

Кинетика и катализ, 2022, T. 63, № 2, стр. 212-222
Биокаталитические гетерогенные процессы низкотемпературного синтеза моноэфиров диолов
Г. А. Коваленко a, *, Л. В. Перминова a, М. В. Шашков a, А. Б. Беклемишев a, b
a ФГБУН ФИЦ Институт катализа им. Г.К. Борескова СО РАН,
630090 Новосибирск, просп. Акад. Лаврентьева, 5, Россия
b НИИ биохимии ФИЦ ФТМ СО РАН
630117 Новосибирск, ул. Тимакова, 2, Россия
* E-mail: galina@catalysis.ru
Поступила в редакцию 02.07.2021
После доработки 14.08.2021
Принята к публикации 14.08.2021
- EDN: ICRBUL
- DOI: 10.31857/S0453881122020046
Аннотация
Проведены исследования процессов низкотемпературного синтеза сложных эфиров гептановой (энантовой, С7:0) кислоты и различных диолов с участием гетерогенных биокатализаторов, приготовленных путем адсорбционной иммобилизации рекомбинантной липазы rPichia/lip на макропористом углеродном аэрогеле. В качестве субстратов изучены диолы, различающиеся длиной углеродного скелета (от 2 до 6 атомов С), положением ОН-группы и изомерией углеродного скелета, такие как 1,2-этандиол (этиленгликоль) и его олигомеры (ди- и три-), 1,2-пропандиол (пропиленгликоль), 1,3-пропандиол, 1,4-бутандиол, 1,6-гександиол, а также 2-этил-1,3-гександиол. Реакцию этерификации и синтеза моноэфиров гептановой кислоты проводили в реакторах периодического действия в очень мягких условиях (20 ± 2°С, 1 бар). Свойства приготовленных биокатализаторов, такие как ферментативная активность, субстратная специфичность и операционная стабильность, были исследованы в зависимости от строения молекулы диола и природы органического растворителя (хлороформ, гексан, ацетон). Обнаружено, что короткоцепочечные диолы С2–С4 необратимо ингибировали иммобилизованную липазу, и биокатализатор полностью инактивировался в течение 1–3 реакционных циклов. Максимальное значение активности, равное 83 ЕА/г, и конверсию кислоты, равную 94% за 24 ч, наблюдали в реакции этерификации гептановой кислоты 1,6-гександиолом, при этом в изученных условиях доля моноэфира составила более 99%. Была обнаружена корреляция между активностью приготовленных биокатализаторов и длиной молекулы симметричных диолов; так, скорость реакции этерификации повышалась при увеличении расстояния между концевыми ОН-группами. Поскольку хлороформ инактивировал адсорбированную rPichia/lip, то были подобраны условия реактивации биокатализаторов путем замены реакционной среды, а именно, растворителя на гексан и диола на бутанол.
ВВЕДЕНИЕ
Сложные эфиры органических кислот и спиртов являются ценными продуктами органического синтеза и широко востребованы на рынке. Эти продукты находят применение в различных областях промышленности, включая парфюмерную (душистые вещества), косметическую (эмоллиенты), пищевую и фармацевтическую отрасли, а также производство моющих средств, лаков, красок и синтетических моторных топлив (поверхностно-активные вещества).
Эфиры жирных кислот и многоатомных спиртов (алифатических полиолов) обладают высокими чистящими, моющими, а также эмульгирующими, диспергирующими и стабилизирующими свойствами. Их используют в производстве пластмасс в качестве мономеров и пластификаторов. Кроме того, данные соединения являются одним из компонентов синтетических смазочных масел. Добавление к нефтяной основе технически обоснованного количества эфира с целью получения так называемого сложноэфирного масла приводит к существенному улучшению его эксплуатационных параметров, таких как повышение смазочных и вязкостно-температурных характеристик, снижение температуры застывания [1]. Высокое качество сложноэфирных смазочных материалов служит основой для разработки базовых авиационных и редукторных масел, а также универсальных и всесезонных (в том числе, арктических) моторных масел.
Высококачественные полусинтетические масла отечественного производства были разработаны в 70–80 гг. XX в. [1, 2]. В маслах 1-го поколения использовали сложные эфиры алифатических двухосновных кислот и одноатомных спиртов. Диэфирные масла имели высокий индекс вязкости (до 160) и были работоспособны в интервале температур от –60 до +175°С. Наиболее широкое применение нашел диизооктиловый эфир себациновой кислоты (диоктил себацинат, ДОС) [1]. Себациновая кислоты представляет собой двухосновную предельную карбоновую кислоту (декандиовая кислота, НООС–(СН2)8–СООН). Масло, приготовленное на основе высокоочищенной нефтяной базовой жидкости и ДОС, работало при температуре –50 до +50°С. Полностью синтетическое масло с добавлением ДОС, предназначенное для форсированных транспортных дизелей специального назначения, позволяло осуществлять холодный запуск при –30°С. Диоктил себацинат как пластификатор является продуктом, востребованным полимерной промышленностью.
В маслах 2-го поколения, разработанных для самолетов с газотурбинным двигателем, летающих с двукратным превышением скорости звука, в качестве основы применяют сложные эфиры многоатомных спиртов. В качестве таких спиртов используют полиолы неопентана (2,2-диметилпропан, С(СН3)4), триметилолэтан, триметилолпропан, пентаэритрит (2,2-бис(гидроксиметил) пропан-1,3-диол, C(CH2OH)4), а также их димеры (ди-триметилолпропан, ди-пентаэритрит). Масла 2-го поколения превосходят масла 1-го поколения по смазочным характеристикам, летучести, термостабильности и ряду других показателей [1, 2]. Например, масла 2-го поколения работоспособны в интервале температур от –60 до +200°C. Примером добавки в такие масла является продукт реакции этерификации пентаэритрита и монокарбоновых кислот (или их смесей) алифатического ряда, нормального строения, например, синтетических С5–С9 жирных кислот, получаемых окислением нефтяных парафинов. Наиболее ценные продукты – эфиры энантовой (С7:0) и пеларгоновой (С9:0) кислот. Важное практическое значение имеют также несимметричные сложные эфиры димерных многоатомных спиртов (ди-триметилолпропана, ди-пентаэритрита) и сравнительно доступных низкомолекулярных карбоновых кислот С5–С6.
Первая и единственная промышленная установка по производству сложных эфиров синтетических жирных кислот и пентаэритрита была построена и пущена в эксплуатацию в 1961 г. В Уфе по технологии, разработанной заводом им. Шаумяна. Процесс химической этерификации проводили при температуре 160–210°C. Воду, образовавшуюся в реакции, удаляли путем подачи азота при интенсивном перемешивании реакционной среды. В оптимальных условиях степень конверсии кислот достигалa 98%. При использовании в качестве гомогенных катализаторов сильнокислотных или щелочных реагентов наблюдались как высокая коррозия оборудования, так и сложности с очисткой целевого продукта. Применяемый для повышения полноты этерификации избыток карбоновых кислот полностью терялся вместе со щелочными стоками. Процесс характеризовался достаточно низким выходом целевого продукта (~70%), в котором содержались спирты с непрореагировавшими ОН-группами, что снижало термоокислительную стабильность продукта. При химическом синтезе всегда получалась смесь несимметричных эфиров неустановленного состава. Начиная с 2002 г. в России отсутствует производство сложных эфиров полиолов и практически остановлено производство химически чистых индивидуальных веществ: монопента-эритритолов, ди- и три-пентаэритритолов, а также низкомолекулярных синтетических жирных кислот С5–С9, без которых невозможен выпуск высококачественных смазочных материалов для современной техники [2].
Ферментативный “зеленый” синтез разнообразных сложных эфиров с участием липаз (гидролаза эфиров глицерина) (К.Ф. 3.1.1.3) в качестве активных компонентов катализаторов привлекают пристальное внимание специалистов в области органического синтеза благодаря уникальным свойствам этих ферментов, прежде всего, наличию хемо-, регио- и стереоспецифичности катализируемых химических реакций. На сегодняшний момент липазы и гетерогенные биокатализаторы на их основе являются многотоннажными промышленными продуктами; например, компания “NOVO” (novozymes.com) является лидером в производстве и продаже гомогенных и гетерогенных биокатализаторов, обладающих активностью липазы, в том числе, предназначенных для органического синтеза. Следует отметить, что в настоящее время получены кристаллические формы липаз бактериального происхождения, расшифрованы их аминокислотные последовательности, промоделированы третичные структуры их белковых молекул, что значительно ускоряет генно-инженерные работы по конструированию рекомбинантных (мутантных) высокопродуктивных штаммов, продуцирующих липазы с заданными свойствами. Липазы обладают уникальной среди ферментов способностью сохранять ферментативную активность в органических растворителях, практически не содержащих воды (известно, для работы практически всех ферментов необходима водная буферная среда). В научных обзорах [3–9] описаны различные липазы и их каталитические свойства, а также области их практического применения. Данные, представленные в [3–9], позволяют сделать вывод о том, что “зеленые” биокаталитические процессы, включая ферментативный синтез сложных эфиров, являются достойной альтернативой органическому синтезу, поскольку дают возможность проводить реакции при низких температурах (ниже 50°С) без образования побочных продуктов.
В настоящее время опубликовано небольшое число научных работ по исследованию “зеленого” синтеза эфиров многоатомных спиртов, в том числе, диолов. В работе [10] была изучена реакция синтеза эфиров лауриновой кислоты (С12:0) и симметричного 1,4-бутиленгликоля с участием панкреатической липазы. В оптимальных условиях, при соотношении реагентов кислота : диол = = 1 : 5, конверсия лауриновой кислоты составила 78% при 30°С за 24 ч [10]. При использовании липазы в трех реакционных циклах (кратность равна 2), конверсия кислоты уменьшилась с 78 до 27% [10], т.е. скорость реакции этерификации в третьем реакционном цикле упала почти в 3 раза. В работе [11] авторы исследовали реакцию этерификации пропионовой кислоты 2-этил-1,3-гександиолом, протекающую в гексане с участием биокатализатора Lipozyme® IM (“NOVO Nordisk Bioindustrials, Inc.”), приготовленного путем иммобилизации липазы из Mucor miehei на макропористом полимере. Для удаления воды (продукта реакции) применяли ионообменную смолу марки Dowex, реакцию проводили в реакторе с движущимся слоем мелкодисперсного биокатализатора (а simulated moving bed reactor (SMBR)). Конверсия кислоты в изученных условиях составила 79–93%, выход моноэфира – 25–44% при температуре 22 ± 2°С [11].
Интересная реакция ферментативного синтеза полиэфиров описана в работе [12]. Реакцию переэтерификации и одновременной полимеризации между диэфирами C4–C10 дикарбоновых кислот и алифатическими диолами C4–C8 проводили с участием биокатализатора Novozym® 435 (“NOVO”), приготовленного иммобилизацией липазы В из Candida antarctica на метакриловом полимере. В данной реакции были успешно синтезированы различные полиэфиры, например, из диэфиров с длиной внутренней углеродной цепи от 4 атомов углерода (С4) (сукцинат) до С10 (себацинат) и диолов с длиной цепи от С4 (1,4-бутандиол) до С8 (1,8-октандиол). Синтезированные в работе [12] полиэфиры имели молекулярную массу от 6000 до 13 000 Да, конверсия составила ≥90%. Так, в реакции между диметил адипинатом и 1,8-октандиолом при 85°С за 24 ч получили продукт с молекулярным весом 7141 Да [12].
В работе [13] была исследована реакция синтеза эфиров насыщенных жирных кислот (каприновой С10:0, лауриновой С12:0 и пальмитиновой С16:0) и полиэтиленгликоля ПЭГ-400, протекающая в смеси бензола с гексаном при участии панкреатической липазы. В оптимальных условиях при соотношении субстратов (кислота : ПЭГ = 1 : 1.8) образовывался сложный эфир, при этом значения конверсии каприновой, лауриновой и пальмитиновой кислот составили 80, 78 и 44% соответственно при 25°С за 48 ч [13].
Биокатализаторы, приготовленные путем адсорбционной иммобилизации рекомбинантной липазы rPichia/lip на макропористом углеродном аэрогеле (МУА), ранее были изучены в реакции этерификации насыщенных жирных кислот С4–С18 алифатическими спиртами С2–С16 [14–17]. Было показано, что скорость реакции этерификации гептановой (энантовой, С7:0) кислоты с участием иммобилизованной rPichia/lip является максимальной независимо от природы первичного алифатического спирта С3–С16 и состава органического растворителя или смесей растворителей [16, 17].
В настоящей работе проведены сравнительные исследования биокатализаторов, приготовленных путем адсорбции липазы rPichia/lip на МУА, в процессах низкотемпературного синтеза эфиров гептановой кислоты С7 с различными диолами, различающимися длиной (от 2 до 6 атомов С), изомерией углеродного скелета и положением ОН-группы, такими как 1,2-этандиол (этиленгликоль) и его олигомеры (ди- и три-), 1,2-пропандиол (пропиленгликоль), симметричные диолы, содержащие концевые первичные ОН-группы – 1,3-пропандиол, 1,4-бутандиол, 1,6-гександиол, а также разветвленный 2-этил-1,3-гександиол. Для проведения реакции этерификации использовали органические растворители (гексан, хлороформ, ацетон), выбор которых определялся, прежде всего, растворимостью субстратов и продуктов ферментативной реакции. Были проведены сравнительные исследования по влиянию структуры диолов на свойства приготовленных биокатализаторов, а именно, на их ферментативную этерифицирующую активность, субстратную специфичность и операционную стабильность, в периодическом процессе низкотемпературного синтеза моноэфиров гептановой кислоты и различных диолов.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Гетерогенные биокатализаторы получали путем адсорбции рекомбинантной липазы T. lanuginosus (обознач. rPichia/lip) на макропористом углеродном аэрогеле (МУА), описанном в работе [15]. Для этого навеску адсорбента МУА (1 вес. ч.) заливали буферным раствором (0.02 М фосфатный буфер, рН 7.0) липазы (100 об. ч.) с начальной концентрацией белка 2.9 мг/мл, и при периодическом перемешивании выдерживали в течение 1 сут при 20 ± 2°С. Затем раствор липазы декантировали, биокатализатор промывали буфером, удаляли излишки влаги фильтровальной бумагой и сушили до суховоздушного состояния в тонком стационарной слое в условиях окружающей среды (20 ± 2°С, 1 бар) в течение 1–2 сут. Величину адсорбции липазы (мг/г) рассчитывали, исходя из концентрации белка в растворе до и после адсорбции. Концентрацию белка в растворе измеряли при помощи красителя Кумасси G-250 методом [18], для построения градуировочного графика использовали бычий сывороточный альбумин (“SIGMA”). Величина адсорбции составила 100 ± 25 мг/г.
Реакцию этерификации проводили при 20 ± ± 2°С в реакторах смешения периодического действия, а именно, в герметично закрываемых сосудах (виалах) объемом 20 см3. Высушенные биокатализаторы (0.01 г) заливали реакционной средой (3.0 мл), содержащей субстраты – кислоту и диол в хлороформе, и интенсивно перемешивали в течение реакции. Было найдено, что растворимость диолов в хлороформе была максимальной по сравнению с другими растворителями (гексаном, диэтиловым эфиром, ацетоном); содержание биокатализатора в данной реакционной среде, составило 2.2 мас. %. Биокатализаторы, потерявшие активность в хлороформе, реактивировали в среде гексана в реакции синтеза бутил гептаноата (контрольная реакция). Поскольку плотность гексана в 2 раза ниже, чем у хлороформа, то содержание биокатализатора в среде было равно 4.2 мас. %.
В реакции этерификации начальная концентрация гептановой С7 кислоты составляла 0.25 ± ± 0.03 моль/л; бутанол и диолы вносили в двух- и трехкратном молярном избытке соответственно; в случае диолов на каждую –СООН приходилось шесть ОН-групп. В качестве растворителей использовали гексан, хлороформ и ацетон (табл. 1). Начальную скорость реакции определяли по линейному участку кинетической кривой убыли гептановой кислоты, концентрацию которой анализировали методом титриметрии с помощью этанольного раствора NaOH с известной молярностью (0.0256 ± 0.0006 моль л–1) с применением фенолфталеина как индикатора точки эквивалентности. Скорость реакции и активность биокатализаторов рассчитывали и выражали, соответственно, в мкмоль л–1 с–1 и в единицах активности (ЕА) на 1 г сухого биокатализатора (1 ЕА = = мкмоль мин–1).
Таблица 1.
Свойства биокатализаторов, приготовленных адсорбцией rPichia/lip на макропористом углеродном аэрогеле, в контрольной реакции синтеза бутил гептаноата
| Растворитель | Начальная активность*, ЕА/г | Конверсия кислоты за 6 ч, % | Сохранившая активность после пяти реакционных циклов, % |
|---|---|---|---|
| Гексан | 399.7 ± 28.0 | 93 | 90 |
| Хлороформ | 23.3 ± 2.3 | 40 | <10 |
| Ацетон | 4.0 ± 0.3 | 7 | 20 |
Ферментативную активность биокатализаторов (ЕА/г) измеряли в периодических реакционных циклах. В первом цикле биокатализаторы проходили стадию кондиционирования, при этом их активность увеличивалась в 2–4 раза. Это было обусловлено тем, что внутри биокатализатора аккумулировалась вода, образующаяся в ходе этерификации, при этом формировалось благоприятное водное микроокружение для липазы, адсорбированной на МУА. После окончания этерификации реакционную смесь, содержащую небольшое количество кислоты, спирт и сложный эфир, декантировали. Биокатализаторы многократно промывали растворителем (гексаном), затем выдерживали в растворителе в течение не менее 20 ч для десорбции субстратов и продукта реакции с поверхности МУА. Следующий реакционный цикл проводили с отмытым биокатализатором в среде, состав которой описан выше.
Исходные реагенты и продукты реакции анализировали методом газовой хроматомасс-спектрометрии (ГХ-МС). Для всех анализов была использована высокополярная колонка собственной разработки на основе ионной жидкости N‑пропил-6-метил-хинолиний-бис(трифторметил-сульфонил)имида (10 м × 0.25 мм × 0.2 мкм) [19]. Условия разделения для всех образцов следующие. Температурная программа термостата колонок включала выдержку при 100°С (3 мин), далее программирование со скоростью 10°С/мин до конечной температуры 280°С. Температура испарителя составляла 280°С, скорость потока газа-носителя (гелия) – 1 мл/мин. Условия работы масс-спектрометра такие: электронная ионизация – 70 эВ, температура источника ионизации – 230°С, температура переходной линии – 250°С. Спектр регистрировали в режиме сканирования в диапазоне 40–450 m/z. Для качественного анализа использовали тонкослойную хроматографию (ТСХ), аналогично описанному в работе [14].
Статистическую обработку результатов проводили по критерию Стьюдента c доверительной вероятностью 0.95, количество измерений n = 3–6. Относительная ошибка не превышала 10%.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Как было показано ранее [14–17], скорость реакции ферментативной этерификации гептановой (энантовой, С7:0) кислоты является максимальной по величине независимо от длины углеродного скелета первичного алифатического спирта С3–С16, поэтому эта кислота была выбрана в качестве субстрата для исследований гетерогенных биокатализаторов, полученных адсорбцией rPichia/lip на МУА.
При подборе органического растворителя для приготовления реакционной среды использовали следующие критерии: 1) субстрат ферментативной реакции – диол, и продукты – моно- и диэфиры, хорошо растворимы в реакционной среде; 2) не наблюдается существенная инактивация исследуемых биокатализаторов в течение реакционного цикла; 3) растворители не обладают высокой токсичностью, неогнеопасны и доступны. Выбор растворителя также определялся достижением практически важных целевых показателей, таких как: i) максимальная скорость реакции этерификации с участием диолов, или, соответственно, максимальная конверсия кислоты за минимальное время; ii) максимальный выход целевого продукта – МОНОэфира (без образования диэфира); iii) максимальная продуктивность биокатализатора, прежде всего, за счет его высокой операционной стабильности и многократного использования в реакционных циклах.
Ранее в работах [16, 20] для реакции синтеза эфиров этилен- и пропиленгликолей были выбраны и изучены следующие растворители: ацетон и смесь ацетона с гексаном в соотношении 3 : 1 (об. ч). Как показали исследования [16], ацетон инактивировал rPichia/lip. Так, скорость реакции синтеза эфира гептановой кислоты и гексадеканового (цетилового, С16) спирта уменьшалась почти на порядок, если ацетон использовали в качестве сорастворителя в гексане (1/4 : 1, об. ч) [16]. Из табл. 1 также видно, что скорость реакции синтеза бутил гептаноата в ацетоне на два порядка ниже по сравнению со скоростью этерификации в гексане. Кроме того, биокатализаторы, работающие в ацетонсодержащих средах, обладали сравнительно низкой операционной стабильностью и в течение шести реакционных циклов теряли до 80% первоначальной активности [16]. Таким образом, смеси гексана и ацетона были далеки от оптимальных с точки зрения влияния на ферментативную активность иммобилизованной липазы. В настоящей работе в качестве растворителя был выбран хлороформ, в котором все изученные диолы и синтезированные эфиры хорошо растворялись. Скорость реакции этерификации в хлороформе была в ~17 раз ниже по сравнению со скоростью этерификации в гексане (табл. 1). Хлороформ, аналогично ацетону, значительно инактивировал активный компонент биокатализатора, а именно, rPichia/lip, что приводило к полной потере биокаталитической активности в течение 3–4 реакционных циклов (табл. 1). Однако, как показано ниже, инактивация липазы в хлороформе протекала обратимо, т.е. активность биокатализаторов частично или полностью восстанавливалась при замене хлороформа на гексан.
В настоящей работе были исследованы свойства приготовленных биокатализаторов в реакции синтеза эфиров гептановой кислоты и различных диолов (табл. 2). Максимальная начальная скорость реакции наблюдалась при этерификации с участием 1,6-гександиола; при выбранном молярном соотношении субстратов (кислота : диол = 1 : 3) доля моноэфира составила 99% при практически полной конверсии кислоты, равной 94% за 24 ч (табл. 2).
Таблица 2.
Начальная активность биокатализаторов в реакции этерификации гептановой кислоты и различных диолов в хлороформе
| Диол | Начальная активность биокатализатора, ЕА/г |
Конверсия кислоты, % (за х ч) | Доля моноэфира, % (за х ч) |
|---|---|---|---|
| Симметричные, первичные ОН-группы | |||
| 1,2-Этандиол | <1 | 2 (24 ч) 18.5 (168 ч) |
100.0 (24 ч) 99.3 (168 ч) |
| 1,3-Пропандиол | <1 | 15 (24 ч) 71 (168 ч) |
96.5 (24 ч) 90.2 (168 ч) |
| 1,4-Бутандиол | 80.5 | 25* (2 ч) | |
| 1,6-Гександиол | 83.7 | 94.4 (24 ч) | 98.6 (24 ч) |
| Несимметричные, первичная и вторичная ОН-группы | |||
| 1,2-Пропандиол | <1 | 25 (24 ч) 69 (168 ч) |
98.6 (168 ч) |
| 2-Этил-гексан-1,3-диол | 67.6 | 67.8 (6 ч) | 97.9 (6 ч) |
| Этиленгликоли, первичные ОН-группы | |||
| Этиленгликоль | <1 | 2 (24 ч) 18.5 (168 ч) |
100.0 (24 ч) 99.3 (168 ч) |
| Ди-этиленгликоль | 25.7 | 53.6 (7 ч) | 98.3 (7 ч) |
| Три-этиленгликоль | 58.0 | 84.1 (7 ч) | 96.5 (7 ч) |
Скорость синтеза эфиров гептановой кислоты с короткоцепочечными диолами С2–С3, содержащими как первичные, так и вторичные ОН-группы, была чрезвычайно низкой (близкой к нулю); конверсия кислоты выше 50% регистрировалась через 7 сут. Проведенные анализы методами ГХ-МС и ТСХ показали, что в изученных условиях синтезировались преимущественно моноэфиры диолов (рис. 1). В случае 1,4-бутандиола реакция протекала в течение первых 2 ч, начальная активность была рассчитана из начального линейного участка кинетической кривой и составила 80.5 ЕА/г (табл. 2). Затем реакция останавливалась на конверсии кислоты 25%, и дальнейшего изменения в величине конверсии не наблюдалось в течение последующих 1–2 сут. Было также обнаружено, что диолы С2–С4 необратимо инактивировали липазу rPichia/lip, поскольку биокатализаторы не восстанавливали этерифицирующую активность после реактивации в контрольной реакции синтеза бутил гептаноата в гексане. Так, после первого реакционного цикла синтеза эфиров гептановой кислоты и 1,4-бутандиола активность (А) биокатализатора во втором цикле составила лишь 0.5% от его первоначальной (А0) активности, А = 1.1 ЕА/г от А0 =210.1 ЕА/г. Коротко-цепочечные диолы С2-С4 имеют 3 класс опасности, и, вероятно, способны инактивировать различные ферменты, в том числе, жизненно важные, такие как, липазы (wikipedia.org).
Рис. 1.
ГХ-МС- и ТСХ-анализ реакционных смесей, в которых использовали 1,2-этандиол (а), 1,3-пропандиол (б), 1,2-пропандиол (в): 1 – исходный диол, 2 – гептановая кислота, 3, 3а – моноэфир, 4 – диэфир.
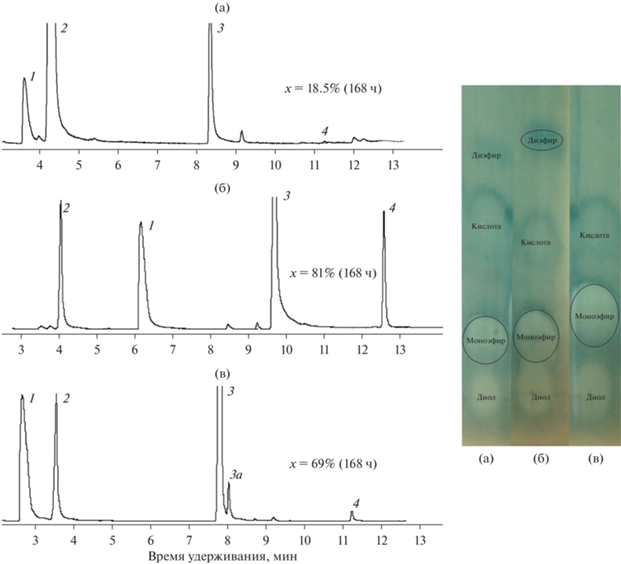
Более длинные диолы, 1,6-гександиол и разветвленный 2-этил-гексан-1,3-диол, этерифицировались с заметными скоростями (табл. 2). В изученных условиях при конверсии гептановой кислоты выше 70% доля моноэфиров 1,6-гександиола и 2-этил-гексан-1,3-диола составили 99 и 98% соответственно (табл. 2, рис. 2). Обнаруженные этиловые и бутиловые эфиры гептановой кислоты (рис. 2, пики 1, 1а) образовывались из примесей этанола и бутанола, присутствующих в реактивах. При этерификации 2-этилгексан-1,3-диола этерифицировалась только первичная ОН-группа (рис. 2б, пик 4).
Рис. 2.
ГХ-МС-анализ реакционных смесей, в которых использовали 1,6-гександиол (а) и 2-этилгексан-1,3-диол (б): 1, 1а – примесные эфиры (этил гептаноат, бутил гептаноат), 2 – гептановая кислота, 3 – исходный диол, 4, 4а – моноэфир, 5 – диэфир.
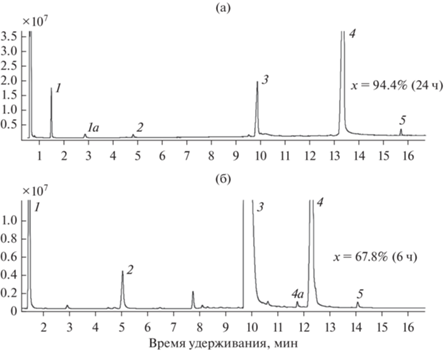
Как было показано ранее, вторичные и третичные ОН-группы спиртов практически не участвуют в этерификации, катализируемой rPichia/lip; скорость реакции, соответственно, на два порядка ниже или равна нулю по сравнению с первичными ОН-группами [17]. На примере бутанола и его изомеров было продемонстрировано, что вторичные и третичные спирты не являются ни субстратами, ни ингибиторами rPichia/lip, в отличии от соответствующих диолов С2–С4, которые, как отмечалось выше, необратимо ингибируют/инактивируют липазу. В настоящей работе подтверждено, что вторичная группа 2-этил-гексан-1,3-диола практически не участвовала в реакции (рис. 2б, пик 4 и 4а).
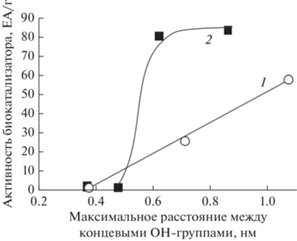
В представленной работе авторами было высказано предположением о том, что важную роль при этерификации гептановой кислоты играет размер молекулы диола, а именно, максимальное расстояние между первичными концевыми ОН-группами. Действительно, наблюдалась корреляция между активностью приготовленных биокатализаторов и данным расстоянием: чем оно больше, тем выше этерифицирующая активность приготовленных биокатализаторов (рис. 3). Для этиленгликоля и его олигомеров наблюдается практически линейная зависимость (рис. 3, кривая 1), анализ которой позволил предложить следующий механизм синтеза сложных эфиров с участием липазы rPichia/lip. Из обзорной литературы [7, 21–23] известно, что в активном центре (а.ц.) липазы находятся остатки трех аминокислот Ser–Glu–His (серин–глутаминовая кислота–гистидин), участвующих в катализе реакции гидролиза триглицеридов по кислотно-основному механизму. Липаза T. lanuginosus и аналогичная ей липаза rPichia/lip относятся к типу RmL, для которых характерно следующее строение активного центра: а. ц. с каталитической триадой аминокислот расположен близко к поверхности ферментной глобулы; для субстрата-кислоты и его ацильной группы имеется узкая щель, для субстрата-спирта – широкая щель а. ц. [24]. С помощью программы HyperChem® были рассчитаны максимальные расстояния (Lmax, нм) между концевыми ОН-группами в “вытянутой” молекуле диолов, а также максимальное расстояние между крайними протонами в молекуле гептановой кислоты. При этерификации первый субстрат (S1), гептановая кислота (Lmax = 1.0 нм), входит в активный центр липазы (Е) и образует промежуточный фермент-субстратный комплекс ЕS1, к которому подходит второй субстрат (S2), в данной случае, диол. Если максимальная длина диола больше глубины залегания активного центра, то фермент-субстратный комплекс ЕS1S2 является продуктивным и “распадается” на Е и продукты – сложный эфир и воду. Если диол короткоцепочечный, то его вторая ОН-группа входит в а.ц.; в этом случае возможно взаимодействие этой группы с остатками аминокислот стенок щелей а.ц., глубина которого оценивается в 0.6–1.0 нм (область максимальной активности на рис. 3, кривая 2). В результате, С2–С3-диолы либо деформируют а.ц., либо блокируют выход продуктов реакции, прежде всего, эфира. Длина молекул С4–С6-диолов сравнима с глубиной а.ц., катализ протекает эффективно, особенно с 1,6-гександиолом (рис. 3). Интересно, что для олигомеров этиленгликоля (ди- и три-) наблюдалась линейная зависимость от максимальной длины молекулы (рис. 3).
Рис. 3.
Этерифицирующая начальная активность биокатализаторов в зависимости от максимального расстояния между концевыми ОН-группами в молекуле симметричных диолов (2, ■) и этиленгликоля и его олигомеров (1, ○).
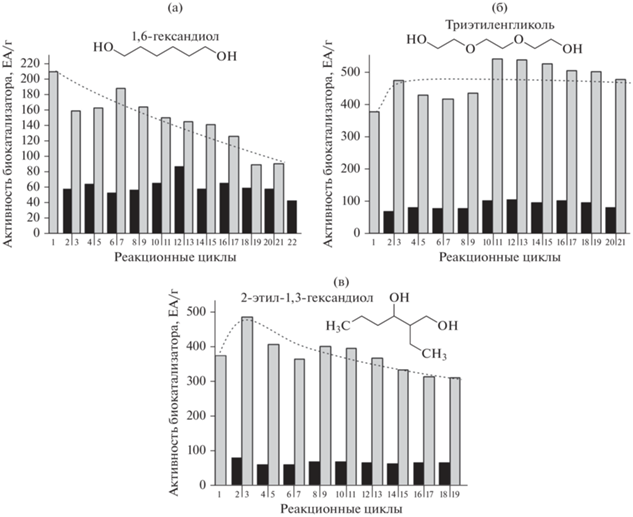
Была исследована стабильность приготовленных биокатализаторов в периодическом процессе синтеза моноэфиров различных диолов. Как отмечалось выше, при работе в хлороформе биокатализаторы полностью инактивировались за 1–3 реакционных цикла, поэтому мы изучили возможность их реактивации путем замены реакционной среды, а именно, растворителя хлороформа на гексан и диола на н-бутанол. Так, после каждого реакционного цикла синтеза моноэфира диола в хлороформе, следующий цикл проводили в гексане в реакции синтеза бутил гептаноата. Обнаружено, что при такой замене реакционной среды активность биокатализаторов частично или полностью восстанавливалась и даже увеличивалась (в 1.1–1.3 раза) по сравнению с первоначальной активностью (рис. 4б). В реакции с участием диэтиленгликоля после 2-х реакционных циклов в хлороформе, биокатализатор так и не восстановил активность, потеряв, как отмечалось выше, более 99% от ее первоначальной величины. При использовании в качестве субстрата триэтиленгликоля полученные биокатализаторы отличались сравнительно высокой операционной стабильностью как в реакции синтеза его моноэфиров, так и в синтезе бутил гептаноата (рис. 4б, серые колонки, пунктирная линия). При использовании 1,6-гесандиола биокатализатор частично реактивировался, и его активность медленно падала от цикла к циклу (рис. 4а, черные колонки), в то время как в реакции синтеза бутил гептаноата инактивация биокатализатора протекала быстрее (рис. 4а, серые колонки, пунктирная линия). В случае диола с разветвленным углеродным скелетом биокатализатор инактивировался медленнее по сравнению с линейным диолом (рис. 4а и 4в). Так, за 20 реакционных циклов он сохранил 40 и 80% первоначальной активности при синтезе моноэфиров 1,6-гександиола и 2-этил-1,3-гексан- диола соответственно (рис. 4а и 4в). Таким образом, используя один и тот же биокатализатор, путем смены реакционной среды можно синтезировать различные эфиры, например, моноэфиры диолов и бутиловые эфиры гептановой кислоты.
Рис. 4.
Активность биокатализаторов в реакции этерификации гептановой кислоты 1,6-гександиолом (а), триэтиленгликолем (б) и 2-этил-1,3-гександиолом (в). Условия реакции: 20 ± 2°С, 0.1 г биокатализатора, 3.0 мл среды. Состав среды в четных реакционных циклах (черные колонки): 0.25 М кислота, 0.75 М диол, хлороформ. Состав среды в нечетных реакционных циклах (серые колонки): 0.25 М кислота, 0.5 М бутанол, гексан.
ЗАКЛЮЧЕНИЕ
Исследованы процессы низкотемпературного синтеза сложных эфиров гептановой (энантовой, С7:0) кислоты и различных диолов с участием гетерогенных биокатализаторов, приготовленных путем адсорбционной иммобилизации рекомбинантной липазы rPichia/lip на макропористом углеродном аэрогеле. В качестве субстратов изучены диолы, различающиеся длиной углеродного скелета (от 2 до 6 атомов С), изомерией углеродного скелета и положением ОН-группы. Реакцию этерификации проводили в реакторах периодического действия в хлороформе в очень мягких условиях (20 ± 2°С, 1 бар). Свойства приготовленных биокатализаторов, такие как ферментативная активность, субстратная специфичность и операционная стабильность, были изучены в зависимости от строения молекулы диола и природы органического растворителя (хлороформа, гексана, ацетона).
Было обнаружено, что короткоцепочечные диолы С2–С4 необратимо инактивировали/ингибировали иммобилизованную липазу rPichia/lip, и биокатализатор полностью инактивировался в течение 1–3 реакционных циклов. Максимальное значение активности, равное 83 ЕА/г, и практически полная конверсия кислоты были измерены в реакции этерификации гептановой кислоты и 1,6-гександиола, при молярном соотношении (кислота : диол = 1 : 3) доля синтезированного моноэфира составила 99%. Была обнаружена корреляция между увеличением активности приготовленных биокатализаторов и удлинением молекулы симметричных диолов с концевыми ОН-группами. Поскольку хлороформ инактивировал адсорбированную rPichia/lip, то были подобраны условия реактивации биокатализаторов путем смены реакционной среды, и именно, замены хлороформа на гексан, а диола на бутанол. В этом случае биокатализаторы сохраняли этерифицирующую активность на высоком уровне в течение 20 реакционных циклов. Таким способом с помощью одного и того же биокатализатора, приготовленного адсорбцией рекомбинантной липазы rPichia/lip на макропористом углеродной аэрогеле, были синтезированы различные эфиры гептановой кислоты, такие как бутиловые эфиры и моноэфиры диолов.
Список литературы
Мамарасулова З.В., Громова В.В. // Химическая промышленность. 2006. Т. 83. № 5. С. 251.
Тонконогов Б.П., Попова К.А., Хурумова. А.Ф. // Химическая технология топлива и высокоэнергетических веществ. 2015. Т. 278. № 1. С. 109.
Jaeger K.-E., Egge T. // Current Opinion in Biotechnology. 2002. V. 13. C. 390.
Villeneuve P., Muderhwa J.M., Graille J., Haas M.J. // J. Mol. Catal. B: Enzym. 2000. № 9. P. 113.
Rodrigues R.C., Fernandez-Lafuente R. // J. Mol. Catal.B: Enzym. 2010. V. 64. P. 1.
Rodrigues R.C., Fernandez-Lafuente R. // J. Mol. Catal. B: Enzym. 2010. V. 66. P. 15.
Fernandez-Lafuente R. // J. Mol. Catal. B: Enzym. 2010. V. 62. P. 197.
Contesini F.J., LopesD.B., Macedo G.A., Nascimento M., Carvalho P. // J. Mol. Catal. B: Enzym. 2010. V. 67. P. 163.
Безбородов А.М., Загустина Н.А. // Прикл. Биохим. Микробиол. 2014. Т. 50. № 4. С. 347.
Зиновьева М.Е., Гамаюрова В.С., Шнайдер К.Л., Назаренко Е.В. // Вестник технологического Университета. 2015. Т. 18. № 1. С. 100.
Meissner J.P., Carta G. // Ind. Eng. Chem. Res. 2002. V. 41. № 19. P. 4722.
Pellis A., Comerford J.W., Maneffa A.J., Sipponen M.H., Clark J.H., Farmer T.J. // Eur. Polymer J. 2018. V. 106. P. 79.
Гамаюрова В.С., Зиновьева М.Е., Калачева Н.В., Шнайдер К.Л. // Катализ в промышленности. 2015. Т. 15. № 2. С. 73. https://doi.org/10.18412/1816-0387-2015-2-73-78
Коваленко Г.А., Перминова Л.В., Беклемишев А.Б., Мамаев А.Л., Патрушев Ю.В. // Катализ в промышленности. 2017. Т. 17. № 5. С. 399. https://doi.org/10.18412/1816-0387-2017-5-399-406
Kovalenko G.A., Perminova L.V., Krasnikov D.V., Kuznetsov V.L. // J. Porous Mater. 2018. V. 25. P. 1017. https://doi.org/10.1007/s10934-017-0512-0
Коваленко Г.А., Перминова Л.В. // Катализ в промышленности. 2020. Т. 20. № 4. С. 313. https://doi.org/10.18412/1816-0387-2020-4-313-322
Перминова Л.В., Коваленко Г.А., Чуканов Н.В., Патрушев Ю.В. // Изв. АН. Серия химическая. 2017. № 11. С. 2194.
Bearden J.C., Jr. // Biochim. Biophys. Acta. 1978. V. 533. P. 525.
Shashkov M.V., Sidelnikov V.N., Bratchikova A.A. // Anal. Lett. 2020. V. 53(1). P. 84.
Kovalenko G.A., Perminova L.V., Beklemishev A.B. // Catal. Today. 2021. V. 379. P. 36.https://doi.org/10.1016/j.cattod.2020.11.018
Rodrigues R.C., Fernandez-Lafuente R. //J. Mol. Catal. B: Enzym. 2010. V. 66. P. 15. https://doi.org/10.1016/j.molcatb.2010.03.008
Albayati S.H., Masomian M., Ishak S.N.H., Ali M.S.M., Thean A.L., Shariff F.M., Noor N.D.M., Rahman R.N.Z.R.A. // Catalysts. 2020. V. 10. P. 747. https://doi.org/10.3390/catal10070747
Contesini F.J., Davanço M.G., Borin G.P., Vanegas K.G., Cirino J.P.G., Melo R., Mortensen H., Hilden K., Campos D.R., Carvalho P. // Catalysts. 2020. V. 10. P. 1032. https://doi.org/10.3390/catal10091032
Naik S., Basu A., Soikia R., Madan D., Paul P., Chaterjii R., Brask J., Svenden A. // J. J. Mol. Catal. B: Enzym. 2010. V. 65. P. 18.
Дополнительные материалы отсутствуют.
Инструменты
Кинетика и катализ