

Кинетика и катализ, 2022, T. 63, № 2, стр. 178-186
О влиянии ионной силы на кинетику окисления сульфита в присутствии Mn(II)
А. Н. Ермаков *
Институт энергетических проблем химической физики им. В.Л. Тальрозе ФИЦ ХФ им. Н.Н. Семенова РАН
119334 Москва, Ленинский просп., 38, корп. 2, Россия
* E-mail: polclouds@yandex.ru
Поступила в редакцию 30.06.2021
После доработки 06.10.2021
Принята к публикации 27.10.2021
- EDN: PUXYLZ
- DOI: 10.31857/S0453881122020022
Аннотация
Проведено рассмотрение имеющихся в литературе данных о влиянии ионной силы растворов на кинетику кислородного окисления сульфита в кислых растворах (рН 1.3–4.5) в присутствии ионов марганца(II). Найдено, что согласованная картина воздействия ионной силы возникает, если рассматривать процесс в рамках вырождено-разветвленного цепного механизма с участием в качестве инициатора ионов примесного железа. Такой подход в согласие с экспериментом привел к выявлению 2-х разных режимов этого процесса и позволил получить явные выражения для наблюдаемых констант скорости реакции. В работе приводятся новые выражения, описывающие негативное действие ионной силы (≈10–3–1.5 моль/л) на константы скорости реакции в обоих режимах.
ВВЕДЕНИЕ
Окисление молекулярным кислородом растворов сульфита – исторически первая цепная жидкофазная реакция [1]. В присутствии ионов переходных металлов (ПМ) этот процесс находит сегодня широкое применение при сероочистке дымовых газов [2]. Ведущее место он занимает и в самоочищении атмосферы от естественных и антропогенных выбросов диоксида серы (т.н. кислые дожди и др.) [3]. Актуальность не теряют исследования токсикологического действия сульфита [4], в том числе при вдыхании диоксида серы [5].
Для толкования данных о кинетике окисления сульфита (ОС) в скрубберных установках, каплях облаков или в биологических системах и управления этими процессами необходимо учитывать, что они осуществляются в растворах с ненулевой ионной силой (μ, моль/л). Имеющиеся сведения, хотя и указывают на негативное влияние μ на наблюдаемые константы скорости реакции (kнабл), остаются несогласованными. Так, например, при катализе ионами марганца ОС в избытке ионов металла в [6] сообщалось о драматическом спаде kнабл с ростом μ (≈ 0–1 моль/л). В [7] в близких концентрационных условиях снижение kнабл с увеличением μ в противовес оказывается намного меньшим. Корректность этих и других оценок влияния μ на kнабл и их сравнение затрудняет недостаток знания природы свободно-радикального (СР) окисления сульфита [8], в особенности применительно к катализу этого процесса ионами ПМ. Так, в [6] и [7] вопреки близости условий опытов наряду с различиями влияния μ на kнабл сообщалось и о разных частных порядках реакции по ионам марганца. Целью настоящего исследования было, опираясь на результаты авторов о свободно-радикальном механизме катализа ионами переходных металлов процесса окисления сульфита [9, 10] и имеющиеся в литературе данные, оценить реальные масштабы влияния ионной силы на кинетику этой реакции в присутствии ионов марганца.
О МЕХАНИЗМЕ РЕАКЦИИ
Ранее кинетику ОС в присутствии ионов марганца удалось непротиворечиво описать в рамках вырождено-разветвленного цепного механизма с участием в качестве переносчиков цепи радикалов ${\text{S}}{{{\text{O}}}_{{3 \div {{5}^{ - }}}}}$ и Mn(III) [9, 10]. Каталитическая активность ионов марганца связывалась при этом с неизбежными примесями ионов железа в растворах в концентрации (2–100) × 10–8 моль/л [9]. Вместе они образуют синергическую пару, в которой ионы Mn(II) многократно усиливают каталитические свойства примесных ионов Fe [9]. Синергизм их действия обусловлен ускорением лимитирующей стадии продолжения цепи (III) в отсутствие ионов ПМ за счет более быстрой реакции (X) [7, 11] (табл. 1), а также смещением распределения ионов железа по зарядовым формам (ζMn = [Fe(III)]/[Fe(II)]) в пользу каталитически активного Fe(III) – участника стадии инициирования цепей (I) [12].
Таблица 1.
Механизм катализа ионами марганца окисления сульфита
| № | Реакция | Константа скорости |
|---|---|---|
| I | Fe(OH)SO3H+ → Fe2+ + ${\text{SO}}_{3}^{{ - \centerdot }}$ + H2O | *0.2 |
| II | ${\text{SO}}_{3}^{{ - \centerdot }}$ + O2 → ${\text{SO}}_{5}^{{ - \centerdot }}$ | 2.5 × 109 |
| IIIа | ${\text{SO}}_{5}^{{ - \centerdot }}$ + HSO3 → ${\text{HSO}}_{5}^{ - }$ + ${\text{SO}}_{3}^{{ - \centerdot }}$ | 3.4 × 103 |
| IIIb | ${\text{SO}}_{5}^{{ - \centerdot }}$ + ${\text{HSO}}_{3}^{ - }$ → ${\text{SO}}_{4}^{{2 - }}$ + ${\text{SO}}_{4}^{{ - \centerdot }}$ + H+ | 2 × 102 |
| IV | ${\text{SO}}_{4}^{{ - \centerdot }}$ + ${\text{HSO}}_{3}^{ - }$ → ${\text{SO}}_{4}^{{2 - }}$ + ${\text{SO}}_{3}^{{ - \centerdot }}$ + H+ | 7.5 × 108 |
| Va | ${\text{SO}}_{5}^{{ - \centerdot }}$ + ${\text{SO}}_{5}^{{ - \centerdot }}$ → SO4-⋅+ ${\text{SO}}_{4}^{{ - \centerdot }}$ + O2 | 8.7 × 107 |
| Vb | ${\text{SO}}_{5}^{{ - \centerdot }}$ + ${\text{SO}}_{5}^{{ - \centerdot }}$ → ${{{\text{S}}}_{2}}{\text{O}}_{8}^{{2 - }}$ + O2 | 1.3 × 107 |
| VI | ${\text{HSO}}_{5}^{ - }$ + ${\text{HSO}}_{3}^{ - }$ + H + → 2${\text{SO}}_{4}^{{2 - }}$ + 3H+ | **107 |
| VII | Fe2+ + ${\text{SO}}_{5}^{{ - \centerdot }}$$\xrightarrow{{{{{\text{H}}}^{ + }}}}$ Fe3+ + ${\text{HSO}}_{5}^{ - }$ | 3.2 × 106 |
| VIII | Fe2+ + ${\text{SO}}_{4}^{{ - \centerdot }}$ → Fe3+ + ${\text{SO}}_{4}^{{2 - }}$ | 3.0 × 108 |
| IX | Fe2+ + ${\text{HSO}}_{5}^{ - }$ → Fe3+ + ${\text{SO}}_{4}^{{ - \centerdot }}$ + OН– | 3.5 × 104 |
| X | Mn2+ + ${\text{SO}}_{5}^{{ - \centerdot }}$$\xrightarrow{{{{{\text{H}}}^{ + }}}}$Mn(III) + ${\text{HSO}}_{5}^{ - }$ | 2.0 × 108 |
| XI | Mn(III) + ${\text{HSO}}_{3}^{ - }$ → Mn2+ + ${\text{SO}}_{3}^{{ - \centerdot }}$ + H+ | 1.3 × 106 |
| XII | Mn(III) + ${\text{SO}}_{5}^{{ - \centerdot }}$$\xrightarrow{{{{{\text{H}}}^{ + }}}}$ Mn(IV) + ${\text{HSO}}_{5}^{ - }$ | ~1 × 108 |
| XIII | Mn(III) + Mn(III) → Mn2+ + Mn(IV) | ~1 × 105 |
Баланс редокс-свойств растворов в отношении пары ионов Fe(III)/Fe(II) определяет при этом конкуренция автокаталитической реакции (VI) и реакции разветвления (IX) цепи, табл. 1. На это указывают результаты прямого контроля ζMn в аэрированных сульфитных растворах (рН 3.8, [S(IV)] = 2 × 10–4, [Fe]0 = 1.8 × 10–6 и [Mn(II)] = = (0.18–1.8) × 10–6 моль/л) согласно [13], в соответствии с которыми концентрация 3-валентных ионов железа при увеличении концентрации ионов марганца в указанных пределах возрастает более чем вдвое. Это положительное действие ионов марганца на ζMn и скорость ОС сказывается уже при [Mn(II)] = 1.8 × 10–7 моль/л!
В [9] отмечалось, что на конкуренцию (VI), (IX) оказывают влияние соотношение компонентов (α = ${{\left[ {{\text{Mn(II)}}} \right]} \mathord{\left/ {\vphantom {{\left[ {{\text{Mn(II)}}} \right]} {\left[ {{\text{HSO}}_{3}^{ - }} \right]}}} \right. \kern-0em} {\left[ {{\text{HSO}}_{3}^{ - }} \right]}}$) и кислотность раствора (рН). От того, какая из реакций преобладает, зависят концентрации промежуточного ${\text{HSO}}_{5}^{ - }$ и переносчиков цепи в растворах, а вместе с этим ζMn и режим ОС в целом [9, 10]. Свидетельством значимости конкуренции (VI), (IX) в динамике каталитического расходования сульфита выступают и данные работ [14, 15], в которых в прямых экспериментах впервые сообщалось о существовании 2-х разных режимов реакции: избыток сульфита над ионами металла (wVI/wIX$ \gg $ 1 и α $ \ll $ 1) и избыток ионов марганца над сульфитом (wVI/wIX$ \ll $ 1 и α ≥ 1), где wi – скорости реакций (VI) и (IX), табл. 1.
ИЗБЫТОК СУЛЬФИТА
Основной формой ионов примесного железа в кислых растворах и в избытке сульфита (α $ \ll $ 1) являются неактивные 2-х валентные ионы железа, т.е. ζMn ≈ 0 и [Fe(II)] ≈ [Fe]0 [9, 10]. Окислительные свойства растворов в отношении Fe(II) выражены при этом достаточно слабо. Это определяет низкую скорость инициированиия цепей (I) и медленное протекание ОС в целом. Причина – высокие $\left[ {{\text{HSO}}_{3}^{ - }} \right]$ и, как следствие, малые α и низкие рН, способствующие преобладанию реакции (VI) над разветвлением цепей (IX). Лимитирующим звеном продолжения цепей является реакция (X), а их обрыв происходит в квадратичном процессе (Vb) [16].
Для скорости ОС в стационарных условиях с учетом вышесказанного в [10] было найдено:
(1)
$~{{w}_{{{\text{Mn}}}}} = {\text{ }}2{{k}_{{{\text{IX}}}}}k_{{\text{X}}}^{2}{{[{\text{Mn}}({\text{II}})]}^{2}}{{[{\text{Fe}}]}_{0}}{\text{/}}{{k}_{{{\text{Vb}}}}}{{k}_{{{\text{VI}}}}}[{\text{HSO}}_{3}^{ - }].$Выражение (1) в явном виде демонстрирует влияние конкуренции реакций (VI), (IX) на wMn и указывает одновременно на замедление сульфитом реакции. Признаки ингибирования сульфитом реакции находим, привлекая к рассмотрению данные опытов [17] и др. В отсутствие фоновых электролитов и при постоянстве [Mn(II)] в [17], например, вопреки 0-ому по сульфиту порядку реакции(!), сообщалось о ~2.5-кратном возрастании wMn при снижении [S(IV)] от 6 × 10–2 до 10–2 моль/л.
Этим данным противоречит, однако, найденный в [17] прирост wMn при контролируемом снижении ионной силы растворов в присутствии фоновых электролитов (HCl, NaCl и NaClO4) и при постоянстве концентраций ионов марганца и сульфита. Указанный факт снимает, казалось, противоречие между 0-ым порядком по сульфиту и чувствительностью wMn к изменениям [S(IV)]. Прирост wMn скорости ОС при снижении концентрации сульфита в [17] всецело связан, казалось, с уменьшающейся при этом ионной силой растворов.
Из сравнения данных опытов [17] следует, однако, что изменение содержания фоновых электролитов, эквивалентное изменению концентрации сульфита в опытах [17], сопровождает примерно вдвое бóльший прирост wMn. Этот результат ставит под сомнение вывод о том, что сульфит оказывает влияние на скорость реакции лишь как электролит. Не согласуется с его ролью как электролита и оставленное без внимания в [17] различие темпов прироста wMn(μ) при наращивании [Mn(II)] в растворах с разным содержанием сульфита, притом что реакция в рассматриваемых условиях демонстрирует квадратичный рост wMn с увеличением концентрации ионов металла (wMn(μ) ∼ [Mn(II)]2). Темпы прироста wMn(μ) при постоянстве ионной силы (${\mu \mathord{\left/ {\vphantom {\mu {\left[ {{\text{HSO}}_{3}^{ - }} \right]}}} \right. \kern-0em} {\left[ {{\text{HSO}}_{3}^{ - }} \right]}}$ $ \gg $ 1) оказываются тем более высокими, чем ниже содержание сульфита, указывая на торможение им реакции. Подобный эффект – ослабление торможения сульфитом реакции в похожих условиях при постоянстве ионной силы раствора (0.05 моль/л) – прослеживается и по данным [18].
Из сказанного следует, что выявленный здесь по данным [17, 18] рост скорости реакции (wMn(μ) ≈ ≈ [Mn(II)]2) с уменьшением концентрации сульфита следует связывать с ослаблением ингибирования сульфитом реакции. Причиной служит замедление автокаталитической реакции (VI) и рост скорости разветвления цепей (IX) при снижении концентрации сульфита. Это оборачивается возрастанием концентрации Fe(III) и увеличением скорости инициирования, а вместе с этим и скорости ОС в целом.
Как сообщалось в [9], отрицательный порядок по S(IV) по данным [17], см. выше, равен ∼ –0.4, но рассматриваемые результаты относятся к различным рН, т.к. эти опыты проводили в отсутствие буферных добавок. Однако в [17] было показано также отсутствие заметного влияния [Н+] на wMn в диапазоне рН ∼ 1.3–3 и при близких α. Используя вместо [S(IV)] зависящую от рН концентрацию ${\text{HSO}}_{3}^{ - }$, найдем, что измеренные в опытах [17] значения wMn при разнящихся рН оказываются близкими к $ \sim {\kern 1pt} {{\left[ {{\text{HSO}}_{3}^{ - }} \right]}^{{ - 1}}}.$ Для изменения концентрации сульфита по ходу процесса в этих рамках будем иметь:
Отсюда явствует и наблюдавшееся в ряде работ постоянство скорости реакции wMn ≈ k_1(набл)[Mn(II)]2/ /[S(IV)]0, т.е. кажущийся 0-ой порядок реакции по сульфиту при том, что порядок по нему является в действительности отрицательным. Тот факт, что wMn при этом не увеличивается по ходу реакции, обусловлен тем, что эксперименты [17] и другие подобные опыты проводили при концентрациях кислорода в растворе, меньших начального содержания сульфита. Контроль wMn в таких условиях может производится лишь при невысоких конверсиях сульфита, т.е. в условиях, когда зафиксировать рост wMn со снижением концентрации сульфита практически не представляется возможным. Различить нулевой и отрицательный порядок по виду кинетических кривых в таких условиях, по крайней мере, затруднительно.
Возвращаясь к обсуждению ослабления ингибирования сульфитом реакции, вызванного ускорением разветвления цепей (IX), отметим, что прирост концентрации Fe(III) и скорости инициирования реакции при снижении $\left[ {{\text{HSO}}_{3}^{ - }} \right]$ продолжается до тех пор, пока в растворе сохраняется избыток сульфита над ионами марганца, т.е. пока [Fe(II)] ≈ [Fe]0. Переход же к избытку ионов марганца приводит к инверсии распределения валентных форм ионов примесного железа, что вызвано резким ускорением окисления Fe(II) в реакциях с ${\text{HSO}}_{5}^{ - }$ (IX) и с переносчиками цепи (VII), (VIII). С ускорением этих реакций и связано отмеченное выше нарастание [Fe(III)] с увеличением α по данным [13]. Бóльшая часть ионов железа в избытке ионов марганца, переходит в окисленную форму; [Fe(III)] ≈ [Fe]0 и ζMn ≈ 1. В таких условиях в [7] вместо 2-го наблюдается нулевой по ионам металла порядок реакции. Нулевой по ионам металла порядок реакции в их избытке отмечен по данным опытов [14, 19] и при более высоких концентрациях сульфита, чем в [7].
В литературе выражение для wMn в избытке сульфита обычно приводится в форме: wMn = = kнабл[Mn(II)]2 [14, 17, 19]. Учет ускорения наработки ${\text{HSO}}_{5}^{ - }$ в присутствии ионов марганца (X), (XI) и связанное с этим усиление окислительных свойств сульфитных растворов естественным образом приводят ко 2-ому порядку реакции по марганцу, уравнение (1). Об этом говорят и данные наших прямых экспериментов [9, 20]. Контроль скорости реакции при применении в качестве инициатора проникающей радиации показал, что введение ионов марганца сопровождает рост скорости расходования сульфита. Несмотря на избыток сульфита, на опыте в таких условиях наблюдался 1-ый по ионам металла порядок реакции. Полученный результат показывает, что ионы марганца ОС, участвующие в стадии продолжения цепей в условиях “железного” инициирования, ускоряют и стадию зарождения радикалов. Использование радиации позволило, таким образом, разделить действие ионов марганца(II) в условиях “железного” инициирования на две составляющие: участие в продолжении и инициировании цепей за счет увеличения наработки ${\text{HSO}}_{5}^{ - }$ (X), (XI) и связанного с ускорением этих реакций усиления окислительных свойств растворов.
Сравнивая k_1(набл)/[S(IV)]0 и kнабл приходим к
Здесь kVI = $k_{{{\text{VI}}}}^{0}$ × 10–рН, где $k_{{{\text{VI}}}}^{0}$ = 107 л2 моль–2 с–1 [21]. Вид приведенного выражения в явном виде раскрывает влияние μ на k_1(набл) в избытке сульфита. Оно является результатом чувствительности комбинации констант скорости реакций (IX), (X), (Vb), (VI) к изменению ионной силы растворов. Доминирование ионов железа в форме Fe(II) в таких условиях [9, 10] избавляет от необходимости учета их участия в комплексообразовании с фоновыми электролитами. Это обусловлено не только меньшей склонностью к комплексообразованию Fe(II) в сравнении с Fe(III) [8]. Присутствующие в экстремально низких концентрациях ионы Fe(III), участвуя в инициировании (III), табл. 1, приводят к формированию лишь начальной концентрации переносчиков цепи – радикалов ${\text{SO}}_{3}^{ - },$ активирующих в реакциях (II) и (X) ионы марганца. В отсутствие переносчиков цепи ионы марганца, как отмечалось выше, каталитически неактивны в отношении ОС. Эти реакции вместе с (IX) и (IV) ведут к росту концентраций переносчиков цепи в растворе. Их стационарные концентрации, определяющие динамику реакции в целом (wMn), достигаются по завершению периода роста их концентраций, т.е. по окончанию периода индукции реакции (τi) [17]. Формирование комплексов ионов примесного железа с фоновым электролитом лишь удлиняет τi, но не меняет wMn. С неизменностью wMn в рассматриваемых условиях и связано отсутствие в растворах с избытком сульфита отличий действия добавок сульфатов в качестве фонового электролита в сравнении с действием других фоновых электролитов [6, 17]. Для корректного сопоставления данных разных авторов о влиянии μ на k_1(набл) в избытке сульфита при заданном уровне μ достаточно, поэтому, учитывать лишь различия рН и [Fe]0.
Из данных [17] о wMn ([Mn(II)] = 10–4, [S(IV)] = = 10–2 моль/л) в растворах с известным содержанием ионов примесного железа ([Fe]0 = 5 × 10–7 моль/л) и при μ ≈ 0 находим: $k_{{{\text{набл}}}}^{*}$ = = wMn[${\text{HSO}}_{3}^{ - }$]/[Mn(II)]2[Fe]0 ≈ 5 × 106 л моль–1 с–1 (звездочка обозначает вид выражения для наблюдаемой константы скорости с учетом выражения (1)). Принимая, что kVI = $k_{{{\text{VI}}}}^{0}$ × 10–рН, приходим к k_1(набл) = $k_{{{\text{набл}}}}^{*}$[H+] ≈ 3.2 × 104 с–1. Отсюда для опытов [15] (рН 2) будем иметь, например, $k_{{{\text{набл}}}}^{*}$ = = k_1(набл)/10pH = 3.2 × 106 л моль–1 с–1. Для ${{{{{[{\text{Fe}}]}}_{0}}} \mathord{\left/ {\vphantom {{{{{[{\text{Fe}}]}}_{0}}} {\left[ {{\text{HSO}}_{{\text{3}}}^{ - }} \right]}}} \right. \kern-0em} {\left[ {{\text{HSO}}_{{\text{3}}}^{ - }} \right]}}$ в этих опытах (kнабл = 300 л моль–1 с–1, $\left[ {{\text{HSO}}_{{\text{3}}}^{ - }} \right]$ ≈ 6 × 10–4 моль/л, μ ≈ 0) находим ${{{{{[{\text{Fe}}]}}_{0}}} \mathord{\left/ {\vphantom {{{{{[{\text{Fe}}]}}_{0}}} {\left[ {{\text{HSO}}_{{\text{3}}}^{ - }} \right]}}} \right. \kern-0em} {\left[ {{\text{HSO}}_{{\text{3}}}^{ - }} \right]}}$ = ${{{{k}_{{{\text{набл}}}}}} \mathord{\left/ {\vphantom {{{{k}_{{{\text{набл}}}}}} {\left[ {k_{{{\text{набл}}}}^{*}} \right]}}} \right. \kern-0em} {\left[ {k_{{{\text{набл}}}}^{*}} \right]}}$ = 300/3.2 × 106 ≈ 9.4 × × 10–5 и [Fe]0 ≈ 9.4 × 10–5 6 × 10–4 ≈ 6 × 10–8 моль/л. Найденное значение близко к величинам [Fe]0, оцененным нами ранее в [22] применительно к т. н. “некаталитическому” окислению сульфита. Используя найденные [Fe]0 и опытные kнабл при бóльших μ, приходим к зависимости $k_{{{\text{набл}}}}^{*}$ от μ по экспериментальным результатам этой и других рассматриваемых ниже работ.
В графическом виде пересчитанные к рН 2 значения $k_{{{\text{набл}}}}^{*}$ в зависимости от μ1/2/(1 + μ1/2) в соответствии с данными [17] показаны на рис. 1. Здесь приведено рассчитанное нами значение $k_{{{\text{набл}}}}^{*}$ и при μ = 0.5 моль/л по результатам опытов [17]. С этими $k_{{{\text{набл}}}}^{*}$ находятся в неплохом согласии вычисленные аналогичным образом значения $k_{{{\text{набл}}}}^{*}$ по результатам опытов [6] (рН 2, α ≈ 1.5 × 10–2). Эксперименты в работе [6] проводили в присутствии фоновых электролитов в диапазоне μ от ~0 до 1 моль/л (рис. 1). Кроме того, на рис. 1 представлены пересчитанные к рН 2 результаты наших вычислений $k_{{{\text{набл}}}}^{*}$ по данным группы работ [14, 19, 23], в которых вариации ионной силы обусловлены кондиционированием рабочих растворов. Хотя изменения ионной силы в этих опытах относительно невелики, прослеживаемый спад $k_{{{\text{набл}}}}^{*}$ с увеличением (μ1/2/(1 + μ1/2)) указывает на выраженное негативное ее влияние, подтверждая тем самым полученные нами значения $k_{{{\text{набл}}}}^{*}$ по данным опытов [6, 17].
Рис. 1.
Катализ ионами марганца окисления сульфита. Избыток сульфита над ионами металла. Влияние ионной силы растворов на наблюдаемую константу скорости каталитического окисления сульфита ($k_{{{\text{набл}}}}^{*},$ л моль–1 с–1): T = 298 K, pH 1.8–4, ионная сила растворов μ = 0.001–1.3 моль/л; ⚪ – [15], ◻ – [17], ☆ – [19], ▽ – [23], △ – [25)], ◇ – [24] без добавок сульфатов, ⊕ – [24] с добавками сульфатов.
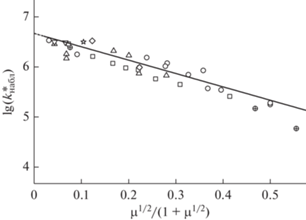
На рис. 1 приведены также вычисленные нами $k_{{{\text{набл}}}}^{*}$ по результатам экспериментов [24] с добавками сульфата калия. Эти добавки мы рассматривали как компонент ионной силы растворов. Найденный спад $k_{{{\text{набл}}}}^{*}$ с ростом (μ1/2/(1 + μ1/2)) близок к наблюдаемому в [6, 17]. Подобным образом подходили и к рассмотрению опытов [25] с добавками серной кислоты (≤0.1 моль/л). Но, в отличие от их толкования в [6], здесь принимали во внимание и рост кислотности растворов, и обусловленный этим сдвиг равновесия ионизации диоксида серы (SO2 + H2O ⇆ ${\text{HSO}}_{3}^{ - }$ + H+). С учетом этого при сопоставимых μ получаем близкие к рассчитанным в [6] значения $k_{{{\text{набл}}}}^{*}$ в работах [24, 25].
Экстраполяция найденных здесь $k_{{{\text{набл}}}}^{*}$ к μ1/2/(1 + μ1/2) → 0 ведет к $k_{{{\text{набл}}}}^{*}$(μ ≈ 0) ≈ 5 × × 106 л моль–1 с–1. Используя известные константы скорости элементарных реакций, входящих в выражение (1), выполним оценку константы скорости реакции (X). Ее абсолютное значение колеблется в пределах (3–20 000) × 106 л моль–1 с–1 [11, 26]. С учетом приводимого $k_{{{\text{набл}}}}^{*}$(μ ≈ 0) находим правдоподобное ее численное значение: kXI ≈ ≈ 1.5 × 107 л моль–1 с–1.
С чем связано найденное в работе негативное влияние μ на $k_{{{\text{набл}}}}^{*}$ в указанных условиях? Как отмечалось, такой режим окисления сульфита определяет конкуренция реакций (VI) и (IX), смещенная в кислых растворах и в избытке сульфита к (VI). Поскольку константа скорости реакции (VI) при рассматриваемых рН и изменении μ от 0 до 1 моль/л остается практически неизменной [27], решающим фактором найденного в работе негативного брутто влияния μ на $k_{{{\text{набл}}}}^{*}$ в избытке сульфита является, по-видимому, реакция разветвления цепи (IX).
Подытоживая сказанное, можно утверждать, что согласованную в целом картину негативного влияния ионной силы на наблюдаемую константу скорости реакции $\left( {k_{{{\text{набл}}}}^{*}} \right)$ в избытке сульфита удается получить лишь в рамках вырождено-разветвленного цепного механизма каталитического ОС, инициированного ионами примесного железа. Таким образом удается учесть влияние неконтролируемых изменений в опытах [Fe]0, а также корректировать воздействие рН на $k_{{{\text{набл}}}}^{*}$(μ). Усредненная по результатам всех наблюдений, эта зависимость имеет вид:
Численное значение $k_{{{\text{набл}}}}^{*}$(μ ≈ 0) оказывается при этом приблизительно на три порядка бóльшим, чем в опытах с фоновыми электролитами [6, 17]. В опытах [6, 17] для зависимости константы скорости (л моль–1 с–1) от μ сообщалось: lg(kнабл) = = 2.83–4.07μ1/2/(1 + μ1/2). Различия приведенных выражений обусловлено учетом в настоящей работе содержания примесных ионов железа в растворах ([Fe]0), а также воздействия рН на $k_{{{\text{набл}}}}^{*}$(μ). С этим связано и заметно меньшее влияние ионной силой на $k_{{{\text{набл}}}}^{*}$(μ) в сравнение с наблюдаемым в [6, 17] на kнабл.
ИЗБЫТОК ИОНОВ МЕТАЛЛА
Основная валентная форма ионов примесного железа в кислых растворах и в избытке ионов металла – каталитически активные ионы Fe(III), т.е. ζMn ≈ 1 и [Fe(III)] ≈ [Fe]0 [10]. Такая асимметрия распределения их валентных форм обусловлена замедлением расходования ${\text{HSO}}_{5}^{ - }$ в реакции с сульфитом (VI) и ростом скорости разветвления цепей (IX). Лимитирующим звеном продолжения цепей является реакция (XI), а их обрыв происходит по реакции (XIII) [21]. Для скорости реакции в стационарных условиях в рамках рассматриваемого здесь цепного механизма [28] приходим к выражению:
(2)
${{w}_{{{\text{Mn}}}}} = 2{{k}_{{{\text{XI}}}}}{{\left[ {{\text{HSO}}_{3}^{ - }} \right]}^{{3/2}}}{{({{k}_{{\text{I}}}}\chi {\kern 1pt} *{{\left[ {{\text{Fe}}} \right]}_{0}}{\text{/}}{{k}_{{{\text{XIII}}}}})}^{{1/2}}}.$Здесь χ* = ${\chi \mathord{\left/ {\vphantom {\chi {\left[ {{\text{HSO}}_{3}^{ - }} \right]}}} \right. \kern-0em} {\left[ {{\text{HSO}}_{3}^{ - }} \right]}},$ где χ = = [Fe(OH)SO3H+]/[Fe(III)] – доля11 сульфитных комплексов ионов 3-х валентного железа в их общем содержании в растворе в виде: Fe3+, FeOH2+, ${\text{FeHSO}}_{3}^{{2 + }},$ Fe(OH)SO3H+, ${\text{Fe(OH)}}_{2}^{ + },$ ${\text{FeSO}}_{3}^{ + }$ и Fe(ОН)SO3, а KI (см. сноску) – константы равновесия их образования [8], табл. 2. О полуторным по сульфиту и нулевом по ионам марганца порядкам реакции: wMn = k3/2_набл(μ ≈ 0) ${{\left[ {{\text{HSO}}_{3}^{ - }} \right]}^{{{3 \mathord{\left/ {\vphantom {3 2}} \right. \kern-0em} 2}}}}$, где k3/2_набл(μ ≈ 0) = 12 л1/2 моль–1/2 с–1 – усредненное по результатам систематизации известных данных значение наблюдаемой константы скорости реакции при μ ≈ 0 и Т ≈ 298 К [28].
Таблица 2.
Равновесия ионизации, гидролиза и комплексообразования компонентов при каталитическом окислении сульфита (Т = 298 К, μ ≈ 0 [8])
| № | Равновесие | Константа равновесия KI |
|---|---|---|
| I | SO2(aq) ⇆ ${\text{HSO}}_{3}^{ - }$ + H+ | 1.39 × 10–2 моль/л |
| II | ${\text{HSO}}_{3}^{ - }$ ⇆ SO32- + H+ | 6.24 × 10–8 моль/л |
| III | [Fe(H2O)6]3+ ⇆ [Fe(H2O)5(OH)]2+ + H+ | 6 × 10–3 л/моль |
| IV | [Fe(H2O)5(OH)]2+ + ${\text{HSO}}_{3}^{ - }$ ⇆ [Fe(H2O)4(HSO3)(OH)]+ | (6–10) × 102 л/моль |
| V | [Fe(H2O)5(OH)]2+⇆ [Fe(H2O)4(OH)2]+ + H+ | 7 × 10–4 моль/л |
| VI | [Fe(H2O)6]3+ + ${\text{HSO}}_{3}^{ - }$ ⇆ [Fe(H2O)5(HSO3)]2+ + H2O | 72 л/моль |
| VII | [Fe(H2O)6]3+ + ${\text{SO}}_{3}^{{2 - }}$ ⇆ [Fe(H2O)5(SO3)]+ + H2O | 7.3 × 106 л/моль |
| VIII | [Fe(H2O)5(OH)]2+ + ${\text{SO}}_{3}^{{2 - }}$ ⇆ [Fe(H2O)4(SO3)] + H2O | ≈2 × 107 л/моль |
| IX | [Fe(H2O)6]3+ + ${\text{SO}}_{4}^{{2 - }}$ ⇆ [Fe(H2O)5(SO4)]+ + H2O | ≈2.5 × 102 л/моль |
Вид выражения (2) показывает, что полуторный по сульфиту порядок является отражением лимитирующей роли реакций с участием сульфита в инициировании (I) и в продолжения цепи (XI). Для концентрации сульфита по ходу реакции находим: ${{\left[ {{\text{HSO}}_{3}^{ - }} \right]}_{t}}$ = ${{{{{\left[ {{\text{HSO}}_{3}^{ - }} \right]}}_{0}}} \mathord{\left/ {\vphantom {{{{{\left[ {{\text{HSO}}_{3}^{ - }} \right]}}_{0}}} {{{{\left( {1 + {{k}_{{{3 \mathord{\left/ {\vphantom {3 {2\_{\text{набл}}}}} \right. \kern-0em} {2\_{\text{набл}}}}}}}\left[ {{\text{HSO}}_{3}^{ - }} \right]_{0}^{{{1 \mathord{\left/ {\vphantom {1 2}} \right. \kern-0em} 2}}}t} \right)}}^{2}}}}} \right. \kern-0em} {{{{\left( {1 + {{k}_{{{3 \mathord{\left/ {\vphantom {3 {2\_{\text{набл}}}}} \right. \kern-0em} {2\_{\text{набл}}}}}}}\left[ {{\text{HSO}}_{3}^{ - }} \right]_{0}^{{{1 \mathord{\left/ {\vphantom {1 2}} \right. \kern-0em} 2}}}t} \right)}}^{2}}}}.$ Здесь ${{\left[ {{\text{HSO}}_{3}^{ - }} \right]}_{t}}$ и ${{\left[ {{\text{HSO}}_{3}^{ - }} \right]}_{0}}$ – концентрации сульфита в момент времени t и в начальный момент соответственно, моль/л. Отсюда явствует и наблюдавшееся в большинстве работ постоянство скорости реакции по ходу реакции, т.е. кажущийся 0-ой по сульфиту порядок при том, что порядок по нему полуторный [28].
Для выявления влияния μ на kнабл(μ) наряду с различиями рН и [Fe]0 необходимо учитывать связывание Fe(III) в комплексы с сульфитом и компонентами фоновых электролитов. Отметим, что за исключением данных работ [6] и [7] в литературе отсутствуют результаты исследования кинетики реакции в избытке ионов металла, в которых бы концентрации фонового электролита (хлориды, сульфаты и перхлораты щелочных и щелочноземельных металлов) превышали бы суммарную концентрацию ионов марганца и сульфита.
Так, в [7] в избытке ионов металла (α ≈ 25, [S(IV)] = 2.3 × 10–5 моль/л, рН 2.4) сообщалось об относительно слабом негативном влиянии ионной силы на kнабл(μ) в диапазоне μ = 0–1 моль/л. В качестве фонового электролита использовался NaClO4, а источником ионов марганца служил Mn(ClO4)2. Рассчитанные kнабл(μ) по кинетическим кривым, отнесенным в работе к 1-ому по сульфиту порядку, указывали на их снижение примерно вдвое от ~0.06 до ~0.03 с–1. Авторами [28] было отмечено, что данные работы [7] отвечают в действительности полуторному по сульфиту порядку, на что указало неожиданно высокое соотношение скоростей реакций в [7] и [15] в избытке ионов марганца (wMn[7]/wMn[15] ≈ 180). И это несмотря на одинаковый (1-ый!) по сульфиту порядок и лишь ∼30-ти кратное превышение концентрации сульфита в [7] в сравнении с [15].
Здесь цифры в подстрочнике отражают номера ссылок на источники. Пересчет найденных kнабл(μ) [15] к полуторному по сульфиту порядку приводит к k3/2_набл = ${{k}_{{{\text{набл}}}}}(\mu ){{[{\text{Mn(II)}}]} \mathord{\left/ {\vphantom {{[{\text{Mn(II)}}]} {{{{\left[ {{\text{HSO}}_{3}^{ - }} \right]}}^{{{1 \mathord{\left/ {\vphantom {1 2}} \right. \kern-0em} 2}}}}}}} \right. \kern-0em} {{{{\left[ {{\text{HSO}}_{3}^{ - }} \right]}}^{{{1 \mathord{\left/ {\vphantom {1 2}} \right. \kern-0em} 2}}}}}}$ ≈ 13 л1/2 моль–1/2 с–1, что находится в согласии с усредненным значением этой константы по данным многих наблюдений в избытке ионов марганца.
Учет влияния ионной силы на наблюдаемую константу скорости реакции в рассматриваемых условиях производили, опираясь на вычисления:
При этом принимали во внимание изменения с ионной силой констант равновесия процессов ионизации диоксида серы, гидролиза и комплексообразования ионов 3-х валентного железа (Ki), в том числе с фоновыми электролитами, табл. 2.
Вычисленные таким образом значения $k_{{{3 \mathord{\left/ {\vphantom {3 2}} \right. \kern-0em} 2}}}^{*}(\mu )$ по данным [7] в зависимости от μ1/2/(1 + μ1/2) представлены на рис. 2. В расчетах учитывали приводимое в этой работе [Fe]0 ≈ 2 × 10–8 моль/л, а также то, что в рассматриваемых условиях χ* ≈ ≈ KIV/(1 + [H+]/KIII). В пользу достаточно слабого влияния ионной силы на kнабл(μ) в избытке ионов металла свидетельствуют и результаты проточных опытов [14], рис. 2. Эти эксперименты выполняли при концентрациях сульфита ~2 × 10–3 моль/л. Поэтому в опытах с избытком ионов металла использовались бóльшие, чем в [7], концентрации ионов марганца. Их источником служил сульфат марганца, содержание которого определяет и ионную силу растворов в этих экспериментах. Как и в [7], в работе [14] сообщалось о близком к 0-ому по ионам металла порядке реакции при [MnSO4] ≥ 3 × 10–3 моль/л, хотя рост [MnSO4] до значения ~0.06 моль/л сопровождался увеличением скорости от ~6 × 10–4 до ≈1.5 × 10–3 моль л–1 с–1. При расчетах $k_{{{3 \mathord{\left/ {\vphantom {3 2}} \right. \kern-0em} 2}}}^{*}(\mu )$ по данным [14] учитывали формирование комплексов Fe(OH)SO3H+, ${\text{FeHSO}}_{3}^{{2 + }},$ ${\text{FeSO}}_{3}^{ + },$ а также ${\text{FeSO}}_{4}^{ + },$ табл. 2. Вычисленные значения $k_{{{3 \mathord{\left/ {\vphantom {3 2}} \right. \kern-0em} 2}}}^{*}(\mu )$ не обнаруживают, однако, отклонения от хода данных [7] и при максимальном содержании сульфата марганца (≈0.06 моль/л, μ ≈ ≈ 0.24 моль/л), рис. 2, что говорит, казалось, о незначительной роли сульфатов. Отсутствие их влияния как комплексообразователя в [14] может маскировать отмеченный выше рост wMn с увеличением [MnSO4], хотя нельзя исключать также воздействия неконтролируемых колебаний содержания ионов примесного железа в растворах ((2–100) × 10–8 моль/л) [9]. Аналогичным образом в настоящей работе были получены $k_{{{3 \mathord{\left/ {\vphantom {3 2}} \right. \kern-0em} 2}}}^{*}(\mu )$ по данным опытов по окислению SO2 в каплях малых размеров, находившихся в контакте c воздухом, содержащем примесь диоксида серы [29]. Источником ионов металла в этих опытах, как и в [14], служили растворы сульфата марганца. Их концентрации превосходили таковую сульфита, обусловленную растворением диоксида серы. Найденное при этом изменение $k_{{{3 \mathord{\left/ {\vphantom {3 2}} \right. \kern-0em} 2}}}^{*}(\mu )$ с ростом μ в [29] по абсолютной величине и темпам спада близко к найденным в [14], что исключает диффузионные ограничения в этих опытах, рис. 2. Согласие вычисленных нами величин $k_{{{3 \mathord{\left/ {\vphantom {3 2}} \right. \kern-0em} 2}}}^{*}(\mu )$ с найденными по результатам экспериментов в проточных условиях [14] указывает на невыраженное воздействие на кинетику реакции связывания сульфатами примесных ионов 3-х валентного железа. Похожий, хотя и не прямой, вывод о невлиянии сульфита можно сделать и по данным работы [6]. В этой публикации сообщалось о близких по абсолютной величине наблюдаемых константах скорости реакции в присутствии хлоридов и сульфатов, использовавшихся в опытах в качестве фоновых электролитов. При этом отмечалось, однако, и значительно более сильное негативное влияние μ на наблюдаемую константу скорости реакции, рис. 2, что связано, по-видимому, с нестационарным протеканием реакции, см. ниже.
Рис. 2.
Катализ ионами марганца окисления сульфита. Избыток ионов металла над сульфитом. Влияние
ионной силы растворов на наблюдаемую константу скорости каталитического окисления
сульфита ($k_{{{3 \mathord{\left/ {\vphantom {3 2}} \right. \kern-0em} 2}}}^{*}(\mu ),$ л1/2 моль–1/2 с–1): T = 298 K, pH 2–4, ионная сила растворов μ = 0.001–1.3 моль/л; ⚪ – [7], △ – [14], ◇ – [6, 15]. Серым цветом ( ) показаны значения констант в опытах по гетерофазному окислению сульфита в каплях
[29]. Пунктирная линия и окружающие ее точки (◇) при μ ≥ 0.1 моль/л отвечают данным работы
[6].
) показаны значения констант в опытах по гетерофазному окислению сульфита в каплях
[29]. Пунктирная линия и окружающие ее точки (◇) при μ ≥ 0.1 моль/л отвечают данным работы
[6].
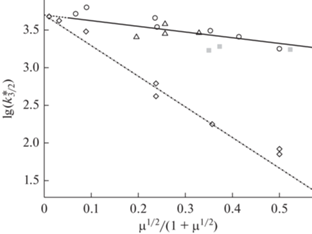
Экстраполяция данных рассматриваемых здесь работ о $k_{{{3 \mathord{\left/ {\vphantom {3 2}} \right. \kern-0em} 2}}}^{*}(\mu )$ к μ1/2/(1 + μ1/2) → 0 приводит к $k_{{{3 \mathord{\left/ {\vphantom {3 2}} \right. \kern-0em} 2}}}^{*}(\mu \approx 0)$ ≈ 2.5 × 105 л1/2 моль–1/2 с–1, см. продолжение линии 1 на рис. 2. К близкой величине ($k_{{{3 \mathord{\left/ {\vphantom {3 2}} \right. \kern-0em} 2}}}^{*}(\mu \approx 0)$ ≈ 1.5 × 105 л1/2 моль–1/2 с–1) приходим, рассчитывая константу скорости этой реакции с использованием известных при μ ≈ 0 значений констант скорости элементарных реакций (I), (III), а также оцененную нами ранее величину k(XIII) [20]. Согласие найденных таким образом k3/2(μ ≈ 0) указывает на корректность применяемого здесь подхода для оценки влияния ионной силы на кинетику реакции в избытке ионов марганца.
С представленными на рис. 2 данными контрастируют результаты опытов [6], см. пунктирную линию 2 и окружающие ее точки. При похожих условиях (α ≈ 15, [S(IV)] = 10–6 моль/л, рН 2) в этой публикации сообщалось о первых по сульфиту и ионам марганца порядках реакции и выраженном влиянии ионной силы на kнабл (л моль–1 с–1). Как уже отмечалось, факт регистрации первого по сульфиту порядка в работах [6] и [7] находится в противоречии с вычисленным нами соотношением скоростей расходования в них сульфита wMn[7]/wMn[6] ≈ 180 и с подобным их соотношением по данным опытов других авторов. Рассчитанные нами значения $k_{{{3 \mathord{\left/ {\vphantom {3 2}} \right. \kern-0em} 2}}}^{*}(\mu )$ по результатам экспериментов [6] приведены на рис. 1. Видно, что значения $k_{{{3 \mathord{\left/ {\vphantom {3 2}} \right. \kern-0em} 2}}}^{*}(\mu )$ по данным [6] и [7] близки лишь при невысоких μ.
При μ > 0.2 моль/л численные значения $k_{{{3 \mathord{\left/ {\vphantom {3 2}} \right. \kern-0em} 2}}}^{*}(\mu )$ заметно уступают тем, что группируются вокруг линии 1. Как отмечалось, особенностью опытов [6] является использование низких концентраций сульфита (≤10–6 моль/л). Расхождения $k_{{{3 \mathord{\left/ {\vphantom {3 2}} \right. \kern-0em} 2}}}^{*}(\mu )$ по данным [6] не следует, однако, связывать, с усилением действия ионной силы в опытах с низким содержанием сульфита. Их не стоит приписывать и вырождению цепного расходования сульфита при малых его концентрациях. Так, введение Mn(III) в десятитысячной доле от [Mn(II)] оказывается достаточным при [S(IV)] = 2.3 × 10–5 моль/л для полной конверсии сульфита [7]. Причина кажущегося усиления действия ионной силы в [6] видится в неустановившемся, вероятно, режиме реакции в этих экспериментах при повышенных μ (>0.2 моль/л). Стационарный режим реакции требует постоянства ζMn. Но основной формой ионов примесного железа в избытке ионов марганца является Fe(III), что связано с лимитирующей ролью звена их восстановления в реакции с сульфитом (III). В силу медленности этой реакции условия опытов [6] протекание ОС не отвечают, по-видимому, стационарным условиям. Как итог, для усредненной по данным наблюдаемой константы скорости реакции в избытке ионов металла находим: lg($k_{{{3 \mathord{\left/ {\vphantom {3 2}} \right. \kern-0em} 2}}}^{*}(\mu )$) = 3.7 – 0.76μ1/2/(1 + μ1/2). В сравнении с избытком сульфита влияние ионной силы на наблюдаемую константу скорости в избытке ионов металла, как видно, значительно снижается. Это связано с тем, что конкуренция реакций (VI) и (IX) в рассматриваемых условиях смещена к (IX). Как результат, происходит вырождение сильного негативного влияния ионной силы на константу скорости реакции разветвления цепи (IX), а вместе с этим и на наблюдаемую константу скорости ОС в избытке ионов марганца в сравнении таковым в избытке сульфита.
ЗАКЛЮЧЕНИЕ
В представленной работе приводятся результаты критического рассмотрения известных данных о влиянии ионной силы (μ) на кинетику кислородного окисления сульфита в кислых растворах (рН 1.3–4) в присутствии солей марганца(II). Их систематизация и обобщение, проведенные в рамках вырождено-разветвленного цепного механизма этой каталитической реакции, инициируемой ионами примесного железа, указали на существование двух различных режимов реакции окисления сульфита, что находится в согласии и с результатами экспериментов. В работе сообщается о переопределении известных в литературе выражений для зависимости от ионной силы наблюдаемых констант скорости реакции. В избытке сульфита над ионами металла наблюдаемую константу скорости (л моль–1 с–1) характеризует выраженная чувствительность к изменениям ионной силы: lg($k_{{{3 \mathord{\left/ {\vphantom {3 2}} \right. \kern-0em} 2}}}^{*}(\mu )$) = 6.7 – 2.67μ1/2/(1 + μ1/2). Для реакции, протекающей в избытке ионов марганца, найдено существенное ослабление влияния ионной силы на наблюдаемую константу скорости реакции: lg$\left( {k_{{{\text{набл}}}}^{*}} \right)$ = 3.7 – 0.76μ1/2/(1 + μ1/2) в таких условиях. При этом отмечена важность стационарного режима реакции при осуществлении опытов.
Список литературы
Семенов Н.Н. Цепные реакции. 2-е изд., испр. и доп. М.: Наука, 1986. 535 с.
Zhou D.N., Chen L., Li J.J., Wu F. // Chem. Eng. J. 2018. V. 346. P. 726.
Langner J., Rodhe H. // J. Atmos. Chem. 1991. V. 13. № 3. P. 255.
Jameton R.A., Muller J.G., Burrows C.J. // Compt. Rend. Chimie. 2002. V. 5. № 5. P. 461.
Herbarth O., Fritz G., Krumbiegel P., Diez U., Franck U., Richter M. // Environ. Toxicol. 2001. V. 16. № 3. P. 269.
Martin L.R., Hill M.W. //Atmos. Environ. 1987. V. 21. № 10. P. 2267.
Berglund J., Fronaeus S., Elding L. // Inorg. Chem. 1993. V. 31. № 21. P. 4257.
Brandt Ch., van Eldik R. // Chem. Rev. 1995. V. 95. № 1. P.119.
Ermakov A.N., Purmal A.P. // Kinet. Catal. 2002. V. 43. № 2. P. 249.
Yermakov A.N., Purmal A.P. // Progr. React. Mech. 2003. V. 28. № 3. P. 189.
Berglund J., Elding L.I., Buxton G.V., McGowan S., Salmon G.A. // J. Chem. Soc. Faraday Trans. 1994. V. 90. № 21. P. 309.
Brandt Ch., van Eldik R. // Transit. Met. Chem. 1998. V. 23. № 6. P. 667.
Grgić I., Berčič G. // J. Atmos. Chem. 2001. V. 39. № 2. P. 155.
Coughanowr D.R., Krause F.E. // Ind. Eng. Chem. Fund. 1965. V. 4. № 1. P. 61.
Martin L.R., Hill M.W. // J. Phys. E: Sci. Instrum. 1987. V. 20. № 11. P. 1383.
Yermakov A.N., Zhitomirsky B.M., Poskrebyshev G.A., Sosurakov D.M. // J. Phys. Chem. 1993. V. 97. № 41. P. 10712.
Huss A., Jr., Lim P.K., Eckert C.A. // J. Phys. Chem. 1982. V. 86. № 21. P. 4224.
Connick R.E., Zhang Y.-X. // J. Inorg. Chem. 1996. V. 35. № 16. P. 4613.
Pasiuk-Bronikowska W., Bronikowski T. // Chem. Engng. Sci. 1981. V. 36. № 1. P. 215.
Yermakov A.N., Poskrebyshev G.A., Purmal A.P. // Prog. React. Kinet. 1997. V. 22. № 2. P. 141.
Betterton E.A., Hoffmann M.R. // J. Phys. Chem. 1988. V. 92. № 21. P. 5962.
Ермаков А.Н. // Кинетика и катализ. 2021. Т. 62. № 5. С. 518.
Hoather R.C., Goodeve C.F. // Trans. Faraday Soc. 1934. V. 30. P. 1149.
Hudson J.L., Erwin J., Catiopovich N.M. Research Report. Kinetics of sulfur dioxide oxidation in aqueous solutions. The University of Illinois Urbana. Illinois 61801. US Environmental Protection Agency EPA-600/7-79-030. 1979. January. P. 82.
Johnstone H.F., Coughanowr D.R. // Ind. Engng. Chem. 1958. V. 50. № 8. P. 1169.
Herrmann H., Jacobi H.W., Raabe G., Reese A., Zellner R. // Fresenius J. Anal. Chem. 1996. V. 355. № 3–4. P. 343.
Warneck P., Mirabel P., Salmon G.A., Eldik R. van, Vinckier C., Wannowius K.J., Zetsch C. Review of the activities and achievements of the EUROTRAC subproject HALIPP (Ed. P. Warneck), Heterogeneous and liquid phase processes. Berlin, Heidelberg: Springer-Verlag, 1996. P. 7.
Ермаков А.Н. // Кинетика и катализ. 2021. Т. 62. № 5. С. 518.
Matteson M.J., Stober W., Luther H. // Ind. Eng. Chem. Fund. 1969. V. 8. № 4. P. 677.
Дополнительные материалы отсутствуют.
Инструменты
Кинетика и катализ