

Кинетика и катализ, 2022, T. 63, № 3, стр. 363-371
Влияние промотирования углеродом на свойства катализатора Co/MgAlO4 синтеза Фишера–Тропша
П. А. Чернавский a, b, c, Г. В. Панкина a, b, Р. В. Казанцев b, С. В. Максимов a, С. Ю. Купреенко a, А. Н. Харланов a, О. Л. Елисеев b, *
a ФГБОУ ВО Московский государственный университет им. М.В. Ломоносова,
Химический факультет
119991 Москва, Ленинские Горы, 1, стр. 3, Россия
b ФГБУН Институт органической химии им. Н.Д. Зелинского РАН
119991 Москва, Ленинский просп., 47, Россия
c ФГБУН Институт нефтехимического синтеза им. Н.С. Топчиева РАН
119991 Москва, Ленинский просп., 29, Россия
* E-mail: oleg@server.ioc.ac.ru
Поступила в редакцию 01.10.2021
После доработки 19.01.2022
Принята к публикации 19.01.2022
- EDN: XVPYBT
- DOI: 10.31857/S0453881122030066
Аннотация
Исследовано влияние углерода на генезис активной фазы кобальта в катализаторах на алюминиево-магниевой шпинели в качестве подложки и на их каталитических свойства в синтезе углеводородов из СО и Н2. Промотирование углеродом проводили двумя независимыми методами: в первом случае углерод наносили на носитель шпинель путем термолиза глюкозы с последующим нанесением кобальта; во втором приготовление катализатора осуществляли пропиткой носителя совместно растворами нитрата кобальта и глюкозы с последующим термолизом. Катализаторы охарактеризованы методами синхронного термического анализа в комплексе с масс-спектроскопией выделяющихся газов, магнитными измерениями in situ, низкотемпературной адсорбции азота, просвечивающей электронной микроскопии. Модифицированные катализаторы демонстрируют существенно бóльшую скорость превращения СО (частоту оборотов реакции) и селективность в отношении целевых жидких углеводородов по сравнению с непромотированным образом.
ВВЕДЕНИЕ
В связи с повышающимся спросом на экологически чистое жидкое топливо и диверсификацию сырьевых ресурсов нефтехимии растет интерес к альтернативным источникам углеводородного сырья, включая возобновляемые – уголь, биомассу, природный газ, твердые бытовые отходы. Среди ряда известных процессов наиболее перспективным в этом отношении является синтез Фишера–Тропша (ФТ), который преобразует синтез-газ любого генезиса в углеводородные фракции с ничтожным содержанием серы, прочих гетероэлементов и ароматических углеводородов [1–5]. В зависимости от применяемых катализаторов и температурного режима синтез ФТ позволяет получать как легкие, так и тяжелые продукты (вплоть до твердых парафинов), содержащие различное количество олефинов и высших спиртов. В низкотемпературном синтезе ФТ, направленном на получение топливных фракций премиального качества, наилучшими считаются кобальтовые катализаторы. Они обладают высокой активностью и селективностью образования линейных алканов, устойчивы к окислению водой, при этом практически не катализируют реакцию водяного газа. В промышленности используются исключительно нанесенные кобальтовые катализаторы. Их обычно готовят пропиткой водным раствором нитрата кобальта и металлов-промоторов тугоплавких синтетических оксидных носителей с развитой поверхностью, таких как SiO2, Al2O3, TiO2 и др. Далее следуют термообработка, в ходе которой соль-прекурсор разлагается до оксида Co3O4, прочно связанного с поверхностью носителя. Перед применением катализаторы активируют водородом при температурах 350–500°С для получения металлического кобальта – активной фазы в синтезе ФТ [6–9].
Главным недостатком оксидных носителей является их недостаточная химическая инертность. В ходе высокотемпературной обработки катализатора активный компонент вступает во взаимодействие с оксидным носителем, образуя смешанные оксиды. Они не восстанавливаются в стандартных условиях активации катализатора и не проявляют каталитическую активность в гидрировании СО [3, 10, 11]. Этого недостатка лишены углеродные носители, такие как активированный уголь, углеродные волокна, нанотрубки, наносферы, графен и др. В последнее время синтетические углеродные материалы привлекают большое внимание в качестве носителей для катализаторов синтеза углеводородов из СО и Н2 [12–17]. Однако малая механическая прочность и высокая стоимость снижают потенциал их практического применения. Решением проблемы может стать использование композитных носителей, в которых на оксидную матрицу наносится углеродный материал. Такой композит обладает высокой прочностью оксида и инертной поверхностью, предотвращая нежелательное образование невосстанавливаемых смешанных оксидов. В качестве источника углерода удобно применять сахарозу [18] или другие доступные углеводы, которые разлагаются до углерода в ходе прокаливания материала в безвоздушной среде. Такой подход был нами опробован ранее для приготовления композитных катализаторов Fe/C–SiO2. На поверхности композитного носителя оксиды железа образовывали более мелкие кластеры с более узким распределением по размерам, чем на поверхности силикагеля. Их восстановление в активную фазу (карбид Хэгга) протекало легче, что обеспечивало лучшие каталитические характеристики в синтезе ФТ [19].
В настоящей работе нами была поставлена цель исследовать промотирование углеродом кобальтовых катализаторов, нанесенных на алюмо-магниевую шпинель. Карбонизацию поверхности проводили, пропитывая носитель водным раствором глюкозы с последующим анаэробным термолизом полученного материала. Катализаторы охарактеризованы методами термогравиметрии, дифференциального термического анализа, температурно-программированное восстановления, магнитными измерениями in situ, низкотемпературной адсорбции азота, просвечивающей электронной микроскопии. Каталитическую активность измеряли в проточном реакторе высокого давления с неподвижным слоем катализатора.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Катализаторы готовили методом пропитки носителя водным раствором гексагидрата нитрата кобальта(II). В качестве носителя использовали шпинель MgAl2O4 (Sasol PURALOX MG 30) состава 71.2 мас. % Al2O3, 27.7 мас. % MgO с удельной поверхностью 23 м2/г. Источником углерода была глюкоза.
Промотирование катализатора углеродом осуществляли двумя независимыми методами. В первом случае углеродная подложка формировалась при термолизе глюкозы, нанесенной на шпинель, после чего кобальт наносили методом пропитки полученного носителя. Термолиз нанесенной на шпинель глюкозы проводили после предварительной сушки в роторном испарителе при 80°С с последующим нагревом в токе азота при скорости нагрева 10°С/мин до Т = 450°С с экспозицией в течение 2 ч. После пропитки синтезированного материала водным раствором нитрата кобальта катализатор снова сушили в роторном испарителе при 80°С и затем прокаливали в токе азота при 450°С в течение 2 ч. Во втором случае катализатор был приготовлен путем совместной пропитки MgAl2O4 растворами нитрата кобальта и глюкозы с последующим термолизом. Условия сушки и последующего термолиза были те же, что и в первом способе.
В обоих катализаторах мольное отношение Со/глюкоза составляло 8. Взятое для пропитки количество Co(NO3)2 · 6H2O соответствовало массе кобальта 15% от массы носителя (шпинели или карбонизированной шпинели соответственно). Далее в тексте катализаторы обозначены как Co/MA в отсутствии глюкозы, Co/G/MA в случае последовательного нанесения глюкозы и кобальта и (CoG)/MA в случае совместного нанесения.
Прокаленные катализаторы анализировали на содержание кобальта методом масс-спектрометрии с индуктивно-связанной плазмой (ИСП-МС) на спектрометре ICPE-9800 (“Shimadzu”, Япония).
Термический анализ выполняли на приборе синхронного термического анализа STA 449 C Jupiter (“Netzsch”, Германия). Для исследования методом термогравиметрии высушенные при 80°С катализаторы помещали в измерительную ячейку. Скорость нагрева до температуры 600°С в токе Ar составляла 10°С/мин. Регистрировали ионные токи, соответствующие выделению NO, NO2 и CO2. Для определения содержания углерода в катализаторах проводили термогравиметрический анализ в токе воздуха, нагревая образцы до 1000°С со скоростью 10°С/мин. По потере веса образца судили о содержании углерода.
Низкотемпературную адсорбцию азота выполняли на сорбтометре ASAP 2010N. (“Micrometrics”, США). Образцы были откачаны при 300°C в течение 4 ч перед измерениями. Площадь поверхности рассчитывали из уравнения Брунауэра–Эммета–Теллера (БЭТ), объемы пор были определены с применением модели Барретта–Джойнера–Халенды (BJH).
Исследования с помощью просвечивающей электронной микроскопии (ПЭМ) осуществляли на микроскопе JEM-2100 F (“JEOL”, Япония) с разрешением 0.1–0.2 нм и ускоряющим напряжением 200 кВ. Перед анализом образец тщательно растирали в агатовой ступке в этаноле и оставляли закрытым на несколько часов. Затем каплю надосадочной жидкости наносили на полимерную пленку, расположенную на медной сетке. Распределение частиц по размерам получали визуальным анализом микрофотографий с использованием программного обеспечения “Image-Pro Plus 6.0”. Гистограммы строили на основании измерения размеров совокупности от 300 до 500 частиц. Рассчитывали среднечисленные логнормального распределения линейных размеров частиц Co3O4.
Для проведения магнитных измерений исследуемые катализаторы восстанавливали в токе водорода при 450°С до установления постоянного значения намагниченности непосредственно в измерительной ячейке магнитометра в поле 3 кЭ. После охлаждения до комнатной температуры были получены зависимости намагниченности от величины магнитного поля (петли гистерезиса). Путем экстраполяции значений намагниченности к нулевому полю были рассчитаны намагниченности насыщения и определены количества металлического кобальта в катализаторах после восстановления. Значение намагниченности насыщения для кобальта принимали равным 165 А/м при 25°С.
Каталитическую активность в синтезе углеводородов исследовали в стальном реакторе (внутренний диаметр – 14 мм) с неподвижным слоем и нисходящим потоком газа. Навеску катализатора в количестве 400 мг смешивали с 5 см3 кварцевого песка для предотвращения локальных перегревов и загружали в изотермическую зону реактора. Катализаторы активировали в токе водорода при атмосферном давлении и температуре 450°С в течение 4 ч, после чего подавали в реактор синтез-газ состава: 27% CO, 64% H2, 9% Ar (внутренний стандарт). Давление в реакторе устанавливали 2 МПа, температуру плавно поднимали в течение 16 ч от 180 до 210°С, после чего выдерживали при этой температуре до достижения псевдостационарного режима. Каталитическую активность оценивали через 48 ч работы в этих условиях. Частоту оборотов вычисляли как отношение количества молекул СО, превращенных за 1 с на поверхностном атоме металлического Со. Состав газообразных продуктов (углеводороды C1–C4 и CO2) определяли на газовом хроматографе Кристаллюкс-4000M (“Мета-хром”, Россия) с двумя насадочными стальными колонками: 1.5 м × 2.1 мм с фазами HayeSep (дивинилбензол 60%, этиленгликольдиметакрилат 40%) и молекулярные сита NaX; газ-носитель – гелий, детектор – катарометр. Селективность образования углеводородов С5+ рассчитывали по разности от общего баланса массы углерода в СО и газообразных продуктах С1–С4 и СО2. Возможное закоксовывание поверхности катализатора в балансе углерода не учитывали, принимая во внимание низкую температуру процесса и высокое соотношение Н2/СО = 2.4 в сырье.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Согласно данным ИСП-МС содержание кобальта в приготовленных катализаторах составляет около 11 мас. %. Термогравиметрический анализ в ходе разложения в токе воздуха позволил оценить содержание углерода в образцах. В катализаторе, полученном методом последовательного нанесения, оно оказалось существенно выше, чем при совместной пропитке глюкозой и нитратом кобальта (табл. 1).
Таблица 1.
Содержание Со и углерода в катализаторах
| Катализатор | Содержание Со, мас. % | Содержание С, мас. % |
|---|---|---|
| Co/MA | 11.0 ± 0.7 | 0 |
| Co/G/MA | 11.0 ± 0.5 | 16 |
| (CoG)/MA | 10.0 ± 0.5 | 4 |
Результаты масс-спектроскопии образующихся при нагреве образцов катализаторов газов представлены в форме температурных зависимостей ионных токов, соответствующих выделению NO (m/e = 30), NO2 (m/e = 46) и CO2 (m/e = 44) (рис. 1). Ионный ток для CO (m/e =28) на порядок меньше, чем для NO, NO2 и CO2, поэтому на рис. 1 он не показан. Промотирование шпинели глюкозой приводит к существенному изменению характера выделения NO и NO2 при термолизе. Количество образующегося NO более чем на порядок меньше для катализатора (CoG)/MA. Профили выделения СО2, NO и NO2 для образца Co/G/MA совпадают. Профили выделения NO и СО2 для катализатора (CoG)/MA значительно отличаются, и при температуре выше 300°С наблюдается дополнительное образование СО2. Кроме того, в масс-спектре отсутствует сигнал, относящийся к NO2. Это указывает на различие механизмов термолиза в катализаторах Co/G/MA и (CoG)/MA. Процесс выделения NO при термолизе практически завершается при температуре 400°С для всех катализаторов. Совпадение профилей выделения NO и СО2 можно объяснить протеканием реакции окисления углерода диоксидом азота, образующимся при термолизе нитрата кобальта:
Рис. 1.
Зависимости ионных токов от температуры термолиза для катализаторов Co/G/MA (а), (CoG)/MA (б) и Co/MA (в).
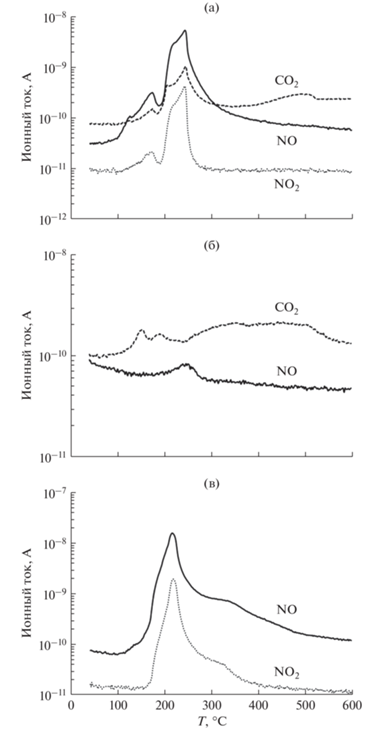
В процессе термолиза (CoG)/MA не наблюдается выделения NO2, однако при термолизе Co/G/MA он образуется в заметных количествах. Мы предполагаем, что в случае (CoG)/MA весь NO2 расходуется на реакцию окисления углерода в CO2.
Промотирование носителя углеродом приводит к значительным текстурным изменениям в катализаторе: удельная поверхность возрастает почти на порядок, в три раза увеличивается объем пор и в два раза уменьшается их средний диаметр. При совместном нанесении кобальта и глюкозы изменения не столь существенны, однако удельная поверхность становится больше в два раза (табл. 2).
Таблица 2.
Текстурные свойства катализаторов по результатам низкотемпературной адсорбции азота
| Катализатор | Удельная поверхность, м2/г | Объем пор, см3/г | Диаметр пор, Å |
|---|---|---|---|
| Co/MA | 14 | 0.028 | 78.5 |
| Co/G/MA | 103 | 0.093 | 36 |
| (CoG)/MA | 27 | 0.049 | 71 |
Результаты просвечивающей электронной микроскопии в виде фотографий и гистограмм распределений по размерам приведены на рис. 2. Промотирование носителя углеродом способствует заметному снижению среднего размера частиц оксида кобальта, при этом само распределение по размерам становится ýже. В катализаторе (CoG)/MA средний размер частиц в два раза меньше, чем в Со/МА. Состав поверхности, показанной на рис. 2, определяли методом энергодисперсионной рентгеновской спектроскопии, совмещенной с ПЭМ. Средние размеры частиц Co3O4 для образца Со/МА составляют 30 нм, для Со/G/MA – 20 нм и для (CoG)/MA – 15 нм.
Рис. 2.
Электронные микрофотографии и распределение частиц по размерам: Со/МА (а), Со/G/MA (б) и (CoG)/MA (в).
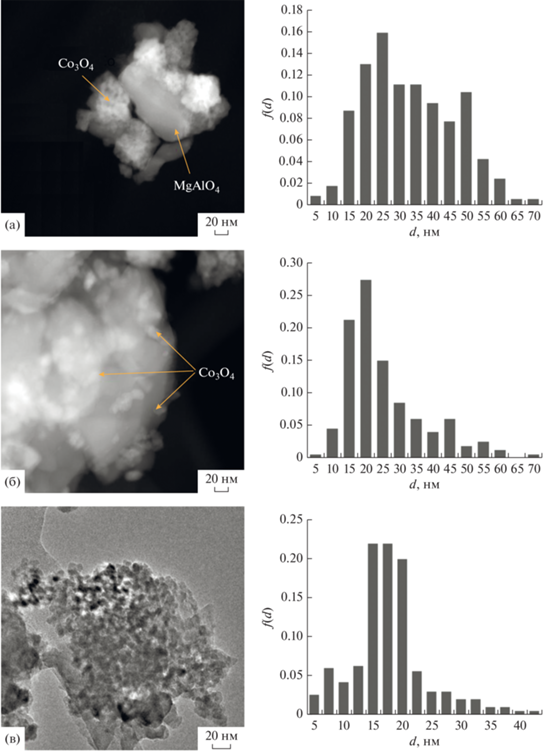
При окислении глюкозы нитрат-ионом образуется глюконовая кислота, продукты полимеризации которой являются хорошими хелатирующими агентами [18]. Известно, что при приготовлении пропиточных катализаторов из раствора, содержащего хелатные комплексы, повышается дисперсность нанесенного металла [20]. Вероятно, благодаря этому эффекту в (CoG)/MA преобладают относительно мелкие частицы Co3O4. Уменьшение размера частиц в Со/G/MA по сравнению с Со/МА может быть обусловлено значительной разницей в удельной поверхности исходной шпинели и ее карбонизированной формы G/MA.
Зависимости намагниченности активированных в водороде катализаторов от величины магнитного поля (петли гистерезиса) показаны на рис. 3. Содержание металлического кобальта в образцах было вычислено из величин намагниченности насыщения. На основании магнитных измерений, данных ИСП-МС и ПЭМ были рассчитаны степень восстановления кобальта и его активная поверхность в катализаторе (табл. 3).
Рис. 3.
Зависимости намагниченности активированных в водороде катализаторов от величины магнитного поля (петли гистерезиса) для катализаторов Со/МА (1), Со/G/MA (2), (CoG)/MA (3).
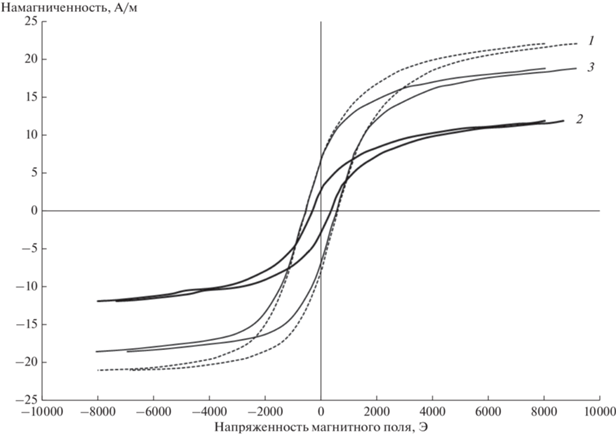
Таблица 3.
Степень восстановления кобальта и его поверхность в катализаторе
| Катализатор | Степень восстановления Со, % | Удельная поверхность Co0, м2/г |
|---|---|---|
| Co/MA | 97.3 | 2.2 |
| Co/G/MA | 54.5 | 1.2 |
| (CoG)/MA | 84.9 | 1.8 |
На рис. 4 приведены зависимости намагниченности образцов при восстановлении катализаторов в токе водорода при 450°С в ячейке магнитометра. Эта величина отражает превращение Со3О4 в Со. Из вида полученных кривых следует, что присутствие углерода на поверхности носителя существенно ускоряет процесс восстановления.
Рис. 4.
Зависимости намагниченности образца от времени для катализаторов Со/G/MA (1), Co/MA (2) и (CoG)/MA (3).
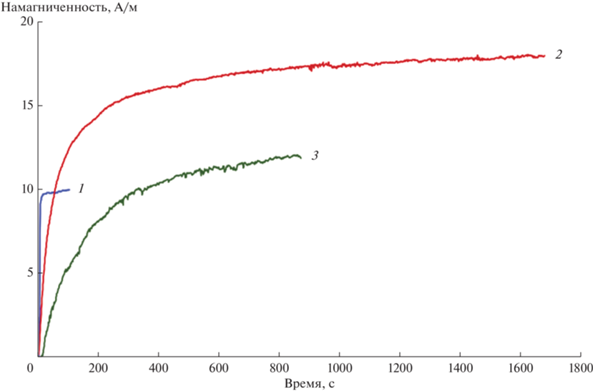
Известно, что крупные кристаллиты оксида кобальта в ходе активации водородом легче восстанавливаются до металла, чем мелкие [2, 3]. По-видимому, этим объясняется высокая восстанавливаемость кобальта в Co/MA по сравнению с Co/G/MA. Однако, несмотря на то что в (CoG)/MA преобладают мелкие кристаллиты оксида кобальта, степень восстановления его при активации в водороде также высокая. По-видимому, восстановление оксида кобальта в этом образце глюкозой и продуктами ее разложения начинается уже на стадии прокаливания катализатора в токе азота.
Все катализаторы после активации водородом показали активность в синтезе углеводородов. Для испытаний была выбрана температура 210°С, что типично для низкотемпературного синтеза ФТ. Расход синтез-газа регулировали таким образом, чтобы достичь степени превращения СО ~10%. Это позволяет корректно сравнивать удельную активность и селективность образцов при близком парциальном давлении реагентов и образующихся в слое катализаторов паров воды. Известно, что продуктовая вода оказывает влияние на активность и селективность Со-катализаторов синтеза ФТ [21–24]. Поэтому рекомендуется проводить сопоставление каталитических свойств при одинаковой степени превращения СО, что обеспечивает приблизительно равное парциальное давление паров воды в реакторе [25, 26].
Регистрируемая в первые часы работы конверсия практически не меняется в течение последующих 35–40 ч, что говорит о быстром достижении псевдостационарного режима. Селективность по CH4, углеводородам С2–С4, С5+ и СО2 также остается постоянной после первых 8 ч каталитических испытаний в пределах погрешности хроматографического анализа. Промотирование поверхности углеродом существенно повышает активность нанесенного кобальта: частота оборотов реакции на Co/G/MA оставляет 1.1 с–1 против 0.6 с–1 для образца сравнения Co/MA. Катализатор, приготовленный совместной пропитки носителя растворами нитрата кобальта и глюкозы с последующим термолизом, демонстрирует незначительное повышение активности. Важно отметить, что промотированные катализаторы показали рост селективности по целевым жидким углеводородам и снижение метанообразования по сравнению с Co/MA (табл. 4).
Таблица 4.
Каталитические испытания в синтезе ФТ
| Катализатор | Конверсия СО, % | Селективность, мол. % | Частота оборотов, с–1 | ||
|---|---|---|---|---|---|
| CH4 | C5+ | CO2 | |||
| Co/MA | 10.0 | 5.6 | 85.7 | 1.0 | 0.6 |
| Co/G/MA | 8.4 | 3.7 | 88.5 | 1.1 | 1.1 |
| (CoG)/MA | 10.0 | 4.6 | 88.9 | 0.4 | 0.7 |
Следует принять во внимание, что данные о структуре поверхности получены для активированного в водороде катализатора, в то время как каталитические характеристики сняты после нескольких десятков часов работы. В процессе взаимодействия с синтез-газом активная поверхность могла, вообще говоря, трансформироваться, например, из-за окисления кристаллитов металлического кобальта парами продуктовой воды и/или спекания этих частиц [25]. Однако в условиях низкотемпературного синтеза за столь небольшое время эти изменения, по-видимому, невелики [27, 28].
Хорошо известно, что активная фаза кобальтовых катализаторов синтеза углеводородов из СО и Н2 – металлический кобальт, находящийся в высокодисперсном состоянии на поверхности носителя [6–9]. При этом частота оборотов реакции практически не зависит от дисперсности металла при размере его кристаллитов более 8 нм [25]. Возможной причиной повышенной активности Co/G/MA и (CoG)/MA является адсорбция СО поверхностным углеродом, что увеличивает его концентрацию у активных центров – кристаллитов металлического кобальта. Известно, что СО обратимо сорбируется активированным углем [29]. Не исключено также, что контакт углерода с кристаллитами кобальта облегчает образование поверхностных карбидов – частиц, инициирующих рост углеводородной цепи согласно принятому карбидному механизму синтеза ФТ [1].
ЗАКЛЮЧЕНИЕ
Таким образом, предложенное нами промотирование нанесенного катализатора углеродом посредством обработки раствором глюкозы с последующим ее термолизом приводит к заметному уменьшению среднего размера нанесенных частиц Co3O4 и сужению распределения по размерам частиц. Восстанавливаемость кобальта снижается по сравнению с непромотированным образцом. Скорость превращения СО на активных центрах катализатора (частота оборотов) увеличивается благодаря промотированию углеродом, причем максимальный эффект достигается для образца, в котором кобальт нанесен на предварительно модифицированную углеродом поверхность.
Список литературы
Dry M.E. // Catal. Today. 2002. V. 71. P. 227.
Iglesia E. // Appl. Catal. A: Gen. 1997. V. 161. P. 59.
Khodakov A.Yu., Chu W., Fongarland P. // Chem. Rev. 2007. V. 107. P. 1692.
Чернавский П.А., Елисеев О.Л., Казанцев Р.В., Панкина Г.В., Строкова Н.Е., Лапидус А.Л. // Кинетика и катализ. 2018. Т. 59. № 6. С. 795. (Chernavskii P.A., Eliseev O.L., Kazantsev R.V., Pankina G.V., Strokova N.E., Lapidus A.L. // Kinet. Catal. 2018. V. 59. № 6. P. 828.)
Яковенко Р.Е., Зубков И.Н., Нарочный Г.Б., Папета О.П., Денисов О.Д., Савостьянов А.П. // Кинетика и катализ. 2020. Т. 61. № 2. С. 278. (Yakovenko R.E., Zubkov I.N., Narochniy G.B., Papeta O.P., Denisov O.D., Savost’yanov A.P. // Kinet. Catal. 2020. V. 61. № 2. P. 310.)
de la Pena O’Shea V.A., Homs N., Fierro J.L.G., de la Piscina P.R. // Catal. Today. 2006. V. 114. P. 422.
Yan Z., Bukur D.B., Goodman D.W. // Catal. Today. 2010. V. 160. P. 39.
Rausch A.K., van Steen E., Roessner F. // J. Catal. 2008. V. 253. P. 111.
Rønning M., Tsakoumis N.E., Voronov A., Johnsen R.E., Norby P., van Beek W., Borg Ø., Rytter E., Holmen A. // Catal. Today. 2010. V. 155. P. 289.
Wielers A.F.H., Kock A.J.H.M., Hop C.E.S.A., Geus J.W., van Der Kraan A.M. // J. Catal. 1989. V. 117. P. 1.
Mogorosi R.P., Fischer N., Claeys M., van Steen E. // J. Catal. 2012. V. 289. P. 140.
Chen Y., Wei J., Duyar M.S., Ordomsky V.V., Khodakov A.Y., Liu J. // Chem. Soc. Rev. 2021. V. 50. P. 2337.
Fu T., Li Z. // Chem. Eng. Sci. 2015. V. 135. P. 3.
Cheng Q., Zhao N., Lyu S., Tian Y., Gao F., Dong L., Jiang Z., Zhang J., Tsubaki J., Li X. // Appl. Catal. B: Env. 2019. V. 248. P. 73.
Eschemann T.O., Lamme W.S., Manchester R.L., Parmentier T.E., Cognigni A., Rønning M., de Jong K.P. // J. Catal. 2015. V. 328. P. 130.
Xiong H., Jewell L.L., Coville N.J. // ACS Catal. 2015. V. 5. P. 2640.
Chew L.M., Xia W., Düdder H., Weide P., Ruland H., Muhler M. // Catal. Today. 2016. V. 270. P. 85.
Girardon J.-S., Quinet E., Griboval-Constant A., Chernavskii P.A., Khodakov A.Y. // J. Catal. 2007. V. 248. P. 143.
Eliseev O., Chernavskii P., Kazantsev R., Pankina G., Maslakov K., Lunin B. // Energy Technol. 2019. V. 7. 1800961.
Van Dillen A.J., Teroörde R.J.A.M., Lensveld D.J., Geus J.W., De Jong K.P. // J. Catal. 2003. V. 216. P. 257.
Krishnamoorthy S., Tu M., Ojeda M.P., Pinna D., Iglesia E. // J. Catal. 2002. V. 211. P. 422.
Hilmen A.M., Schanke D., Hanssen K.F., Holmen A. // Appl. Catal. A: Gen. 1999. V. 186. P. 169.
Borg Ø., Storsætera S., Eri S., Wigum H., Rytter E., Holmen A. // Catal. Lett. 2006. V. 107. P. 95.
Tavasoli A., Nakhaeipour A., Sadaghiani K. // Fuel Proc. Technol. 2007. V. 88. P. 461.
Borg Ø., Eri S., Blekkan E.A., Storsæter S., Wigum H., Rytter E., Holmen A. // J. Catal. 2007. V. 248. P. 89.
Borg Ø., Dietzel P.D.C., Spjelkavik A.I., Tveten E.Z., Walmsley J.C., Diplas S., Eri S., Holmen A., Rytter E. // J. Catal. 2008. V. 259. P. 161.
Saib A.M., Moodley D.J., Ciobîca I.M., Hauman M.M., Sigwebela B.H., Weststrate C.J., Niemantsverdriet J.W., van de Loosdrecht J. // Catal. Today. 2010. V. 154. P. 271.
Savost’yanov A.P., Eliseev O.L., Yakovenko R.E., Narochniy G.B., Maslakov K.I., Zubkov I., Soromotin V.N., Kozakov A.T., Nicolskii A.V., Mitchenko S.A. // Catal. Lett. 2020. V. 150. P. 1932.
Tamon H., Kitamura K., Okazaki M. // AIChE J. 1996. V. 42. P. 422.
Дополнительные материалы отсутствуют.
Инструменты
Кинетика и катализ