформулировал два основных типа механизмов образования кокса:
1) механизм консекутивных реакций ненасыщенных поверхностных соединений;
2) механизм карбидного цикла для металлических катализаторов Fe, Co, Ni.
В качестве выводов монографии он определил основные направления исследований, которые должны были бы в будущем охватить следующие важные вопросы [1]:
– механизмы образования кокса на катализаторах разной природы и связь таких механизмов с каталитически активными компонентами катализаторов;
– механизмы формирования разных морфологических видов кокса и топография его отложений;
– природа отравляющего действия кокса на катализаторы;
– кинетика и оптимизация режимов работы катализаторов с учетом целевого процесса и процесса образования кокса.
Именно этим вопросам посвящена настоящая статья обзорного характера, представляющая некоторые результаты работ автора с сотрудниками в Омском филиале ИК СО РАН.
Прежде всего, проиллюстрируем характер и масштаб влияния коксообразования в некоторых промышленных процессах. Так, при каталитическом крекинге около 3–5% сырья (вакуумного газойля) превращается в кокс и сжигается во время регенерации катализатора. Только в США это составляет 15–18 млн тонн вакуумного газойля в год, что почти идентично мощности всех установок крекинга в России.
В процессе риформинга бензинов закоксование катализатора обуславливает проведение процесса при высоком давлении и в избытке водорода, т.е. в термодинамически неоптимальных условиях. По этой же причине в реакторы загружается 20–50-кратный запас катализатора, обеспечивающий межрегенерационный период 0.5–1 г. [2].
В процессе паровой конверсии метана образование углеродных нитей на металлических катализаторах до сих пор рассматривается как серьезная проблема из-за дезактивации катализатора и возможных повреждений стенок реактора. Повышенное коксообразование является, кроме того, основным препятствием для создания промышленного катализатора “сухого риформинга”, а также и катализаторов для топливных элементов [3].
Известно, что промышленное применение твердокислотных катализаторов алкилирования изобутана бутенами затруднено очень быстрой дезактивацией из-за образования на катализаторе высоконенасыщенных продуктов продуктов олигомеризации [4].
Можно сказать, что формирование коксоподобных отложений на катализаторах становится ключевой проблемой современного катализа, главной целью которого является уже повышение не активности катализатора, а его стабильности и селективности. Именно поэтому для идентификации и характеристики углеродистых отложений используется множество методов [5], таких как TPO, EM, EELS, FTIR, NMR, AES, XRD, XPS, EPR и др.
Исследования структуры кокса создают также основу для целенаправленного синтеза различных углеродных материалов (волокон, “луковиц”, нанопены, нанотрубок и т.п.) с помощью катализаторов. Таким образом, закоксование катализатора из явления, опасного для промышленного катализа, перерождается в нанотехнологию углеродных материалов.
СТРУКТУРА КОКСОВЫХ ОБРАЗОВАНИЙ
Углеродистые отложения растут как дети, т.е. под влиянием “родителей” (катализатор и реакция) и “улицы” (температура, давление и концентрация).
Как правило, коксовые отложения различаются по химическому составу (соотношение Н/С); по молекулярной структуре (алифатические, ароматические, графитовые); по морфологии (2D-пятна, 3D-блоки, нити, трубки, оболочки); по локализации (на кристаллите металла, на носителе, внутри или снаружи кристаллитов цеолита). Разнообразие видов кокса проявляется, например, в кривых температурно-программируемого окисления (ТРО) закоксованых катализаторов (рис. 1).
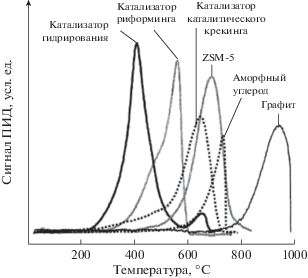
С помощью этой информации можно получить общую формулу CHx кокса (или Н/С) путем измерения общего количества H2O и CO2, выделяющихся в ходе TPO. Например, при изомеризации бутана на ферриерите [6] было обнаружено, что соотношение Н/С уменьшалось со временем и что в ходе анализа TPO оно варьировалось от 4 при 300°C до 0 при 600°C.
При дегидрировании н-гептана “…в коксе, образованном на различных катализаторах Pt–Re, нанесенных на оксид алюминия, выявлена трехмерная укладка ароматических кольцевых структур размером менее 1 нм, которые могут в дальнейшем образовывать более крупные пористые частицы углерода по мере увеличения коксования” [7].
Никелевые и железные катализаторы хорошо известны своей способностью образовывать “усы” (или нитевидный углерод). В паровом риформинге бутана на катализаторах Ni/Al2O3 и Ni/MgO с использованием электронной микроскопии высокого разрешения были обнаружены три вида углеродных отложений: волокна, трубки и оболочки [8]. Отложения, формирующиеся при низких температурах (ниже 500–600°C), состоят из волокон. При более высоких температурах (600–680°C) образуются трубки со слоями углерода, почти параллельными оси трубки. Слоистые оболочки возникали только на крупных металлических частицах размером около 100 нм.
Некоторые типы углеродных волокон, также известные как “нанотрубки”, были открыты еще в начале 50-х гг. Радушкевичем и Лукьяновичем [9, 10] и широко охарактеризованы в последние пятнадцать лет [10]. Нанотрубки имеют графитовые плоскости, параллельные поперечному сечению волокна, тогда как углеродные “усы” – графитовые плоскости, параллельные оси.
В истории углеродных нанотрубок (CNT) одностенные нанотрубки (SWCNT) следует отличать от многостенных (MWCNT) [10]. О формировании SWCNTS впервые было сообщено в 1993 г. [11, 12], но MWCNTS широко исследуются и используются в течение, по крайней мере, шестидесяти лет [10, 13].
МЕХАНИЗМЫ ОБРАЗОВАНИЯ КОКСА
Классификация механизмов дезактивации катализаторов и, в частности, закоксования, а также их анализ подробно изложены в монографии Буянова [1], в ряде обзоров и книг [14, 15] и в многочисленных статьях. Рассмотрим кратко основные механизмы.
1. Механизм “консекутивной схемы”
Механизм предполагает постепенное укрупнение коксовых отложений за счет последовательного присоединения предшественников кокса, которыми являются ненасыщенные углеводороды. Примерами могут служить коксообразование по реакциям типа Дильса–Альдера или образование олигомеров, рассмотренные в нескольких обзорах в сборнике [14], для процессов на катализаторах кислотного типа. Этот механизм чаще других используется для интерпретации экспериментов и наиболее вероятен в низкотемпературных процессах (<300°С), где продукты уплотнения имеют полимерное строение. При средних температурах (300–600°С) “консекутивная схема”, по мнению Буянова [16], реализуется на оксидных катализаторах.
2. Механизм “карбидного цикла”
Механизм характерен для металлов подгруппы железа и их оксидов и отличается разнообразием дезактивирующего действия [1, 16]. Образующиеся карбидные структуры иногда сами могут быть активными центрами [17, 18]. Значительная часть центров в результате распада карбидов снова возвращается в исходное активное состояние, что и обуславливает циклический характер механизма [1, 19]. В результате карбидного цикла формируется преимущественно графитоподобный кокс разнообразной формы и структуры [20–22], что существенно влияет на зависимость активности катализаторов от его концентрации.
3. Механизм “компенсированного распада”
Механизм предложен для металлов платиновой группы [23], но до сих пор он изучен недостаточно. Образование кокса здесь проходит через “ряд промежуточных полуразрушенных форм углеводорода” вплоть до углерода, способного внедряться в приповерхностный слой металла или графитизироваться. Слабое растворение углерода в Pt обнаружено и в других работах [24]. Оно происходит при высоких температурах, а после охлаждения углерод выходит на поверхность. Если для Ni и Fe характерна диффузия углерода внутрь кристаллитов металлов, то на Pt наблюдается диффузия углерода [25] и его предшественников [26] по поверхности. Энергия активации поверхностной диффузии составляет 25–35 ккал/моль [25], что сопоставимо с обычными энергиями активации для многих процессов превращения углеводородов. Существенная особенность углеродных образований на платине – то, что они входят в структуру активных центров некоторых реакций [26]. Другими словами, углерод участвует в формировании активных центров под действием реагирующей смеси.
ПРОСТЕЙШИЕ МАТЕМАТИЧЕСКИЕ МОДЕЛИ
Коксообразование – это типичный нестационарный процесс. Поэтому естественной его моделью является дифференциальное уравнение динамики накопления количества или концентрации кокса на катализаторе (C):
где $\bar {y}$ – вектор мольных долей реагирующих веществ, Т – температура, t – время.Тем не менее, для катализа важна не столько концентрация кокса на катализаторе, сколько его локализация на активных центрах. Поэтому необходимо еще и уравнение динамики активности катализатора (a), т.е. уравнение кинетики дезактивации:
(2)
$\frac{{{\text{d}}a}}{{{\text{d}}t}} = {{f}_{a}}\left( {\bar {y},T,a} \right),\,\,\,\,a(t) = \frac{{r(t)}}{{{{r}^{0}}}},$Комбинацией (1) и (2) получаем связь активности с концентрацией кокса:
(3)
$\frac{{{\text{d}}a}}{{{\text{d}}C}} = \frac{{{{f}_{a}}\left( {\bar {y},T,a} \right)}}{{{{f}_{c}}\left( {\bar {y},T,C} \right)}} = {{f}_{{ac}}}\left( {\bar {y},T,a,C} \right).$Из (3) легко получаются линейная и экспоненциальная зависимости активности катализатора от концентрации кокса, предложенные в [27] и широко используемые в инженерных расчетах:
где γ = kd/kc– отношение констант скоростей дезактивации и коксообразования.Линейная зависимость получается при нулевых порядках скоростей по a и по С:
(5)
${{{\text{d}}a} \mathord{\left/ {\vphantom {{{\text{d}}a} {{\text{d}}t}}} \right. \kern-0em} {{\text{d}}t}} = - {{k}_{{\text{d}}}},\,\,\,\,{{{\text{d}}C} \mathord{\left/ {\vphantom {{{\text{d}}C} {{\text{d}}t}}} \right. \kern-0em} {{\text{d}}t}} = {{k}_{{\text{c}}}},\,\,\,\,{{{\text{d}}a} \mathord{\left/ {\vphantom {{{\text{d}}a} {{\text{d}}C}}} \right. \kern-0em} {{\text{d}}C}} = - \gamma .$Экспоненциальная зависимость соответствует первому порядку по a и нулевому по С:
(6)
${{{\text{d}}a} \mathord{\left/ {\vphantom {{{\text{d}}a} {{\text{d}}t}}} \right. \kern-0em} {{\text{d}}t}} = - {{k}_{{\text{d}}}}a,\,\,\,\,{{{\text{d}}C} \mathord{\left/ {\vphantom {{{\text{d}}C} {{\text{d}}t}}} \right. \kern-0em} {{\text{d}}t}} = {{k}_{{\text{c}}}},\,\,\,\,{{{\text{d}}a} \mathord{\left/ {\vphantom {{{\text{d}}a} {{\text{d}}C}}} \right. \kern-0em} {{\text{d}}C}} = - {{\gamma }}a.$Из (5) и (6) следует, что оба уравнения (4) формально соответствуют росту концентрации кокса независимо от уровня активности катализатора, в то время как активность падает с ростом С. При этом кокс откладывается либо монослойно, либо “лавинообразно”, т.е. сразу толстым слоем [28].
Несмотря на простоту, формулы (4) часто используются при обработке экспериментов, так как испытания (особенно промышленные) проводятся в узком интервале активности, и не допускается ее падение ниже 60–70% от начальной.
МОДЕЛИ СТАДИЙНЫХ МЕХАНИЗМОВ
Уравнения кинетики дезактивации
Для современного уровня знаний о механизмах катализа и коксообразования недостаточны упрощенные зависимости (4). Все чаще для исследований (и даже для практики) требуются более детальные, чем (5) и (6), уравнения общего вида (1) и (2). Понятно, что они должны выводиться на основе стадийных механизмов как основных реакций, так и образования кокса.
Для описания кинетики дезактивации (2) широко используются феноменологические уравнения степенного типа, предложенные в [30] и [31]:
(7)
$\begin{gathered} \frac{{{\text{d}}a}}{{{\text{d}}t}} = - {{k}_{{\text{d}}}}y_{i}^{m}{{a}^{h}},\,\,\,\,\frac{{{\text{d}}a}}{{{\text{d}}t}} = - {{f}_{y}}\left( {{{y}_{i}},T} \right){{a}^{d}}, \\ d = 1 + \frac{{h - 1}}{m}, \\ \end{gathered} $Строгий метод вывода уравнений типа (2) на основе стадийных механизмов реакций и дезактивации, основанный на принципе квазистационарности, предложен в [32, 33], а также детально описан в [2] с многочисленными примерами. В случае линейных механизмов (где скорость любой стадии rn линейна относительно покрытия Θn, т.е. rn = kn yi Θn) выведено универсальное уравнение кинетики дезактивации [33]:
(8)
$\frac{{{\text{d}}a}}{{{\text{d}}t}} = - {{r}^{0}}\frac{{{{w}_{{\text{d}}}}}}{{{{w}_{{\text{j}}}}}}a + {{w}_{{\text{s}}}}(1 - a),\,\,\,\,a = \frac{r}{{{{r}^{0}}}},$Стадия саморегенерации присутствует в большинстве процессов, сопровождающихся коксообразованием. Она предусматривает частичную регенерацию блокированных центров компонентами самой реакционной среды. Обычно это водород (в процессах гидрирования, изомеризации, риформинга бензинов, гидрокрекинга, гидроочистки) или водяной пар (в паровой конверсии метана, дегидрирования олефинов и т.п.). Именно с целью саморегенерации Н2 и Н2О присутствуют как правило в избытке.
Проиллюстрируем вывод уравнения (8) на примере простого двухстадийного механизма:

Здесь A – исходное вещество; B – продукт; H – водород; D – дезактивирующая примесь, Z – промежуточные соединения на поверхности катализатора.
Скорости стадий: r1 = k1yAΘ0; r2 = k2yBΘ0 – k–2ΘB; rd = kdyDΘB; rs = ksyHΘD.
Веса стадий: w1 = k1yA; w2 = k2yB; w–2 = k–2; wd = = kdyD; ws = ksyH.
Согласно принципа квазистационарности скорость реакции намного больше скорости дезактивации. Поэтому реакция всегда успевает подстроиться под изменения, вызванные дезактивацией. Следствием этого являются соотношения:
(10a)
$\begin{gathered} {\text{при}}\,\,t = 0{\text{:}}\,\,{{\Theta }}_{{\text{0}}}^{{\text{o}}} + {{\Theta }}_{{\text{B}}}^{{\text{o}}} = 1, \\ {\text{при}}\,\,t > 0{\text{:}}\,\,\,\,{{{{\Theta }}}_{{\text{0}}}} + {{{{\Theta }}}_{{\text{B}}}} = 1 - {{{{\Theta }}}_{{\text{D}}}} \\ \end{gathered} $(10b)
${{{{\Theta }}}_{{\text{0}}}} = {{\Theta }}_{{\text{0}}}^{{\text{o}}}\left( {1 - {{{{\Theta }}}_{{\text{D}}}}} \right),\,\,\,\,{{{{\Theta }}}_{{\text{B}}}} = {{\Theta }}_{{\text{B}}}^{{\text{o}}}\left( {1 - {{{{\Theta }}}_{{\text{D}}}}} \right).$Если 1-я стадия лимитирующая, то 2-я быстрая и обратимая, а, значит, равновесная. Тогда r2 → 0, откуда ΘB = bByBΘ0, где bB = k2/k–2 – константа адсорбционного равновесия.
Вследствие баланса (10а) ${{\Theta }}_{{\text{0}}}^{{\text{o}}} + {{b}_{{\text{B}}}}{{y}_{{\text{B}}}}{{\Theta }}_{{\text{0}}}^{{\text{o}}} = 1;$ ${{{{\Theta }}}_{{\text{0}}}} + {{b}_{{\text{B}}}}{{y}_{{\text{B}}}}{{{{\Theta }}}_{{\text{0}}}} = 1 - {{{{\Theta }}}_{{\text{D}}}},$ поэтому:
(11)
${{\Theta }}_{{\text{0}}}^{{\text{o}}} = \frac{1}{{1 + {{b}_{{\text{B}}}}{{y}_{{\text{B}}}}}},\,\,\,\,{{\Theta }}_{{\text{B}}}^{{\text{o}}} = \frac{{{{b}_{{\text{B}}}}{{y}_{{\text{B}}}}}}{{1 + {{b}_{{\text{B}}}}{{y}_{{\text{B}}}}}}.$Общая скорость реакции равна скорости лимитирующей стадии r = r1 = k1yAΘ0, тогда вследствие (10b):
(12)
${{r}^{0}} = {{k}_{{\text{1}}}}{{y}_{{\text{A}}}}\frac{1}{{1 + {{b}_{{\text{B}}}}{{y}_{{\text{B}}}}}},\,\,\,\,r = {{k}_{{\text{1}}}}{{y}_{{\text{A}}}}\frac{1}{{1 + {{b}_{{\text{B}}}}{{y}_{{\text{B}}}}}}\left( {1 - {{{{\Theta }}}_{{\text{D}}}}} \right).$Поэтому отношение текущей и начальной скоростей реакции (равное активности) пропорционально доле активных центров, что справедливо для линейных механизмов:
(13)
$a = \frac{r}{{{{r}^{0}}}} = 1 - {{{{\Theta }}}_{{\text{D}}}},\,\,\,\,{\text{d}}a = --{\text{d}}{{{{\Theta }}}_{{\text{D}}}}.$Соответствующее уравнение для ΘD запишем на основе схемы механизма (9):
(14)
$\frac{{{\text{d}}{{{{\Theta }}}_{{\text{D}}}}}}{{{\text{d}}t}} = {{w}_{{\text{d}}}}{{{{\Theta }}}_{{\text{B}}}} - {{w}_{{\text{s}}}}{{{{\Theta }}}_{{\text{D}}}} = {{w}_{{\text{d}}}}{{\Theta }}_{{\text{B}}}^{{\text{o}}}\left( {1 - {{{{\Theta }}}_{{\text{D}}}}} \right) - {{w}_{{\text{s}}}}{{{{\Theta }}}_{{\text{D}}}}.$Используя (13), получим уравнение для активности:
(15)
$\frac{{{\text{d}}a}}{{{\text{d}}t}} = - {{w}_{{\text{d}}}}{{\Theta }}_{{\text{B}}}^{{\text{o}}}a + {{w}_{{\text{s}}}}\left( {1 - a} \right).$Выразим ${{\Theta }}_{{\text{B}}}^{{\text{o}}}$ через r0, используя (11) и (12). Легко видеть, что после подстановки ${{\Theta }}_{{\text{B}}}^{{\text{o}}}$ в уравнение (15) оно будет соответствовать уравнению (8), в котором wj = w1/bByB:
(16)
$\frac{{{\text{d}}a}}{{{\text{d}}t}} = - {{k}_{{\text{d}}}}{{y}_{{\text{D}}}}\frac{{{{b}_{{\text{B}}}}{{y}_{{\text{B}}}}}}{{1 + {{b}_{{\text{B}}}}{{y}_{{\text{B}}}}}}a + {{k}_{{\text{s}}}}{{y}_{{\text{H}}}}\left( {1 - a} \right).$Наличие саморегенерации обеспечивает падение активности не до нуля, а до некоторого установившегося значения as при t → ts, когда скорости rd и rs сравняются: kdyDΘA = ksyHΘD.
Тогда, при t → ts, правые части уравнений (15) и (16) становятся равными нулю:
Если теперь подставить ws в (15), то получим уравнение дезактивации в виде:
(17)
$\frac{{{\text{d}}a}}{{{\text{d}}t}} = - {{w}_{{\text{d}}}}{{\Theta }}_{{\text{B}}}^{{\text{o}}}\frac{{a - {{a}_{{\text{s}}}}}}{{1 - {{a}_{{\text{s}}}}}} = - {{k}_{{\text{d}}}}{{y}_{{\text{D}}}}\frac{{{{b}_{{\text{B}}}}{{y}_{{\text{B}}}}}}{{1 + {{b}_{{\text{B}}}}{{y}_{{\text{B}}}}}}\frac{{a - {{a}_{{\text{s}}}}}}{{1 - {{a}_{{\text{s}}}}}}.$В такой форме уравнение становится удобнее для обработки экспериментов, так как as более наглядный параметр, чем ws, потому что является асимптотой на графике a = f(t), рис. 2.
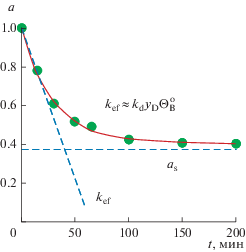
Механизм (9) применим для реакций дегидрирования, например, для дегидрирования циклогексана в бензол. Эта реакция широко используется как модельная для изучения различных катализаторов риформинга бензинов Pt/γ-Al2O3 или Pt–Re/γ-Al2O3, Pt–Sn/γ-Al2O3 [34–36]:
(I)
${{{\text{C}}}_{{\text{6}}}}{{{\text{H}}}_{{12}}} + {{{\text{Z}}}_{0}} = {\text{Z}}{\kern 1pt} --{\kern 1pt} {{{\text{C}}}_{{\text{6}}}}{{{\text{H}}}_{6}} + 3{{{\text{Н}}}_{2}},\,\,\,\,{{r}_{1}} = {{k}_{1}}{{y}_{{\text{A}}}}{{\Theta }_{0}},$(II)
$Z{\kern 1pt} --{\kern 1pt} {{{\text{C}}}_{6}}{{{\text{H}}}_{6}} = {\text{ }}{{{\text{C}}}_{{\text{6}}}}{{{\text{H}}}_{6}} + {\text{ }}{{{\text{Z}}}_{0}},\,\,\,\,{{r}_{2}} = {{k}_{2}}{{y}_{{\text{B}}}}{{\Theta }_{0}}--{{k}_{{--2}}}{{\Theta }_{{\text{B}}}}.$В типичных условиях протекания этой реакции, т.е. при температуре 300–350°С и разбавлении сырья водородом 3–5 моль/моль, дезактивация катализатора практически не наблюдается. Она становится заметной либо при замене большей части водорода на инертный газ, либо при добавлении в сырье других углеводородов, таких как метилциклопентан (MCP), парафины или олефины. Последнее соответствует процессу риформинга бензинов, состоящих, в основном, из парафинов и нафтенов. Кроме того, дезактивацию исследуют при повышенной температуре 350–380°С.
Так, например, в [37, 38] использовали смесь состава: циклогексан (СН) – 65%, метилциклопентан (МСР) – 25%, н-гексан (n-C6) – 7.5%, бензол (B) – 2.5%, Н2/сырье = 5 моль/моль. Испытания проводили в проточном безградиентном реакторе, при температуре 360°С. В этих условиях влияние обратной реакции, а также адсорбционного равновесия, незначительно [36]. Поэтому уравнение скорости реакции (12) сводится к уравнению первого порядка:
(18)
${{r}^{0}} \approx {{k}_{{\text{1}}}}{{y}_{{\text{A}}}},\,\,\,\,r = {{r}^{0}}\left( {1 - {{{{\Theta }}}_{{\text{D}}}}} \right) \approx {{k}_{{\text{1}}}}{{y}_{{\text{A}}}}a,$(19)
$\frac{{{\text{d}}a}}{{{\text{d}}t}} = - {{k}_{{\text{d}}}}{{y}_{{\text{D}}}}\frac{{a - {{a}_{{\text{s}}}}}}{{1 - {{a}_{{\text{s}}}}}}.$Наконец, уравнение материального баланса в лабораторном безградиентом реакторе дает возможность выразить активность катализатора через измеряемые концентрации:
(20)
$\begin{gathered} {{y}_{{\text{A}}}} - y_{{\text{A}}}^{{\text{o}}} = - {{\tau }}{{k}_{{\text{1}}}}{{y}_{{\text{A}}}}a,\,\,\,\,\frac{{y_{{\text{A}}}^{{\text{o}}} - {{y}_{{\text{A}}}}}}{{y_{{\text{A}}}^{{\text{o}}}}} = {{\tau }}{{k}_{{\text{1}}}}\frac{{{{y}_{{\text{A}}}}}}{{y_{{\text{A}}}^{{\text{o}}}}}a, \\ X = {{\tau }}{{k}_{{\text{1}}}}(1 - X)a, \\ \end{gathered} $На свежем катализаторе, т.е. при t = 0: a = 1, X = X o и, следовательно:
(21)
$\begin{gathered} \frac{{{{X}^{{\text{o}}}}}}{{1 - {{X}^{{\text{o}}}}}} = {{\tau }}{{k}_{{\text{1}}}},\,\,\,\,a = \frac{{1 - {{X}^{{\text{o}}}}}}{{{{X}^{{\text{o}}}}}}\frac{X}{{1 - X}}, \\ {{a}_{{\text{s}}}} = \frac{{1 - {{X}^{{\text{o}}}}}}{{{{X}^{{\text{o}}}}}}\frac{{{{X}_{{\text{s}}}}}}{{1 - {{X}_{{\text{s}}}}}}. \\ \end{gathered} $Формулы (21) позволяют преобразовать экспериментальную зависимость изменения конверсии во времени X(t) в экспериментальную зависимость изменения активности во времени a(t), рис. 3.
Рис. 3.
Дезактивация катализатора Pt/Al2O3 в реакции дегидрировании циклогексана [2]. Точки – эксперимент, сплошная линия – расчет по уравнению (22).

Концентрация метилциклопентана практически не менялась в ходе опыта yD = 25 ± 2% [37, 38], поэтому в (19) kdyD ≈ const = $k_{{\text{d}}}^{*},$ а уравнение (19) решается аналитически:
(22)
$a(t) = {{a}_{{\text{s}}}} + (1 - {{a}_{{\text{s}}}})\exp \left\{ { - \frac{{k_{{\text{d}}}^{{\text{*}}}}}{{1 - {{a}_{{\text{s}}}}}}t} \right\}.$Для большинства промышленных процессов уравнения материального баланса и кинетики дезактивации не сводятся к простым уравнениям типа (19) и (20). Тем не менее, ряд типичных моделей (включая и нелинейные) для реакторов идеального смешения и идеального вытеснения выведены в [2, 39] и продемонстрировано их применение.
Уравнения кинетики коксообразования
Рассмотрим модель полислойного образования кокса [2, 28], которая может быть применена как для консекутивной схемы, так и для механизма компенсированного распада. Она состоит из скоростей стадий, соответствующих скоростям роста (расширения) слоев кокса:
(23m)
${\text{D}} + {{{\text{Z}}}_{0}} \to {{{\text{Z}}}_{1}}~\,\,\,\,{{r}_{1}} = {{k}_{{\text{m}}}}{{y}_{{\text{D}}}}{{\Theta }_{0}} = {{r}_{{\text{m}}}},~$(23p)
${\text{D}} + {{{\text{Z}}}_{1}} \to {{{\text{Z}}}_{2}}\,\,\,\,~{{r}_{2}} = {{k}_{{\text{p}}}}{{y}_{{\text{D}}}}{{\Theta }_{1}},$(23n)
${\text{D}} + {{{\text{Z}}}_{n}}_{{--1}} \to {{{\text{Z}}}_{n}}\,\,\,\,{{r}_{n}} = {{k}_{{\text{p}}}}{{y}_{D}}{{\Theta }_{n}}_{{--1}},~~$Для наглядности представим механизм (23) в виде схемы образования и расширения слоев, рис. 4.

Очевидно, что доли активной и закоксованной поверхностей выражаются формулами:
(24)
${{\Theta }_{{\text{0}}}} = 1 - {{{{\Theta }}}_{{\text{D}}}},\,\,\,\,{{{{\Theta }}}_{{\text{D}}}} = \sum\limits_{n = 1}^N {{{{{\Theta }}}_{n}}} .$Общая скорость накопления кокса складывается из скорости образования “монослоя” и суммы скоростей отложения кокса на последующих слоях:
(25)
$\frac{1}{{{\xi }}}\frac{{{\text{d}}{{C}_{{\text{c}}}}}}{{{\text{d}}t}} = {{k}_{{\text{m}}}}{{y}_{{\text{D}}}}{{{{\Theta }}}_{{\text{0}}}} + {{k}_{{\text{p}}}}{{y}_{{\text{D}}}}\sum\limits_{n = 2}^N {{{{{\Theta }}}_{{n - 1}}}} ,$С учетом (24) уравнение (25) принимает вид:
(26)
$\frac{1}{{{\xi }}}\frac{{{\text{d}}{{C}_{{\text{c}}}}}}{{{\text{d}}t}} = {{k}_{{\text{m}}}}{{y}_{{\text{D}}}}\left( {1 - {{{{\Theta }}}_{{\text{D}}}}} \right) + {{k}_{{\text{p}}}}{{y}_{{\text{D}}}}\left( {{{{{\Theta }}}_{{\text{D}}}} - {{{{\Theta }}}_{N}}} \right).$Изменение активности катализатора a = 1 – ΘD вызывается образованием только первого слоя, а остальные влияют лишь на концентрацию кокса. Поэтому уравнение для ΘD запишется как:
(27)
$\begin{gathered} \frac{{{{C}_{{\text{m}}}}}}{{{\xi }}}\frac{{{\text{d}}{{{{\Theta }}}_{{\text{D}}}}}}{{{\text{d}}t}} = {{k}_{{\text{m}}}}{{y}_{{\text{D}}}}\left( {1 - {{{{\Theta }}}_{{\text{D}}}}} \right),\,\,{\text{или}} \\ \frac{{{{C}_{{\text{m}}}}}}{\xi }\frac{{{\text{d}}a}}{{{\text{d}}t}} = - {{k}_{{\text{m}}}}{{y}_{{\text{D}}}}a, \\ \end{gathered} $Из сравнения уравнений (19) и (27) следует, что kd = ξkm/Cm. Связь активности катализатора с концентрацией кокса можно получить при совместном решении уравнений (26) и (27). Этому мешает входящее в (26) ΘN – доля поверхности, занятой N слоями кокса. Простейший способ обойти эту проблему заключается в предположении, что последнего слоя не существует, ΘN = 0. Это кажется не совсем физичным, но вполне реально при конечном времени работы катализатора. На практике процесс обычно останавливают, если активность снизилась больше чем на 50%.
Поделив (26) на (27), имеем:
(28)
$\begin{gathered} \frac{1}{{{{C}_{{\text{m}}}}}}\frac{{{\text{d}}{{C}_{{\text{c}}}}}}{{{\text{d}}{{{{\Theta }}}_{{\text{D}}}}}} = 1 + \frac{{{{k}_{{\text{p}}}}}}{{{{k}_{{\text{m}}}}}}\frac{{{{{{\Theta }}}_{{\text{D}}}}}}{{1 - {{{{\Theta }}}_{{\text{D}}}}}},\,\,{\text{или}} \\ - \frac{1}{{{{C}_{{\text{m}}}}}}\frac{{{\text{d}}{{C}_{{\text{c}}}}}}{{{\text{d}}a}} = 1 + \frac{{{{k}_{{\text{p}}}}}}{{{{k}_{{\text{m}}}}}}\frac{{1 - a}}{a}. \\ \end{gathered} $После интегрирования получаем уравнение, связывающее активность с концентрацией кокса в предположении бесконечного его накопления:
(29)
$\frac{{{{C}_{{\text{c}}}}}}{{{{C}_{{\text{m}}}}}} = \left( {1 - \varphi } \right)\left( {1 - a} \right) - \varphi \ln a,\,\,{\text{где}}\,\,{{\varphi }} = \frac{{{{k}_{{\text{p}}}}}}{{{{k}_{{\text{m}}}}}}.$Вывод и анализ уравнения (29) опубликованы в [2, 28], а графики представлены на рис. 5. Пример его использования для описания экспериментальных данных приведен на рис. 6.

Рис. 6.
Дезактивация катализатора Cr2O3–Al2O3 в реакции дегидрировании бутена-1. Точки – реконструкция эксперимента из [40], сплошная линия – расчет по уравнению (29).

Варианты уравнения (29) выведены также и для нелинейных механизмов как основной реакции, так и (или) дезактивации [28].
Уравнение (29) включает в себя как частные случаи линейную и экспоненциальную зависимости (4). Если на закоксованной поверхности кокс образуется незначительно, т.е. kp → 0, то φ → 0, и получаем линейную зависимость Сс/Cm = 1 – a или a = 1 – Сс/Cm. Экспоненциальная зависимость получается при равенстве констант скоростей монослойного и полислойного образования кокса kp = km, т.е. φ → 1. Тогда из (29) получаем a = exp(–Сс/Cm).
Другой способ решения уравнения (26) состоит в замене максимального количества слоев N на максимальную концентрацию кокса Cmax, которая доступна для измерения. Для этого в уравнении (25) предполагаем, что Θn– 1 пропорционально (Cm – Сn)/Cm.
Путем дополнительных преобразований, представленных в [28], приходим к уравнению:
(30)
$\begin{gathered} \frac{1}{{{\xi }}}\frac{{{\text{d}}{{C}_{{\text{c}}}}}}{{{\text{d}}t}} = \left( {{{k}_{{\text{m}}}}{{y}_{{\text{D}}}} - {{k}_{{\text{p}}}}{{y}_{{\text{D}}}}} \right)\left( {1 - {{{{\Theta }}}_{{\text{D}}}}} \right) + \\ + \,\,{{{{k}_{{\text{p}}}}{{y}_{{\text{D}}}}\left( {{{C}_{{\max }}} - {{C}_{{\text{c}}}}} \right)} \mathord{\left/ {\vphantom {{{{k}_{{\text{p}}}}{{y}_{{\text{D}}}}\left( {{{C}_{{\max }}} - {{C}_{{\text{c}}}}} \right)} {{{C}_{{\text{m}}}}}}} \right. \kern-0em} {{{C}_{{\text{m}}}}}}. \\ \end{gathered} $Поделив (30) на (27) получаем:
(31)
$ - \frac{1}{{{{C}_{{\text{m}}}}}}\frac{{{\text{d}}{{C}_{{\text{c}}}}}}{{{\text{d}}a}} = \left( {1 - \frac{{{{k}_{{\text{p}}}}}}{{{{k}_{{\text{m}}}}}}} \right){{C}_{{\text{m}}}} + \frac{{{{k}_{{\text{p}}}}}}{{{{k}_{{\text{m}}}}}}\frac{{{{C}_{{\max }}} - {{C}_{{\text{c}}}}}}{a}.$После интегрирования и некоторых преобразований получаем уравнение, связывающее активность с концентрацией кокса с учетом максимального его накопления:
(32)
$\frac{{{{C}_{{\text{c}}}}}}{{{{C}_{{\max }}}}} = 1 - \frac{{{{C}_{{\text{m}}}}}}{{{{C}_{{\max }}}}}a - \left( {1 - \frac{{{{C}_{{\text{m}}}}}}{{{{C}_{{\max }}}}}} \right){{a}^{{{\varphi }}}},\,\,{\text{где}}\,\,{{\varphi }} = \frac{{{{k}_{{\text{p}}}}}}{{{{k}_{{\text{m}}}}}}.$Его можно представить и в другом виде, удобном для сопоставления с уравнением (29):
(33)
$\frac{{{{C}_{{\text{c}}}}}}{{{{C}_{{\text{m}}}}}} = \frac{{{{C}_{{\max }}}}}{{{{C}_{{\text{m}}}}}} - a - \left( {\frac{{{{C}_{{\max }}}}}{{{{C}_{{\text{m}}}}}} - 1} \right){{a}^{{{\varphi }}}}.$Кроме того, соотношение Cmax/Cm = N, т.е. равно числу слоев кокса, следовательно:
(34)
${{{{C}_{{\text{c}}}}} \mathord{\left/ {\vphantom {{{{C}_{{\text{c}}}}} {{{C}_{{\text{m}}}}}}} \right. \kern-0em} {{{C}_{{\text{m}}}}}} = N - a - \left( {N - 1} \right){{a}^{{{\varphi }}}}.$Графики зависимостей, описываемых уравнением (33), представлены на рис. 7.

Видно, что здесь предельными случаями являются две линейные зависимости:
– при φ = 0, из (33): a = 1 – Сс/Cm;
– при φ = 1, из (32): a = 1 – Сс/Cmax.
Линия φ = 1 делит область рис. 7 на две принципиально различные зоны. В первой зоне (при φ < 1) падение активности замедляется с ростом концентрации кокса, что характерно и для модели (29), рис. 5. Такие зависимости превалируют в большинстве реальных процессов.
В другой зоне (при φ > 1), наоборот, с ростом концентрации кокса дезактивация ускоряется. Это происходит потому, что с приближением Сс к Cmax снижается не только доля активной поверхности, но и ее доступность, причем тем быстее, чем больше φ. Экспериментальных наблюдений таких зависимостей очень мало и они ограничиваются цеолитными катализаторами.
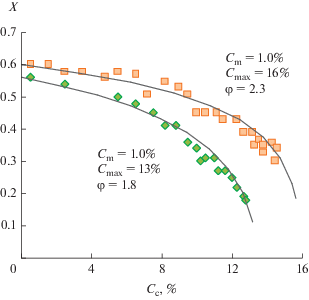
Примером может служить конверсия метанола в олефины (процесс MTO), на катализаторе SAPO-34 [41]. Эксперимент проводили в реакторе TEOM (tapered element oscillating microbalance), который формально можно считать проточным безградиентным реактором, так как навеска катализатора была 5 мг, а весовая удельная скорость сырья WHSV составляла 385 ч–1 [41]. Результаты опытов в виде зависимости конверсии метанола X от концентрации кокса Сс представлены на рис. 8 вместе с результатами расчета по уравнению (33). Так как уравнение (33) выведено для активности a, а экспериментально измерялась конверсия X, то выполнялся пересчет a в X по формулам (21), справедливым для проточного беградиентного реактора. Xo в (21) соответствует начальной точке Сс = 0.
Рис. 8.
Дезактивация катализатора SAPO-34 в реакции конверсии метанола в олефины. Размер кристаллитов катализатора: (■) – 0.25 мкм; (♦) – 2.5 мкм. Точки – реконструкция эксперимента из [41], сплошные линии – расчет по уравнениям (21), (33).
Уравнения (29) и (32), (33), а также их комбинации применялись для описания влияния кокса на оксидных [28] и нанесенных металлических катализаторах [29] и на некоторых цеолитах [42].
КОМПЛЕКСНАЯ МОДЕЛЬ ДЛЯ НАНЕСЕННЫХ КАТАЛИЗАТОРОВ
Закоксование нанесенных металлических катализаторов имеет ряд специфических особенностей, влияющих на кинетику их дезактивации и зависимость активности катализатора от концентрации кокса. Это наиболее ярко проявляется в низкопроцентных металлических катализаторах, используемых в риформинге бензинов (Pt, Pt–Re, Pt–Sn на Al2O3), дегидрировании легких парафинов (Pt–Sn на Al2O3), селективном гидрировании алкинов (Pd–Cu, Pd–Au на Al2O3), изомеризации парафинов (Pt–Cl на Al2O3 и цеолитах).
Имеется множество работ (обзоров и статей), в которых анализируются особенности дезактивации коксом таких катализаторов. Отметим только некоторые, касающиеся механизмов отравляющего воздействия кокса [43, 44], типов коксовых структур, таких как “полиены” [45], “полиарены” [46], графитоподобный кокс [44, 47]. Они отличаются не только структурой, но и характером дезактивирующего действия.
“Полимерный” кокс легко удаляется водородом и поэтому назван в [44] “обратимым”, а графитоподобный – “необратимым”. Это свойство “обратимого” кокса делает возможной частичную саморегенерацию катализатора водородом, обеспечивая длительную его работу в процессах риформинга, изомеризации, гидрирования и дегидрирования [46, 48, 49]. Важной является информация о том, что кокс на платине составляет <1 вес. %, образуется в первый час работы катализатора и далее его количество не увеличивается [44, 48]. В то же время общая концентрация кокса на катализаторе растет, достигая 20–30 вес. % к концу межрегенерационного периода [44, 48]. Ясно, что это происходит за счет отложения кокса на носителе. Подобный механизм “поведения” кокса описан также в [23, 47] под названием механизма компенсированного распада, а также в [43] под названием механизма миграции. Авторы [44] предложили математическую модель накопления “обратимого” и “необратимого” кокса, однако без учета состава реагирующей смеси, кокса на носителе и природы образования графитизированного кокса.
Схема и соответствующая модель, учитывающая эти особенности, предложены в [2, 29].
Здесь Θi – доли поверхности: свободной – Θo; занятой полимерным коксом – Θp; занятой графитизированным коксом – Θg; занятой косом на носителе – Θz; ki – константы скоростей образования кокса; yD, yH – мольные доли коксообразующего вещества и водорода.
Общая концентрация кокса Сс равна сумме концентраций Сс = Сp + Сg + Сz, для каждой из которых, в соответствии со схемой (рис. 9) и аналогично (25) и (26), можем записать:
(35)
$\frac{1}{{{{{{\xi }}}_{{\text{p}}}}}}\frac{{{\text{d}}{{C}_{{\text{p}}}}}}{{{\text{d}}t}} = {{k}_{{\text{p}}}}{{y}_{{\text{D}}}}\left( {1 - {{{{\Theta }}}_{{\text{p}}}} - {{{{\Theta }}}_{{\text{g}}}}} \right) - {{k}_{{\text{s}}}}{{y}_{{\text{H}}}}{{{{\Theta }}}_{{\text{p}}}} - {{k}_{{\text{g}}}}{{{{\Theta }}}_{{\text{p}}}},$(36)
$\frac{1}{{{{{{\xi }}}_{{\text{g}}}}}}\frac{{{\text{d}}{{C}_{{\text{g}}}}}}{{{\text{d}}t}} = {{k}_{{\text{g}}}}{{{{\Theta }}}_{{\text{p}}}},$(37)
$\frac{1}{{{{{{\xi }}}_{{\text{z}}}}}}\frac{{{\text{d}}{{C}_{{\text{z}}}}}}{{{\text{d}}t}} = {{k}_{{\text{z}}}}{{y}_{{\text{D}}}}\left( {1 - {{{{\Theta }}}_{{\text{z}}}}} \right).$
Для вывода уравнений, связывающих концентрации кокса и доли активной поверхности Θo, необходимо учесть три важные особенности рассматриваемой схемы, рис. 9:
1) кокс на носителе образуется независимо, если нет миграции прекурсоров с металла;
2) стадии схемы (рис. 9) имеют различные характеристические времена;
3) стадия графитизации влияет на количество полимерного (“обратимого”) кокса на металле, но не влияет на долю активной поверхности Θo, т.е. на активность.
Прежде всего, из рис. 9 ясно, что активность a пропорциональна Θo = 1 – Θp – Θg. На этапе t < ts медленным процессом графитизации можно пренебречь, тогда правая часть уравнения для Сp (35) формально соответствует правой части уравнения для Θo:
(38)
$ - \frac{{{{C}_{{\text{m}}}}}}{{{{{{\xi }}}_{{\text{p}}}}}}\frac{{{\text{d}}{{{{\Theta }}}_{{\text{o}}}}}}{{{\text{d}}t}} = {{k}_{{\text{p}}}}{{y}_{{\text{D}}}}\left( {1 - {{{{\Theta }}}_{{\text{p}}}} - {{{{\Theta }}}_{{\text{g}}}}} \right) - {{k}_{{\text{s}}}}{{y}_{{\text{H}}}}{{{{\Theta }}}_{{\text{p}}}}.$Так как a = Θo и da = dΘo, то поделив (35) на (38) получаем dСp/da = –Сm и, следовательно,
(39)
$a = {{1 - {{C}_{{\text{p}}}}} \mathord{\left/ {\vphantom {{1 - {{C}_{{\text{p}}}}} {{{C}_{{\text{m}}}}}}} \right. \kern-0em} {{{C}_{{\text{m}}}}}},\,\,{\text{при}}\,\,t < {{t}_{{\text{s}}}}.$При этом уравнение для активности аналогично (19):
(40)
$\frac{{{{C}_{{\text{m}}}}}}{{{{{{\xi }}}_{{\text{p}}}}}}\frac{{{\text{d}}a}}{{{\text{d}}t}} = - {{k}_{{\text{p}}}}{{y}_{{\text{D}}}}\frac{{a - {{a}_{{\text{s}}}}}}{{1 - {{a}_{{\text{s}}}}}},\,\,\,\,{{a}_{{\text{s}}}} = {{{{k}_{{\text{s}}}}{{y}_{{\text{H}}}}} \mathord{\left/ {\vphantom {{{{k}_{{\text{s}}}}{{y}_{{\text{H}}}}} {\left( {{{k}_{{\text{p}}}}{{y}_{{\text{D}}}} + {{k}_{{\text{s}}}}{{y}_{{\text{H}}}}} \right)}}} \right. \kern-0em} {\left( {{{k}_{{\text{p}}}}{{y}_{{\text{D}}}} + {{k}_{{\text{s}}}}{{y}_{{\text{H}}}}} \right)}}.$После установления равновесия между образованием “обратимого” кокса и саморегенерацией, т.е. при t > ts, активность медленно меняется за счет графитизации dΘg = –dΘp, которая смещает это равновесие. Так как при этом as = 1 – Θps, a = = as – Θg и da = –dΘg, то
(41)
$\frac{{{{C}_{{\text{m}}}}}}{{{{{{\xi }}}_{{\text{g}}}}}}\frac{{{\text{d}}a}}{{{\text{d}}t}} = - {{k}_{{\text{g}}}}a\,\,\,\,{\text{при}}\,\,\,\,t \geqslant ~{{t}_{{\text{s}}}}.$Концентрация кокса Сc при этом растет за счет его образования на носителе dСc ≈ dСz, т.е. согласно уравнению (37), в котором Θz можно выразить как Θz = Сc/Сmax:
(42)
$\frac{1}{{{{{{\xi }}}_{{\text{z}}}}}}\frac{{{\text{d}}{{C}_{{\text{c}}}}}}{{{\text{d}}t}} \approx {{k}_{{\text{z}}}}{{y}_{{\text{D}}}}\left( {1 - {{{{C}_{{\text{c}}}}} \mathord{\left/ {\vphantom {{{{C}_{{\text{c}}}}} {{{C}_{{\max }}}}}} \right. \kern-0em} {{{C}_{{\max }}}}}} \right).$Поделив (42) на (41) и проинтегрировав, получаем для небольших концентраций (1–10%):
(43)
$a = {{a}_{{\text{s}}}}\exp \left( { - \frac{{{{C}_{{\text{c}}}} - {{C}_{{\text{s}}}}}}{{{{C}_{{\text{m}}}}}}{{{{\varphi }}}_{{\text{1}}}}} \right),\,\,\,\,{{{{\varphi }}}_{{\text{1}}}} = \frac{{{{{{\xi }}}_{{\text{g}}}}}}{{{{{{\xi }}}_{{\text{z}}}}}}\frac{{{{k}_{{\text{g}}}}}}{{{{k}_{{\text{z}}}}{{y}_{{\text{D}}}}}}.$Рассмотрим, наконец, уравнения динамики накопления кокса (35)–(37). Они не обязательны для описания дезактивации катализатора, для этого достаточны уравнения (16)–(21), (40), (41). Тем не менее, описания кривых накопления кокса во времени служат дополнительной проверкой обоснованности предполагаемых механизмов дезактивации. Для этого правые части уравнений (35)–(37) необходимо выразить не через покрытия Θi, а через концентрации кокса Сi.
Простейший пример – уравнение (42) на интервале t ≥ ts, решение которого имеет вид:
(44)
${{C}_{{\text{c}}}} = {{C}_{{\max }}}\left( {1 - {{e}^{{ - {{\gamma }}t}}}} \right),\,\,\,\,{{\gamma }} = {{{{{{\xi }}}_{{\text{z}}}}{{k}_{{\text{z}}}}{{y}_{{\text{D}}}}} \mathord{\left/ {\vphantom {{{{{{\xi }}}_{{\text{z}}}}{{k}_{{\text{z}}}}{{y}_{{\text{D}}}}} {{{C}_{{\max }}}}}} \right. \kern-0em} {{{C}_{{\max }}}}}{\kern 1pt} .$Решение уравнений для “полимерного кокса” (Cp) и суммарного его содержания (Cc) значительно облегчается если воспользоваться той особенностью, что первые два слагаемых в уравнении (35) равны правой части уравнения (40), см. [2]:
Тогда уравнения для (Cp) и (Cc) приобретают вид:
(45)
$\frac{{{\text{d}}{{C}_{{\text{p}}}}}}{{{\text{d}}t}} = {{{{\xi }}}_{{\text{p}}}}{{k}_{{\text{p}}}}{{y}_{{\text{D}}}}\frac{{a - {{a}_{{\text{s}}}}}}{{1 - {{a}_{{\text{s}}}}}} - {{{{\xi }}}_{{\text{g}}}}{{k}_{{\text{g}}}}{{{{\Theta }}}_{{\text{p}}}},$(46)
$\frac{{{\text{d}}{{C}_{{\text{c}}}}}}{{{\text{d}}t}} = {{{{\xi }}}_{{\text{p}}}}{{k}_{{\text{p}}}}{{y}_{{\text{D}}}}\frac{{a - {{a}_{{\text{s}}}}}}{{1 - {{a}_{{\text{s}}}}}} + {{{{\xi }}}_{{\text{z}}}}{{k}_{{\text{z}}}}{{y}_{{\text{D}}}}\left( {1 - {{{{\Theta }}}_{{\text{z}}}}} \right).$Воспользуемся решением уравнения (40) в следующей форме:
(47)
$\frac{{a - {{a}_{{\text{s}}}}}}{{1 - {{a}_{{\text{s}}}}}} = {{e}^{{ - {{\beta }}t}}},\,\,\,\,{{\beta }} = {{{{{{\xi }}}_{{\text{p}}}}{{k}_{{\text{p}}}}{{y}_{{\text{D}}}}} \mathord{\left/ {\vphantom {{{{{{\xi }}}_{{\text{p}}}}{{k}_{{\text{p}}}}{{y}_{{\text{D}}}}} {{{C}_{{\text{m}}}}}}} \right. \kern-0em} {{{C}_{{\text{m}}}}}}{\kern 1pt} .$Выразим также Θp и Θz через концентрации кокса Θp = Сp/Сm, Θz = Сc/Сmax.
С учетом этих преобразований уравнения (45) и (46) принимают вид:
(48)
$\frac{{{\text{d}}{{C}_{{\text{p}}}}}}{{{\text{d}}t}} = {{{{\xi }}}_{{\text{p}}}}{{k}_{{\text{p}}}}{{y}_{{\text{D}}}}{{e}^{{ - {{\beta }}t}}} - {{{{\xi }}}_{{\text{g}}}}{{k}_{{\text{g}}}}\frac{{{{C}_{{\text{p}}}}}}{{{{C}_{{\text{m}}}}}},$(49)
$\frac{{{\text{d}}{{C}_{{\text{c}}}}}}{{{\text{d}}t}} = {{{{\xi }}}_{{\text{p}}}}{{k}_{{\text{p}}}}{{y}_{{\text{D}}}}{{e}^{{ - {{\beta }}t}}} + {{{{\xi }}}_{{\text{z}}}}{{k}_{{\text{z}}}}{{y}_{{\text{D}}}}\left( {1 - \frac{{{{C}_{{\text{c}}}}}}{{{{C}_{{\max }}}}}} \right).$Они решаются аналитически, в результате чего мы получаем уравнения динамики накопления “полимерного” (Cp) и общего кокса (Cc), которые выведены в [2, 29]:
(50)
${{C}_{{\text{p}}}} = \frac{{{{C}_{{\text{s}}}}}}{{1 - {{{\alpha }} \mathord{\left/ {\vphantom {{{\alpha }} {{\beta }}}} \right. \kern-0em} {{\beta }}}}}\left( {{{e}^{{ - {{\alpha }}t}}} - {{e}^{{ - {{\beta }}t}}}} \right),\,\,\,\,{{\alpha }} = {{{{{{\xi }}}_{{\text{p}}}}{{k}_{{\text{g}}}}} \mathord{\left/ {\vphantom {{{{{{\xi }}}_{{\text{p}}}}{{k}_{{\text{g}}}}} {{{C}_{{\text{m}}}},{\kern 1pt} }}} \right. \kern-0em} {{{C}_{{\text{m}}}},{\kern 1pt} }}$(51)
$\begin{gathered} {{C}_{{\text{c}}}} = \frac{{{{C}_{{\text{m}}}}}}{{1 - {{{\gamma }} \mathord{\left/ {\vphantom {{{\gamma }} {{\beta }}}} \right. \kern-0em} {{\beta }}}}}\left( {{{e}^{{ - {{\gamma }}t}}} - {{e}^{{ - {{\beta }}t}}}} \right) + {{\gamma }}{{C}_{{\max }}}\left( {1 - {{e}^{{ - {{\gamma }}t}}}} \right), \\ {{\gamma }} = {{{{{{\xi }}}_{{\text{z}}}}{{k}_{{\text{z}}}}{{y}_{{\text{D}}}}} \mathord{\left/ {\vphantom {{{{{{\xi }}}_{{\text{z}}}}{{k}_{{\text{z}}}}{{y}_{{\text{D}}}}} {{{C}_{{\max }}}}}} \right. \kern-0em} {{{C}_{{\max }}}}}{\kern 1pt} . \\ \end{gathered} $Здесь Cs соответствует as, а параметры α, β и γ пропорциональны скоростям графитизации (α ~ kg), отложения кокса на металле (β ~ kpyD ) и на носителе (γ ~ kzyD).
Для обоснования применимости предложенных уравнений желательно использовать эксперименты, в которых измерены одновременно отдельные виды кокса. Такие работы очень редкие. Здесь использованы эксперименты, опубликованные в [44], где определяли абсолютный привес кокса (мг). Поэтому данные реконструированы в измененных координатах весовых концентраций, рис. 10. Из рисунка видно, что модель (35)–(37) в виде конечных уравнений (50), (51) качественно и количественно описывает эксперимент.
Рис. 10.
Описание экспериментов накопления кокса на катализаторе Pt/Al2O3 [29]. Точки – реконструкция экспериментов из [44], линии – расчет по уравнениям (50) и (51).

Модель также позволяет связать концентрации различных видов кокса с активностью катализатора в отношении реакций, протекающих как на металле, так и на носителе.
ВАРИАНТ МОДЕЛИ ДЛЯ ЦЕОЛИТНЫХ КАТАЛИЗАТОРОВ
Закоксование цеолитных катализаторов также имеет ряд специфических особенностей. Прежде всего, установлена тесная взаимосвязь скорости образования и удаления углеродистых отложений со структурой и размерами каналов цеолитов [50]. Особенно наглядно это было показано в реакции конверсии метанола на ZSM-5, оффретите и мордените [50].
Комплексное исследование природы коксовых отложений в цеолитах, их структуры и закономерностей формирования выполнены группой авторов [51–53]. Они установили, что в цеолитах нет специфических центров коксообразования, однако модуль цеолита оказывает влияние на структуру кокса. Доказана решающая роль внешней поверхности кристалла цеолита в коксообразовании, найдены условия формирования кокса внутри каналов и установлена необычная структура углеродистых отложений.
Эти работы не были направлены на создание моделей дезактивации, однако в них получены важные соотношения. Так, установлено, что скорость дезактивации обратно пропорциональна наружной поверхности кристаллов цеолита (Sнар) и ее модулю Mпов = (Si/Al)пов, а при одинаковых Mпов прямо пропорциональна диаметру кристаллов (Dк ) [53]:
(52)
$\frac{{{\text{d}}a}}{{{\text{d}}t}}\sim \frac{{{{k}_{{\text{1}}}}}}{{\sqrt {{{S}_{{{\text{нар}}}}}} }},\,\,\,\,\frac{{{\text{d}}a}}{{{\text{d}}t}}\sim \frac{{{{k}_{{\text{2}}}}}}{{\sqrt {{{M}_{{{\text{пов}}}}}} }},\,\,\,\,\frac{{{\text{d}}a}}{{{\text{d}}t}}\sim {{k}_{3}}{{D}_{{\text{к}}}}.$Различные способы учета структуры цеолитов в моделях дезактивации предложены во многих работах. Модель на основе теории перколяции предложена в [54] для цеолитов ZSM-5. Традиционная для этих авторов функция дезактивации Φ (эквивалентная активности) здесь представляется произведением:
где C – концентрация кокса; S – доля незаблокированных коксом активных центров; P – (вероятность перколяции) представляет долю незаблокированных пор.Коэффициент (P) представлен для ZSM-5 в виде:
(54)
$P = {{(1--q{\text{/}}0.5701)}^{{0.454}}}\left( {1{\text{ }} + \sum {{{a}_{k}}} {{q}^{k}}} \right),$Мы же сосредоточимся здесь на тех же вопросах, что рассмотрены в предыдущем разделе, т.е. на типах кокса, их локализации и влиянии на дезактивацию. В качестве примера рассмотрим процесс получения товарного бензина из низкооктановых фракций на цеолитных катализаторах, получивший название “Цеоформинг” [55]. Процесс проводят в безводородной среде, поэтому он сопровождается обильным коксообразованием и значительной дезактивацией катализатора.
Авторы [51–53] с помощью спектральных методов и кинетических экспериментов установили, что в условиях процесса “Цеоформинг” образуется два типа кокса. Первый, высокотемпературный (>420°C) и высококонденсированный – кокс(c), формируется на внешней поверхности цеолитов. Второй, низкотемпературный (<380°C) и слабоконденсированный (полимерный) – кокс(p), откладывается во внутренних полостях кристаллитов. Основными предшественниками кокса в процессе “Цеоформинг” являются олефины и олигомеры.
Установлено ингибирующее влияние адсорбированных олигомеров на скорости реакций и дезактивации, так как активные центры постоянно покрыты промежуточными олигомерами, участвующими как в основных реакциях процесса, так и в реакциях коксообразования.
На основе имевшихся экспериментов [55] и предполагаемого химизма реакций [56] предложена следующая схема превращений агрегированных компонентов [57]:
где n-P – н-парафины; i-P + N – изопарафины и нафтены; G – углеводородные газы С2–С4; Оl – олефины + олигомеры; А – ароматические углеводороды; c, p – два вида кокса.
Согласно кинетике реакций, представленной в [57] и уточненной в [58], уравнения скоростей реакций имеют вид:
(56)
$\begin{gathered} {{r}_{1}} = {\text{ }}{{k}_{1}}(1{\text{ }} - {{{{\Theta }}}_{{\text{L}}}}){{Y}_{{n{\text{ - P}}}}},\,\,\,\,{{r}_{2}} = {\text{ }}{{k}_{2}}(1{\text{ }} - {{{{\Theta }}}_{{\text{L}}}}){{Y}_{{i{\text{ - P}}\,{\text{ + }}\,{\text{N}}}}}, \\ {{r}_{3}} = {\text{ }}{{k}_{3}}(1{\text{ }} - {{{{\Theta }}}_{{\text{L}}}}){{Y}_{{n{\text{ - P}}}}}\left\{ {1 - \frac{{{{Y}_{{i{\text{ - P}}\, + \,{\text{N}}}}}}}{{{{K}_{{\text{R}}}}{{Y}_{{n{\text{ - P}}}}}}}} \right\}, \\ {{r}_{4}} = {\text{ }}{{k}_{4}}{{\Theta }}_{{\text{L}}}^{2},\,\,\,\,{{r}_{5}} = {\text{ }}{{k}_{5}}{{{{\Theta }}}_{{\text{L}}}}{{Y}_{{\text{H}}}}, \\ \end{gathered} $Образование кокса можно представить как реакции олигомеризации и перераспределения водорода, протекающие на поверхности цеолитных катализаторов:
(57a)
$2{{\left[ {{\text{Ol}}} \right]}_{{{\text{ads}}}}} \to {\text{Кокс}}\left( {\text{p}} \right),\,\,\,\,{{r}_{{\text{p}}}} = {\text{ }}{{k}_{{\text{p}}}}{{\Theta }}_{{\text{L}}}^{2},$(57b)
${{\left[ {{\text{Ol}}} \right]}_{{{\text{ads}}}}}~ \to ~{\text{Кокс}}\left( {\text{c}} \right) + {{{\text{H}}}_{2}},\,\,\,\,{{r}_{{\text{c}}}} = {{k}_{{{\text{c}}\,}}}{{{{\Theta }}}_{{\text{L}}}}.$Так как олефины (легкие и особенно олигомеры) прочно адсорбируются на активных центрах катализатора, то ΘL пропорциональна (при низкой температуре 300–440°С) мольной доле олефинов в реакционной смеси ΘL~ bOlYOl. При более высокой температуре (420–500°С) следует использовать равновесную зависимость [58], например Лэнгмюра Θj = bjYj/(1 + bjYj).
При дезактивации катализатора любое покрытие Θj связано с долей дезактивированных центров ΘD, см. (10b). Поэтому для ΘL необходимо записать:
(58)
${{{{\Theta }}}_{{\text{L}}}} = {{\Theta }}_{{\text{L}}}^{{\text{o}}}\left( {1 - {{{{\Theta }}}_{{\text{c}}}}} \right),\,\,\,\,{{\Theta }}_{{\text{L}}}^{{\text{o}}} = \frac{{{{b}_{{{\text{Ol}}}}}{{Y}_{{{\text{Ol}}}}}}}{{1 + {{b}_{{{\text{Ol}}}}}{{Y}_{{{\text{Ol}}}}}}},$Тогда для реакций коксообразования rp и rc можно записать:
(59)
${{r}_{{\text{p}}}} = {{k}_{{\text{p}}}}{{\left( {{{\Theta }}_{{\text{L}}}^{{\text{o}}}} \right)}^{2}}{{\left( {1 - {{{{\Theta }}}_{{\text{p}}}}} \right)}^{2}},\,\,\,\,{{r}_{{\text{c}}}} = {{k}_{{\text{c}}}}{{\Theta }}_{{\text{L}}}^{{\text{o}}}\left( {1 - {{{{\Theta }}}_{{\text{c}}}}} \right),$Из определения относительной активности (aj = ${{{{r}_{j}}} \mathord{\left/ {\vphantom {{{{r}_{j}}} {r_{j}^{0}}}} \right. \kern-0em} {r_{j}^{0}}}$) следует, что ap= 1 – Θp и ac= 1 – Θc. Исходя из этого, можно записать уравнения кинетики дезактивации в виде [58]:
(60)
$\frac{{{\text{d}}{{a}_{{\text{p}}}}}}{{{\text{d}}t}} = - {{k}_{{\text{p}}}}{{\left( {{{\Theta }}_{{\text{L}}}^{{\text{o}}}} \right)}^{2}}a_{{\text{p}}}^{2},\,\,\,\,\frac{{{\text{d}}{{a}_{{\text{c}}}}}}{{{\text{d}}t}} = - {{k}_{{\text{c}}}}{{\Theta }}_{{\text{L}}}^{{\text{o}}}{{a}_{{\text{c}}}}.$В предположении двух типов активных центров, уравнения для скоростей реакций с учетом дезактивации принимают вид [58, 59]:
(61)
$\begin{gathered} {{r}_{1}} = {\text{ }}{{k}_{1}}\left( {1{\text{ }} - {{\Theta }}_{{\text{L}}}^{{\text{o}}}} \right){{Y}_{{n{\text{ - P}}}}}{{a}_{{\text{c}}}},\,\,\,\,{{r}_{2}} = {\text{ }}{{k}_{2}}\left( {1{\text{ }} - {{\Theta }}_{{\text{L}}}^{{\text{o}}}} \right){{Y}_{{i{\text{ - P}}\,{\text{ + }}\,{\text{N}}}}}{{a}_{{\text{c}}}}, \\ {{r}_{3}} = {\text{ }}{{k}_{3}}\left( {1{\text{ }} - {{\Theta }}_{{\text{L}}}^{{\text{o}}}} \right){{Y}_{{n{\text{ - P}}}}}\left\{ {1 - \frac{{{{Y}_{{i{\text{ - P}}\, + \,{\text{N}}}}}}}{{{{K}_{{\text{R}}}}{{Y}_{{n{\text{ - P}}}}}}}} \right\}{{a}_{{\text{c}}}}, \\ {{r}_{4}} = {\text{ }}{{k}_{4}}{{\left( {{{\Theta }}_{{\text{L}}}^{{\text{o}}}} \right)}^{2}}{{a}_{{\text{p}}}},\,\,\,\,{{r}_{5}} = {\text{ }}{{k}_{5}}{{\Theta }}_{{\text{L}}}^{{\text{o}}}{{Y}_{{\text{H}}}}{{a}_{{\text{p}}}}. \\ \end{gathered} $Экспериментальные данные, которые использованы для моделирования дезактивации катализатора, были выполнены Ахметовым и Каратуном [60] в изотермическом реакторе с неподвижным слоем цеолитсодержащего катализатора.
Уравнения материального баланса для интегрального изотермического реактора [59]:
где τ – время контакта; σ – изменение числа молей реагирующей смеси.
Результаты описания экспериментов представлены на рис. 11.
Рис. 11.
Изменение состава продуктов при дезактивации катализатора [58, 59]. Точки – реконструкция экспериментов из [60], линии – расчет по уравнениям (60)–(62); g – весовые доли продуктов реакции.

Конденсированный кокс. Как выше уже было отмечено, этот вид кокса образуется на внешней поверхности кристаллитов цеолита по механизму полислойного отложения (рис. 4, уравнения (29) и (32)).
В данном случае уравнение типа (30) записывается как [61]:
(63)
$\frac{1}{{{{C}_{{\text{m}}}}}}\frac{{{\text{d}}{{C}_{{\text{c}}}}}}{{{\text{d}}t}} = {{k}_{{\text{c}}}}{{\Theta }}_{{\text{L}}}^{{\text{o}}}{{a}_{{\text{c}}}} - {{k}_{{\text{N}}}}{{Y}_{{{\text{Ol}}}}}{{a}_{{\text{c}}}} + {{k}_{{\text{N}}}}{{Y}_{{{\text{Ol}}}}}\frac{{C_{{{\text{max}}}}^{{\text{c}}} - {{C}_{{\text{c}}}}}}{{{{C}_{{\text{m}}}}}}.$Делим его на уравнение дезактивации (60) для этих центров ${{{\text{d}}{{a}_{{\text{c}}}}} \mathord{\left/ {\vphantom {{{\text{d}}{{a}_{{\text{c}}}}} {{\text{d}}t}}} \right. \kern-0em} {{\text{d}}t}} = - {{k}_{{\text{c}}}}\Theta _{{\text{L}}}^{{\text{o}}}{{a}_{{\text{c}}}}$ и интегрируем, как описано в [2, 28]. В результате получаем уравнение аналогичное (32):
(64)
$\frac{{{{C}_{{\text{c}}}}}}{{C_{{{\text{max}}}}^{{\text{c}}}}} = 1 - \frac{{{{C}_{{\text{m}}}}}}{{C_{{{\text{max}}}}^{{\text{c}}}}}{{a}_{{\text{c}}}} - \left( {1 - \frac{{{{C}_{{\text{m}}}}}}{{C_{{{\text{max}}}}^{{\text{c}}}}}} \right)a_{{\text{c}}}^{{{{{{\varphi }}}_{{\text{c}}}}}},\,\,\,\,{{{{\varphi }}}_{{\text{c}}}} = \frac{{{{k}_{{\text{N}}}}{{Y}_{{{\text{Ol}}}}}}}{{{{k}_{{\text{c}}}}{{\Theta }}_{{\text{L}}}^{{\text{o}}}}}.$Ясно, что графики зависимостей aс от Сс совпадают с приведенными на рис. 7.
Полимерный кокс. Он образуется внутри полостей цеолита по механизму монослойного отложения. Так как размер каналов цеолита сопоставим с размерами реагирующих молекул, то монослойное заполнение поверхности пропорционально объемному заполнению полостей цеолита.
Кроме того, поскольку этот кокс “полужидкий” [53], можно предположить, что происходит частичная его регенерация под действием водорода (krYН). Тогда уравнение имеет вид:
(65)
$\frac{1}{{C_{{{\text{max}}}}^{{\text{p}}}}}\frac{{{\text{d}}{{C}_{{\text{p}}}}}}{{{\text{d}}t}} = {{k}_{{\text{p}}}}{{\left( {{{\Theta }}_{{\text{L}}}^{{\text{o}}}} \right)}^{2}}a_{{\text{p}}}^{2} - {{k}_{{\text{r}}}}{{Y}_{{\text{H}}}}{\text{(}}1 - {{a}_{{\text{p}}}}{\text{)}}.$Делим его на уравнение дезактивации (60) для этих центров ${{{\text{d}}{{a}_{{\text{p}}}}} \mathord{\left/ {\vphantom {{{\text{d}}{{a}_{{\text{p}}}}} {{\text{d}}t}}} \right. \kern-0em} {{\text{d}}t}} = - {{k}_{{\text{p}}}}{{\left( {\Theta _{{\text{L}}}^{{\text{o}}}} \right)}^{2}}a_{{\text{p}}}^{2}$ и интегрируем. В результате получаем уравнение, отличное от (29):
(66)
$\begin{gathered} \frac{{{{C}_{{\text{p}}}}}}{{C_{{{\text{max}}}}^{{\text{p}}}}} = 1 - {{a}_{{\text{p}}}} + {{{{\varphi }}}_{{\text{p}}}}\left( {1 - \frac{1}{{{{a}_{{\text{p}}}}}} - {\text{ln}}{{a}_{{\text{p}}}}} \right), \\ {{{{\varphi }}}_{{\text{p}}}} = \frac{{{{k}_{{\text{r}}}}{{Y}_{{\text{H}}}}}}{{{{k}_{{\text{p}}}}{{{\left( {{{\Theta }}_{{\text{L}}}^{{\text{o}}}} \right)}}^{2}}}}. \\ \end{gathered} $Если саморегенерация незначительная, то φp = 0 и ${{C}_{{\text{p}}}} = C_{{\max }}^{{\text{p}}}\left( {1 - {{a}_{{\text{p}}}}} \right).$
Графики зависимостей (66) представлены в [61]. При малой скорости саморегенерации (φp < < 0.1) активность ap линейно снижается с ростом концентрации кокса Сp. Даже при φp = 0.1–0.5 зависимость мало отличается от линейной.
Вся совокупность уравнений материального баланса (62), кинетики реакций (61), дезактивации (60) и коксообразования (63) и (65) использованы в [61] для анализа и описания экспериментов по дезактивации катализатора “Цеоформинга” IC-30-1 в проточном изотермическом реакторе [53]. Сырьем служил газовый конденсат, содержащий 33.2% н‑парафинов, 46.3% изо-парафинов и нафтенов и 20.5% ароматических углеводородов.
Результаты экспериментов и их описания представлены на рис. 12 (динамика изменения состава продуктов) и рис. 13 (динамика накопления кокса).
Рис. 12.
Изменение состава продуктов при дезактивации катализатора “Цеоформинга” IC-30-1 в проточном изотермическом реакторе [61]. Точки – реконструкция экспериментов из [53], линии – расчет по уравнениям (60)–(62).


Если эксперименты проводятся в безградиентном реакторе, то уравнения (60) решаются аналитически, и в результате получаем:
(67)
${{a}_{{\text{p}}}} = {{\left( {1 + {{k}_{{\text{p}}}}{{{\left( {{{\Theta }}_{{\text{L}}}^{{\text{o}}}} \right)}}^{2}}t} \right)}^{{ - 1}}},\,\,\,\,{{a}_{{\text{c}}}} = \exp \left( { - {{k}_{{\text{c}}}}{{\Theta }}_{{\text{L}}}^{{\text{o}}}t} \right).$Так как YOl в ${{\Theta }}_{{\text{L}}}^{{\text{o}}}$ зависит от времени контакта, но слабо меняется со временем [58], то для концентраций кокса можно использовать уравнения (64) и (66).
Такую же модель можно применить и для анализа экспериментов в интегральном реакторе, но при постоянной температуре, а также если градиент YO по слою катализатора не меняется значительно во времени. Именно такой вариант представлен на рис. 14. Два эксперимента проведены при постоянных температурах 460 и 480°С, а третий соответствует температурному режиму, представленному на рис. 13. В этом случае для расчетов использована упрощенная модель, т.е. уравнения (64), (66) и (67).
Рис. 14.
Зависимость концентрации кокса от количества переработанного сырья [61]. Точки – реконструкция экспериментов из [53], линии – расчет по уравнениям (64), (66), (67).

ЗАКЛЮЧЕНИЕ
Для построения надежных кинетических моделей дезактивации коксом важно опираться на конкретные механизмы его образования и механизмы основных реакций, соблюдая, в то же время, необходимые упрощения.
В настоящей работе представлены уравнения дезактивации при полислойном коксообразовании, которое имеет место на многих оксидных и цеолитных катализаторах. Распространенные в литературе линейная и экспоненциальная зависимости активности катализатора от концентрации кокса являются частными случаями этих уравнений.
При закоксовании нанесенных металлических катализаторов процессы дезактивации металла и носителя происходят по различным механизмам. Для учета их взаимного влияния в работе представлены несколько моделей различной детальности. Наиболее полная комплексная модель выведена для нанесенных платиновых катализаторов. Она включает полислойное коксообразование на носителе, быстрое образование на платине “полимерного” кокса, способного к саморегенерации водородом непосредственно в ходе реакции, и медленное превращение его в “графитоподобный” кокс, который удаляется только при окислительной регенерации.
Предложенные модели подтверждены примерами описания кинетических экспериментов.
Список литературы
Буянов Р.А. Закоксование катализаторов. Новосибирск: Наука, 1983.
Островский Н.М. Кинетика дезактивации катализаторов. Москва: Наука, 2001.
De Chen, Lodeng R., Anundskas A., Olsvik O., Holmen A. // Chem. Eng. Sci. 2001. V. 56. P. 1371.
Martinis J.M., Froment G.F. // Ind. Eng. Chem. Res. 2006. V. 45. P. 940.
Querini C.A. Coke Characterization / In Catalysis. The Royal Society of Chemistry. 2004. V. 17. P. 166.
Xu W.Q., Yin Y.G., Suib S.L., O‘Young C.L. // J. Phys. Chem. 1995. V. 99. P. 758.
Querini C.A., Fung S.C. // Catal. Today. 1997. V. 37. P. 277.
Tracz E., Scholz R., Borowiecki T. // Appl. Catal. 1990. V. 66. P. 133.
Радушкевич Л.В., Лукьянович В.М. // Журн. Физ. Хим. 1952. Т. 26. С. 88.
Monthioux M., Kuznetsov V.L. // Carbon. 2006. V. 44. P. 1621.
Iijima S., Ichihashi T. // Nature. 1993. V. 363. P. 603.
Bethune D.S., Kiang C.H., de Vries M.S., Gorman G., Savoj R., Vazquez J., Beyers R. // Nature. 1993. V. 363. P. 605.
Boehm H.P. // Carbon. 1997. V. 35. P. 581.
Deactivation and Poisoning of Catalyst / Chemical Industries. V. 20. Eds: J. Oudar, H. Wise, N.Y.: Marcel Dekker Inc, 1985. 328 p.
Biswas J., Bickle G.M., Gray P.G., Do D.D., Barbier J. // Catal. Rev. Sci. Eng. 1988. V. 30. P. 161.
Буянов Р.А., Чесноков В.В. / Проблемы дезактивации катализаторов. Материалы 2-го Всесоюзн. Совещ., Новосибирск, 1989. С. 3.
Bianchi D., Tau L.M., Borcar S., Bennett C.O. // J. Catal. 1983. V. 84. P. 358.
Cooper B.J., Trimm D.L. // J. Catal. 1980. V. 62. P. 35.
Trimm D.L. // Catal. Rev. Sci. Eng. 1977. V. 16. P. 155.
Yokomizo G.H., Bell A.T. // J. Catal. 1989. V. 119. P. 467.
Sacco A., Geurts F.W., Jablonski G.A., Lee S., Gately R.A. // J. Catal. 1989. V. 119. P. 322.
Chesnokov V.V., Zaikovskii V.I., Buyanov R.A., Molchanov V.V., Plyasova L.M. // Catal. Today. 1995. V. 24. P. 265.
Чесноков В.В., Буянов Р.А., Пахомов Н.А., Зайковский В.И. // Кинетика и катализ. 1991. Т. 32. С. 1494.
Olander D.R., Balooch M. // J. Catal. 1979. V. 60. P. 41.
Martin M.T., Hudson J.B. // J. Vac. Sci. Technol. 1978. V. 15. P. 474.
Davis S.M., Zaera F., Somorjai G.A. // J. Catal. 1982. V. 77. P. 439.
Froment G.F., Bischoff K.B. // Chem. Eng. Sci. 1962. V. 17. P. 105.
Ostrovskii N.M. // Kinet. Catal. 2001. V. 42. P. 317.
Ostrovskii N.M. // Kinet. Catal. 2001. V. 42. P. 326.
Levenspiel O. // J. Catal. 1972. V. 25. P. 265.
Corella J., Asua J.M. // Ind. Eng. Chem. Proc. Des. Develop. 1982. V. 21. P. 55.
Ostrovskii N.M., Yablonskii G.S. // Reac. Kinet. Cat. Lett. 1989. V. 39. P. 287.
Ostrovskii N.M. // Chem. Eng. J. 2006. V. 120. P. 73.
Herz R.K., Gillespie W.D., Petersen E.E., Somorjai G.A. // J. Catal. 1981. V. 67. P. 371.
Jossens L.W., Petersen E.E. // J. Catal. 1982. V. 73. P. 377.
Островский Н.М., Карпова Л.А., Дуплякин В.К. // Кинетика и катализ. 1984. Т. 25. С. 1117.
Островский Н.М., Деманов Ю.К. // Химия и технология топлив и масел. 1991. Т. 42. С. 35.
Ostrovskii N.M., Chalganov E.M., Demanov Yu.K., Kolomytsev Yu.N., Bogomolova O.B. // Reac. Kinet. Cat. Lett. 1990. V. 41. P. 277.
Ostrovskii N.M. // Kinet. Catal. 2005. V. 46. P. 693.
Dumez F.J., Froment G.F. // Ind. Eng. Chem. Proc. Des. Develop. 1976. V. 15. P. 291.
De Chen, Rebo H.P., Holmen A. // Chem. Eng. Sci. 1999. V. 54. P. 3465.
Ostrovski N.M., Rovenskaja S.A., Echevski G.V. // Chem. Ind. Chem. Eng. Quarterly. 2007. V. 13. P. 51.
Trimm D.L. / In Deactivation and poisoning of catalysts. Eds: J. Oudar, H. Wise, Chemical Industries. New-York: Marcel Dekker Inc, 1985. V. 20. P. 151.
Biswas J., Gray P.G., Do D.D. // Appl. Catal. 1987. V. 32. P. 249.
Sarkany A., Lieske H., Szilagyi N., Toth L. // Proc. of VIII Int. Congr. on Catalysis. Berlin. 1984. V. 2. P. 613.
Srivastava R.D., Prasad N.S., Pal A.K. // Chem. Eng. Sci. 1986. V. 41. P. 719.
Боронин А.Н., Бухтияров В.И., Квон Р. // Изв. СО АН СССР. Сер. хим. 1990. Вып. 2. С. 75.
Parera J.M., Figoli N.S., Traffano E.M., Beltramini J.N., Martinelli E.E. // Appl. Catal. 1983. V. 5. P. 33.
Пахомов Н.А., Буянов Р.А., Чесноков В.В. / Тез. докл. 2-го Всесоюзн. совещ. по пробл. дезактивации катализатров. Уфа. 1989. Ч. 2. С. 23.
Dejaifve P., Auroux A., Gravelle P.C., Vedrine J.C., Gabelica Z., Derouane E.G. // J. Catal. 1981. V. 70. P. 123.
Echevskii G.V., Kalinina N.G., Anufrienko V.F., Poluboyarov V.A. // React. Kinet. Catal. Lett. 1987. V. 33. P. 305.
Ечевский Г.В., Харламов Г.В., Полубояров В.А., Калинина Н.Г., Литвак Г.С., Ануфриенко В.Ф. // Кинетика и катализ. 1987. Т. 28. С. 1462.
Ечевский Г.В. Закономерности коксообразования на цеолитах в реакциях синтеза и превращения углеводородов. Дис. … д. х. н., Новосибирск, 1996.
Beyne A.O.E., Froment G.F. // Chem. Eng. Sci. 1990. V. 45. P. 2089.
Степанов В.Г., Литвиненко Н.Г., Ионэ К.Г. // Нефтепереработка и нефтехимия. 1992. № 10. С. 14.
Дорогочинский А.З., Проскурин А.Л., Овчаров С.Н., Крушина Н.Н. Тематич. обзор. М.: ЦНИИТЭнефтехим, 1989. 82 с.
Rovenskaja S.A., Ostrovski N.M. // Chem. Ind. 2003. V. 57. № 9. P. 399.
Ostrovski N.M., Rovenskaja S.A., Echevski G.V. // Chem. Ind. 2004. V. 58. № 3. P. 104.
Ровенская С.А., Островский Н.М. // Химическая технология. 2007. Т. 8. № 11. С. 495.
Ахметов А.Ф., Каратун О.Н. // Нефтепереработка и нефтехимия. 2001. № 1. С. 23.
Ostrovski N.M., Rovenskaja S.A., Echevski G.V. // Chem. Ind. Chem. Eng. Quart. 2007. V. 13. № 2. P. 51.
Дополнительные материалы отсутствуют.
Инструменты
Кинетика и катализ