

Кинетика и катализ, 2022, T. 63, № 4, стр. 506-515
Влияние температуры прокаливания на свойства Mn–Zr–Сe-катализаторов в окислении СО
Т. Н. Афонасенко a, *, Д. В. Глыздова a, В. П. Коновалова b, А. А. Сараев b, Е. Е. Айдаков b, О. А. Булавченко b, **
a Центр новых химических технологий ИК СО РАН
644040 Омск, ул. Нефтезаводская, 54, Россия
b ФГБУН Институт катализа им. Г.К. Борескова СО РАН
630090 Новосибирск, просп. Акад. Лаврентьева, 5, Россия
* E-mail: atnik@ihcp.ru
** E-mail: obulavchenko@catalysis.ru
Поступила в редакцию 15.02.2022
После доработки 23.03.2022
Принята к публикации 24.03.2022
- EDN: IWAOWK
- DOI: 10.31857/S0453881122040013
Аннотация
Изучено влияние температуры прокаливания катализаторов MnOх–ZrO2–CeO2 на их структурные свойства и активность в реакции окисления СО. Исследованы тройные оксидные системы с соотношением катионов Mn : Zr : Се = 0.3 : 0.35 : 0.35, приготовленные методом соосаждения. Согласно данным РФА, ТПВ-Н2 и РФЭС повышение температуры прокаливания от 400 до 800°С вызывает структурную трансформацию твердого раствора MnyZrxCe1 –yO2 – δ. При 400–600°С происходит выход катионов марганца из его структуры в виде высокодисперсных частиц MnOx, при 700–800°С – распад исходного твердого раствора MnyZrxCe1 –yO2 – δ с формированием двух смешанных оксидов на основе CeO2 и ZrO2, а также окристаллизованной фазы Mn3O4. Наибольшая каталитическая активность в реакции окисления СО наблюдается при температурах прокаливания 500–600°С, что, вероятно, обусловлено присутствием марганца как в структуре твердого раствора, так и в виде высокодисперсных частиц MnOх на его поверхности.
ВВЕДЕНИЕ
Среди катализаторов на основе переходных металлов оксиды марганца являются одними из наиболее эффективных в процессах полного окисления углеводородов и СО, а, значит, они перспективны для очистки промышленных выбросов и выхлопных газов автотранспорта. Активность марганцевых катализаторов в вышеуказанных реакциях напрямую коррелирует с их окислительно-восстановительными свойствами, позволяющими обратимо присоединять и отдавать кислород в ходе реакции [1]. Ионы марганца способны легко менять степень окисления, образуя оксиды в различном окисленном состоянии – MnO2, Mn5O8, Mn2O3, Mn3O4, MnO, в результате чего обеспечивается высокая подвижность кислорода.
Комбинирование марганца с оксидом церия дает возможность получать существенно более каталитически активные системы по сравнению с массивными оксидами марганца [2, 3]. Благодаря легкому переходу между Ce3+ и Ce4+ и высокой подвижности ионов O2– в решетке, CeO2 модифицирует окислительно-восстановительную активность марганца, стабилизируя его в более высоких степенях окисления при низких температурах и облегчая миграцию кислорода при повышении температуры [4]. Оксид церия за счет образования лабильныx кислородных вакансий, которые облегчают активацию и перенос кислорода, обладает значительной емкостью для хранения кислорода и сам проявляет активность в окислительных реакциях, например, в окислении сажи [5]. Известно [4, 6], что включение циркония в решетку CeO2 не только улучшает термическую стабильность CeO2, но из-за создания дефектной структуры способствует еще большему возрастанию подвижности кислорода и увеличению его общего количества, которое может быть обратимо обменено между твердым веществом и окружающей атмосферой. Terribille с соавт. [4] установили, что самая высокая степень восстановления и самые низкие температуры восстановления достигаются при эквимолярном отношении церия и циркония (Ce0.5Zr0.5O2).
Свойства катализаторов Mn–Zr–Се широко изучены в реакциях полного окисления O-, Cl-содержащих летучих органических веществ и СО [7–10], окисления сажи [11, 12], селективного удаления NOx аммиаком [13–15]. Марганец в катализаторах Mn–Zr–Се в зависимости от способа приготовления может располагаться как на поверхности твердого раствора ZrO2–CeO2 [15], так и входить в его структуру [7]. В первом случае каталитическая активность Mn–Zr–Се определяется присутствием высокодисперсных частиц MnOx и их сильным взаимодействием с поверхностью носителя, благодаря чему обеспечивается легкость их восстановления [16]. Во втором случае каталитическая активность обусловлена подвижностью решеточного кислорода, которая еще больше возрастает при внедрении ионов Mn в решетку ZrO2–CeO2 с образованием твердого раствора Mn–Zr–Се [17]. Большинство работ по исследованию таких систем посвящены определению влияния соотношения компонентов [7, 15], содержания марганца [9, 16] на их структурные и каталитические свойства, либо сопоставлению свойств Mn–Zr–Се со свойствами оксидов Mn–Ce и Mn–Zr [8, 18, 19]. При этом сведения об изучении поведения катализаторов Mn–Zr–Се при изменении температуры прокаливания весьма ограничены [14, 20], хотя представляют большой интерес. Ранее нами было показано [21], что при варьировании температуры прокаливания MnОх–ZrО2 наибольшая каталитическая активность в реакции окисления СО достигается в результате обработки при 650–700°С, что соответствует пределу существования твердого раствора на основе кубической фазы ZrО2. При этом максимальная каталитическая активность, наблюдаемая для Mn0.4Zr0.6O2–Mn0.6Zr0.4O2, обусловлена, помимо дисперсности частиц самого твердого раствора, присутствием на его поверхности высокодисперсных частиц MnOx, не вошедших в его состав.
Целью настоящей работы являлось изучение структурных превращений, происходящих в системе MnOх–ZrO2–CeO2 при повышении температуры прокаливания, и влияния данных факторов на каталитическую активность образцов в реакции окисления СО.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Приготовление катализаторов
Образцы катализаторов готовили методом осаждения. К совместному раствору солей ZrO(NO3)2, Ce(NO3)3 и Mn(NO3)2 при его постоянном перемешивании постепенно добавляли раствор NH4OH до достижения рН 10. Осаждение осуществляли при 80°С. После завершения осаждения перемешивание полученной суспензии продолжали в течение 1 ч, затем прикапывали к ней Н2О2 в количестве, соответствующем мольному отношению Н2О2 : (Mn + Zr + Се) = 1, что обеспечивало полноту осаждения [20]. После этого суспензию выдерживали без перемешивания в течение 2 ч. Полученный осадок отфильтровывали, промывали водой на фильтре до рН 6–7. Образцы высушивали при 120°С в течение 2 ч, затем прокаливали в муфельной печи при 400–800°С в течение 4 ч. Мольное отношение Mn : Zr : Се в приготовленных образцах составляло 0.3 : 0.35 : 0.35. Образцы фракционировали, для каталитических испытаний использовали фракцию 0.4–0.8 мм.
Образцы были обозначены как Mn–Zr–Се-T, где Т – температура прокаливания.
Физико-химические методы исследования катализаторов
Удельную площадь поверхности катализаторов (Sуд) определяли по методу Брунауэра–Эммета–Теллера (БЭТ) с помощью изотерм адсорбции азота, измеренных при температуре жидкого азота. Исследования проводили с применением автоматизированной системы ASAP 2400 (“Micromeritics Instrument Corp.”, США).
Рентгенофазовый анализ (РФА). Дифрактограммы катализаторов Mn–Ce–Zr с соотношением Mn : Ce : Zr = 30 : 35 : 35 были получены на дифрактометре D8 Advance (“Bruker”, Германия) в диапазоне углов 15°–90° по 2θ с шагом 0.05° и временем накопления 4 с на длине волны CuKα 1.5418Å.
Термопрограммируемое восстановление водородом (ТПВ-Н2) выполняли в кварцевом реакторе с использованием проточной установки с детектором по теплопроводности. Смесь газов (10 об. % H2 в Ar) подавали со скоростью 40 мл/мин. Скорость нагрева от комнатной температуры до 900°С составляла 10°С/мин.
Рентгенфотоэлектронная спектроскопия (РФЭС) было проведена на фотоэлектронном спектрометре Surface Nano Analysis GmbH (“SPECS”, Германия). Спектрометр оснащен полусферическим анализатором PHOIBOS-150-MCD-9, рентгеновским монохроматором FOCUS-500 и источником рентгеновского характеристического излучения XR-50M с двойным Al/Ag-анодом. Для записи спектров использовали немонохроматизированное излучение AlKα (hν = 1486.61 эВ). Калибровку шкалы энергий связи (Есв) осуществляли методом внутреннего стандарта по пику Ce3d3/2-u''' церия, входящего в состав носителя (Есв = 916.7 эВ). Относительные концентрации элементов в зоне анализа определены на основании интегральных интенсивностей РФЭС-линий с учетом сечения фотоионизации соответствующих термов [22]. Для детального анализа выполняли разложение спектров на индивидуальные составляющие. После вычитания фона по методу Ширли [23] экспериментальную кривую раскладывали на ряд пиков, соответствующих фотоэмиссии электронов из атомов в различном химическом окружении. Обработку данных производили с помощью пакета программ CasaXPS [24]. Форма пиков аппроксимирована симметричной функцией, полученной суммированием функций Гаусса и Лоренца.
Каталитические испытания
Образцы испытывали в реакции окисления СО на установке проточного типа в стеклянном реакторе (170 × ∅ 10 мм). Исходная газовая смесь имела состав: 1% СО, 99% воздух, ее общий расход составлял 487 мл/мин. Анализ реакционной смеси до и после реактора осуществлялся на хроматографе ЛХМ-8МД (Россия) с разделением смеси на насадочной колонке, заполненной цеолитом СаА (3 м). С помощью детектора по теплопроводности определяли непрореагировавшее количество СО. Навеску катализатора 0.500 г смешивали с кварцем до объема 3 мл. Температуру в слое катализатора контролировали и регулировали с использованием хромель-алюмелевой термопары, соединенной с терморегулятором “Варта” (Россия). Степень превращения СО (XСО) рассчитывали по формуле:
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Структурные и микроструктурные свойства
На рис. 1 приведены дифракционные картины исследуемых катализаторов. Видно, что для катализаторов, прокаленных при 400–700°С рентгенограммы похожи, наблюдаются широкие пики с максимумами при 2θ = 28.9°, 33.5°, 48.2°, 57.2°, 60.0°, 70.5°, 78.0°, 80.4°, 89.3°, соответствующие рефлексам 111, 002, 022, 113, 222, 004, 133, 024, 224 оксида CeO2 со структурой флюорита (PDF № 431002). Можно заметить, что при 2θ = 36.3° присутствует слабый пик, положение которого характерно для наиболее интенсивного рефлекса Mn3O4 (PDF № 240734). При повышении температуры прокаливания интенсивность рефлекса оксида марганца увеличивается. В случае Mn–Zr–Ce-800 происходят довольно значительные изменения в дифракционных картинах – проявляются рефлексы Mn3O4 и отмечается расщепление рефлексов флюорита.
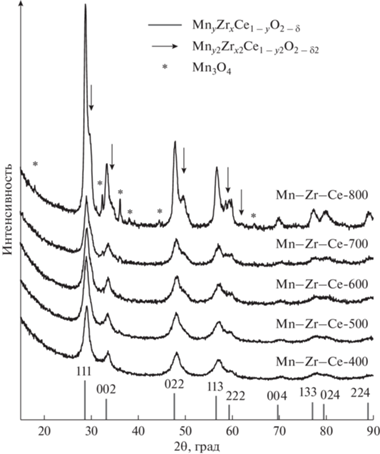
Структурные характеристики катализаторов, рассчитанные методом Ритвельда, приведены в табл. 1. Как видно, значение параметра решетки CeO2 варьируется в диапазоне 5.182(1)–5.371(1) Å, что меньше, чем для “чистого” CeO2, для которого характерна величина 5.411 Å (PDF № 431002). Изменение параметра решетки свидетельствует о формировании твердого раствора типа MnyZrxCe1 –yO2 – δ. Так как ионные радиусы катионов циркония и марганца меньше, у церия (Mn2+ – 0.097 нм, Mn3+ – 0.065 нм, Mn4+ – 0.053 нм, Zr4+ – 0.084 нм, Ce4+ – 0.097 нм), то внедрение этих элементов ведет к наблюдаемому сокращению параметров решетки.
Таблица 1.
Структурные и микроструктурные характеристики катализаторов
| Образец | Фазовый состав | Параметры решетки, Å | ОКР, Å | Sуд, м2/г |
|---|---|---|---|---|
| Mn–Zr–Се-400 | MnyZrxCe1 –yO2 – δ следы Mn3O4 |
5.337(1) – |
50 130 |
146 |
| Mn–Zr–Се-500 | MnyZrxCe1 –yO2 – δ следы Mn3O4 |
5.335(1) – |
50 130 |
110 |
| Mn–Zr–Се-600 | MnyZrxCe1 –yO2 – δ следы Mn3O4 |
5.316(1) – |
50 140 |
88 |
| Mn–Zr–Се-700 | MnyZrxCe1 –yO2 – δ Mn3O4 Mny2Zrx2Ce1 –y2O2 – δ2 |
5.345(1) – 5.182(3) |
60 270 60 |
63 |
| Mn–Zr–Се-800 | MnyZrxCe1 –yO2 – δ Mn3O4 Mny2Zrx2Ce1 –y2O2 – δ2 |
5.371(1) – 5.214(1) |
130 340 80 |
12 |
При повышении температуры прокаливания (Тпрокал) от 400 до 600°С происходит уменьшение параметра решетки от 5.337(1) до 5.316(1) Å, сопровождаемое увеличением содержания оксида марганца, а при 700°С появляется дополнительная фаза. Вероятно, происходит развал твердого раствора, при котором катионы Mn выходят из состава исходного оксида и появляется “новый” твердый раствор типа Mny2Zrx2Ce1 –y2O2 – δ2. Для катализаторов, полученных в результате прокаливания при 700–800°С, параметры решетки равны 5.345(1)–5.371(1) и 5.182(3)–5.214(1) Å, данные величины стремятся к значениям для “чистых” оксидов. Можно предположить, что формируется два смешанных оксида, первый на основе оксида церия, второй – на основе оксида циркония. Кроме того, нельзя исключать некой неоднородности по составу для катализаторов, синтезированных при меньших температурах. С ростом Тпрокал наблюдается закономерное увеличение средних размеров области когерентного рассеяния (ОКР) от 130 до 340 Å для Mn3O4 и от 50 до 130 Å для смешанного оксида.
При повышении температуры прокаливания удельная поверхность образцов Mn–Zr–Ce закономерно уменьшается, коррелируя с фазовыми трансформациями (табл. 1). При 400°С ее значение составляет 146 м2/г. В температурной области преимущественного существования твердого раствора она практически линейно снижается по мере увеличения Тпрокал и при 700°С составляет 63 м2/г, а при 800°С падает до 12 м2/г.
ТПВ-Н2
Метод ТПВ-Н2 был использован для оценки изменения окислительно-восстановительных свойств Mn–Zr–Ce при повышении температуры прокаливания. Как видно из рис. 2, для всех образцов на кривых ТПВ-Н2 в области температур восстановления 650–900°С присутствует широкое гало. Оно характерно для образцов на основе CeO2 и обусловлено частичным восстановлением церия от 4+ до 3+ [18, 25, 26]. Согласно [27, 28] восстановление СеО2 представляет собой двухэтапный процесс удаления кислорода – сначала с поверхности, а затем из объема частицы СеО2, и характеризуется двумя пиками при ~460 и 700°С.

Для катализаторов, прокаленных при 400–700°С, профили ТПВ-Н2 имеют схожий вид. Они представляют собой два частично неразделенных пика в диапазоне 100–500°С. Для Mn–Zr–Ce-400 максимумы пиков наблюдаются при 257 и 366°С. С ростом Тпрокал до 600°С их положение существенно не меняется, однако в области низких температур появляется дополнительная интенсивность, вследствие чего первый пик уширяется. Для Mn–Zr–Ce-700 низкотемпературный пик становится размытым со слабо выраженным максимумом при 267°С, а второй пик смещается до 375°С. Наличие вышеуказанных пиков обусловлено последовательными фазовыми превращениями оксидов марганца, сопровождающимися потерей кислорода в ходе восстановления. Первый пик соответствует переходу MnO2/Mn2O3 → → Mn3O4, а второй – дальнейшему восстановлению Mn3O4 до MnO [14, 17 ]. Но, поскольку, согласно результатам РФА, часть марганца присутствует в составе твердого раствора MnyZrxCe1–yO2–δ, эти пики также могут быть связаны с последовательным изменением степени окисления катионов марганца в структуре MnyZrxCe1–yO2–δ. Ранее нами [29] при изучении фазовой трансформации твердого раствора MnOx–ZrO2, происходящей при его восстановлении, было установлено, что первый пик соответствует восстановлению катионов марганца, находящихся в составе твердого раствора: Mn4+/3+ → Mn3+/2+, а второй – дальнейшему восстановлению марганца до Mn2+: Mn 2+/3+ → → Mn2+, которое сопровождается выходом катионов марганца из объема твердого раствора и их сегрегацией на поверхности.
Для образца Mn–Zr–Се-800 профиль ТПВ-Н2 принципиально отличается от таковых для катализаторов, прокаленных при меньших температурах. Кривая ТПВ-H2 содержит интенсивный пик с максимумом при 485°С, который можно отнести к восстановлению окристаллизованных частиц Mn3O4 (табл. 1). Кроме того, в области ~260°С наблюдается слабое поглощение Н2, которое можно объяснить присутствием незначительного количества ионов марганца Mn4+/3+ в структуре твердых растворов, образовавшихся после распада исходного MnyZrxCe1 –yO2 – δ [21].
Общее поглощение водорода с ростом температуры прокаливания закономерно уменьшается. При 400–600°С оно незначительно изменяется от 2.24 до 2.17 ммоль/г, при 700°С составляет 2.09 ммоль/г, а при 800°С резко снижается до 1.87 ммоль/г.
Сопоставление данных РФА и ТПВ-Н2 позволяет предположить, что возможной причиной появления низкотемпературного поглощения водорода при 150–300°С и “размытия” первого пика восстановления при повышении температуры прокаливания Mn–Zr–Се от 400 до 600°С является присутствие высокодисперсных частиц MnOx, которые могли образоваться на поверхности твердого раствора MnyZrxCe1 –yO2 – δ в ходе его постепенного распада. Gutiérrez-Ortiz с соавт. [30] наблюдали подобные широкие пики на профилях ТПВ-Н2 Mn–ZrO2 в диапазоне температур 100–220°C и относили их к нестехиометрическим дисперсным фазам MnOx на поверхности ZrO2. Разложение профилей ТПВ-Н2 аналитическими функциями показало, что для образца, покаленного при 400°С, доля низкотемпературной компоненты (Твосст ~ 200°С) составляет ~ 3% от общего поглощения водорода. С ростом температуры прокаливания до 500–600°С она увеличивается до ~12%, а при дальнейшем повышении Тпрокал уменьшается. Таким образом, наибольшее значение доли низкотемпературной компоненты в профиле ТПВ-Н2 наблюдается для образцов Mn–Zr–Се-500 и Mn–Zr–Се-600, что соответствует состояниям твердых растворов перед “распадом” и может указывать на формирование высокодисперсных рентгеноаморфных частиц MnOx. Из рис. 2 видно, что с ростом Тпрокал катализаторов профиль ТПВ-Н2 усложняется, на кривых, характеризующих восстановление Mn–Zr–Се-600 и Mn–Zr–Се-700, появляется дополнительная компонента с максимумом при 430°C, возможно, связанная с наложением пика поверхностного восстановления СеО2 с профилем восстановления марганца [31] или с формированием “второго” твердого раствора Mny2Zrx2Ce1 –y2O2 – δ2 и восстановлением катионов марганца из его структуры.
РФЭС
Для оценки изменения электронных свойств и соотношения элементов на поверхности катализаторов Mn–Ce-Zr был использован метод РФЭС. Относительные концентрации (атомные соотношения) элементов в приповерхностном слое образцов, определенные на основании данных РФЭС, представлены в табл. 2. Спектры Zr3d, Ce3d, O1s и Mn2p приведены на рис. 3.
Таблица 2.
Атомные отношения элементов в приповерхностном слое образцов
| Образец | Mn2p3/2 | [O]/[Me] | [Zr]/[Me] | Ce3+, % | [Oa]/ [Oa + Ob] |
|||
|---|---|---|---|---|---|---|---|---|
| [Mn]/[Me] | Mn2+, % | Mn3+, % | Mn4+, % | |||||
| Mn–Zr–Се-400 | 0.274 | 11 | 39 | 50 | 2.37 | 0.25 | 16 | 0.76 |
| Mn–Zr–Се-500 | 0.291 | 11 | 41 | 48 | 2.27 | 0.22 | 11 | 0.80 |
| Mn–Zr–Се-600 | 0.317 | 12 | 41 | 47 | 2.45 | 0.22 | 9 | 0.80 |
| Mn–Zr–Се-700 | 0.369 | 10 | 44 | 46 | 2.21 | 0.21 | 9 | 0.81 |
| Mn–Zr–Се-800 | 0.318 | 21 | 41 | 38 | 2.43 | 0.27 | 14 | 0.77 |

Спектры Zr3d описываются одним дублетом Zr3d5/2–Zr3d3/2 с энергий связи Zr3d5/2 равной 181.9 эВ, что характерно для циркония в состоянии Zr4+ [16]. По мере повышения Тпрокал до 700°С относительное содержание циркония [Zr]/[Mn + + Ce + Zr] на поверхности уменьшается от 0.25 до 0.21, а при 800°С составляет 0.27. Спектры Ce3d исследованных катализаторов в результате спин-орбитального взаимодействия имеют сложную форму. Результаты разложения их на индивидуальные составляющие позволили сделать вывод, что церий находится преимущественно в виде Ce4+, при этом доля ионов Ce3+ с повышением температуры прокаливания от 400 до 700°С снижается с 16 до 9%, а при 800°С возрастает до 14% (табл. 2). В спектре O1s наблюдается несколько пиков с энергией связи в районе 529.2–529.4, 531.5 и 533.4 эВ, относящихся к решеточному кислороду (Оа), адсорбированным кислородсодержащим группам (Оb), а также адсорбированным OH-группам и воде соответственно [17]. Решеточный кислород является преобладающей формой кислорода во всех катализаторах, отношение [Oa]/[Oa + Ob] слабо зависит от температуры прокаливания и варьируется от 0.76 до 0.81.
Спектр Mn2p представляет собой дублет Mn2p3/2–Mn2p1/2, интегральные интенсивности линий которого соотносятся как 2 : 1. Разложение спектра Mn2p исследованных образцов на индивидуальные составляющие показало (рис. 3), что марганец в приповерхностном слое находится в трех состояниях: Mn2+, Mn3+ и Mn4+ с энергией связи Mn2p3/2 равной 639.9, 640.7 и 641.4 эВ соответственно [15, 19]. Доля Mn2+ в образцах, прокаленных при 400–700°С, равна 10–12%, а в катализаторе, прокаленном при 800°С, она возрастает до 21%. Отношение Mn4+/Mn3+ с повышением Тпрокал постепенно сокращается от 1.3 до 0.9. Что касается общего содержания марганца в приповерхностном слое, то, как видно из данных табл. 2, с ростом температуры прокаливания до 700°C соотношение [Mn]/[Mn + Ce + Zr] увеличивается, а при 800°С – уменьшается. Обогащение поверхности катализатора марганцем диапазоне температур прокаливания 400–700°С находится в согласии с данными ТПВ-Н2 и, вероятно, обусловлено формированием высокодисперсных поверхностных частиц MnOx в процессе распада твердого раствора MnyZrxCe1 –yO2 – δ. Снижение значения [Mn]/[Mn + + Ce + Zr] при 800°С вызвано спеканием этих частиц с образованием, по данным РФА, кристаллической фазы Mn3O4.
Каталитические свойства
Результаты каталитических испытаний образцов Mn–Zr–Ce-Т в реакции окисления СО при варьировании Тпрокал представлены на рис. 4. С увеличением температуры прокаливания от 400 до 500°С кривая превращения СО сдвигается в более низкотемпературную область, температура достижения 50%-ной конверсии СО (Т50%) изменяется от 185 до 164°С, что указывает на возрастание активности катализатора. Повышение Тпрокал до 600°С не вызывает значимых изменений каталитических свойств, для Mn–Zr–Ce-500 и Mn–Zr–Ce-600 кривые превращения СО очень близки, значения Т50% совпадают. Дальнейшее повышение Тпрокал приводит к снижению каталитической активности, для Mn–Zr–Ce-700 значение Т50% составляет 178°С. В случае Mn–Zr–Ce-800 кривая превращения существенно смещается в сторону высоких температур, и Т50% возрастает до 314°С. Таким образом, зависимость каталитической активности Mn–Zr–Ce в реакции окисления СО от температуры прокаливания образцов проходит через максимум, где наибольшая каталитическая активность достигается при Тпрокал = = 500–600°С.

Сопоставление результатов каталитических испытаний с данными РФА, РФЭС и ТПВ-Н2 указывает на то, что рост каталитической активности при повышении температуры прокаливания Mn–Ce–Zr от 400 до 600°С, вероятно, обусловлено постепенным распадом твердого раствора MnyZrxCe1 –yO2 – δ с выделением на его поверхности высокодисперсных частиц MnOx, подвижный кислород которых, как известно [16, 32], проявляет высокую активность в реакциях окисления. При увеличении Тпрокал от 600 до 700°С образец лишь незначительно теряет активность в окислении СО, при этом Mn–Ce–Zr-700 более активен, чем Mn–Ce–Zr-400. Появление низкотемпературного поглощения водорода при 150–300°С по результатам ТПВ-H2 (рис. 2), обогащение поверхности катионами марганца по данным РФЭС (Mn/Ce + Zr) и наблюдаемые тенденции в эволюции фазового состава (расслоение твердого раствора MnyZrxCe1–yO2–δ) могут свидетельствовать об образовании высокодисперсных частиц MnOx. Падение каталитической активности, наблюдаемое после прокаливания образца при 800°С, обусловлено присутствием марганца преимущественно в виде окристаллизованных объемных частиц Mn3O4.
Таким образом, изменения каталитической активности Mn–Zr–Ce, наблюдаемые при повышении температуры прокаливания, согласуются с фазовыми трансформациями твердого раствора MnyZrxCe1 –yO2 – δ. По всей видимости, каталитическая активность в Mn–Zr–Ce определяется присутствием марганца как в структуре твердого раствора, так и в виде высокодисперсных частиц MnOx.
ЗАКЛЮЧЕНИЕ
Изучено влияние температуры прокаливания катализатора катионного состава Mn0.3Zr0.35Се0.35 на его структурные свойства и каталитическую активность в реакции окисления СО. Методами РФА и ТПВ-Н2 показано, что после прокаливания при 400°С образуется твердый раствор MnyZrxCe1 –yO2 – δ, а также присутствуют следовые количества Mn3O4. При повышении температуры прокаливания от 400 до 600°С вероятен постепенный распад твердого раствора MnyZrxCe1 –yO2 – δ с выходом катионов марганца из его структуры в виде высокодисперсных частиц MnOx, что обуславливает наблюдаемое увеличение каталитической активности. При 700–800°С твердый раствора MnyZrxCe1 –yO2 – δ распадается с образованием двух смешанных оксидов на основе CeO2 и на основе ZrO2, а также окристаллизованной фазы Mn3O4, что вызывает снижение каталитической активности. Таким образом, наибольшей каталитической активностью в реакции окисления СО обладают образцы, прокаленные при 500–600°С, что, вероятно, связано с присутствием марганца как в структуре твердого раствора, так и в виде высокодисперсных частиц MnOх на его поверхности.
Список литературы
Frey K., Iablokov V., Sáfrán G., Osán J., Sajó I., Szukiewicz R., Chenakin S., Kruse N. // J. Catal. 2012. V. 287. P. 30.
Tang X., Li Y., Huang X., Xu Y., Zhu H., Wang J., Shen W. // Appl. Catal. B: Env. 2006. V. 62. P. 265.
Mousavi S.M., Niaei A., Gómez M.J.I., Salari D., Panahi P.N., Abaladejo-Fuentes V. // Mater. Chem. Phys. 2014. V. 143. P. 921.
Terribile D., Trovarelli A., Leitenburg C., Primavera A., Dolcetti G. // Catal. Today. 1999. V. 47. № 1–4. P. 133.
Machida M., Murata Y., Kishikawa K., Zhang D., Ikeue K. // Chem. Mater. 2008. V. 20. № 13. P. 4489.
Nelson A.E., Schulz K.H. // Appl. Surf. Sci. 2003. V. 210. P. 206.
Azalim S., Franco M., Brahmi R., Giraudon J.-M., Lamonier J.-F. // J. Hazar. Mater. 2011. V. 188. P. 422.
Gallegos M.V., Garbarino G., Colman Lerner J.E., Finocchio E., Busca G., Sambeth J.E., Peluso M.A. // Lat. Am. Appl. Res. 2021. V. 51. № 2. P. 81.
Zhu L., Li X., Liu Z., Yao L., Yu P., Wei P., Xu Y., Jiang X. // Nanomater. 2019. V. 9. P. 675.
Kaplin I.Yu., Lokteva E.S., Bataeva S.V., Maslakov K.I., Fionov A.V., Shumyantsev A.V., Isaikina O.Ya., Kamaev A.O., Golubina E.V. // Pure Appl. Chem. 2021. V. 93. № 4. P. 447.
Alinezhadchamazketi A., Khodadadi A.A., Mortazavi Y., Nemati A. // J. Env. Sci. 2013. V. 25. № 12. P. 2498.
Sánchez Escribano V., Fernández López E., Gallardo-Amores J.M., del Hoyo Martínez C., Pistarino C., Panizza M., Resini C., Buscac G. // Combust. Flame. 2008. V. 153. P. 97.
Cao F., Xiang J., Wang P., Sun L., Hu S., Lei S. // Chem. Eng. J. 2014. V. 243. P. 347.
Sun W., Li X., Mu J., Fan S., Yin Z., Wang X., Qin M., Tade M., Liu S. // J. Colloid Interface Sci. 2018. V. 531. P. 91.
Shen B., Wang Y., Wang F., Liu T. // Chem. Eng. J. 2014. V. 236. P. 171.
Hou Z., Feng J., Lin T., Zhang H., Zhou X., Chen Y. // Appl. Surf. Sci. 2018. V. 434. P. 82.
Long G., Chen M., Li Y., Ding J., Sun R., Zhou Y., Huang X., Han G., Zhao W. // Chem. Eng. J. 2019. V. 360. P. 964.
Shen B., Zhang X., Yao Y., Liu T. // J. Env. Sci. 2013. V. 25. № 4. P. 791.
Rao T., Shen M., Jia L., Hao J., Wang J. // Catal. Commun. 2007. V. 8. P. 1743.
Либерман Е.Ю., Клеусов Б.С., Наумкин А.В., Загайнов И.В., Конькова Т.В., Симакина Е.А., Изотова А.О. // Перспективные материалы. 2020. № 9. С. 75.
Афонасенко Т.Н., Булавченко О.А., Гуляева Т.И., Цыбуля С.В., Цырульников П.Г. // Кинетика и катализ. 2018. Т. 59. №1. С. 127.
Scofield J.H. // J. Electron Spectros. Relat. Phenomena. 1976. V. 8. № 2. P. 129.
Shirley D.A. // Phys. Rev. B. 1972. V. 5. P. 4709.
Fairley N. // www.casaxps.com.
Moretti E., Storaro L., Talona A., Lenarda M., Riello P., Frattini R., del Valle Martínez de Yusoc M., Jiménez-Lópezc A., Rodríguez-Castellónc E., Ternerod F., Caballerod A., Holgadod J.P. // Appl. Catal. B: Env. 2011. V. 102. P. 627.
Azalim S., Brahmi R., Agunaou M., Beaurain A., Giraudon J.-M., Lamonier J.-F. // Chem. Eng. J. 2013. V. 223. P. 536.
Tang L., Yamaguchi D., Burke N., Trimm D., Chiang K. // Catal. Commun. 2010. V. 11. P. 1215.
Jampaiah D., Venkataswamy P., Tur K.M., Ippolito S.J., Bhargava S.K., Reddy B.M. // Z. Anorg. Allg. Chem. 2015. V. 641. № 6. P. 1141.
Bulavchenko O.A., Vinokurov Z.S., Afonasenko T.N., Tsyrul’nikov P.G., Tsybulya S.V., Saraev A.A., Kaichev V.V. // Dalton Trans. 2015. V. 44. P. 15499.
Gutiérrez-Ortiz J.I., De Rivas B., López-Fonseca R., Martín S., González-Velasco J.R. // Chemosphere. 2007. V. 68. P. 1004.
Zhong L., Fang Q., Li X., Li Q., Zhang C., Chen G. // Appl. Catal. A: Gen. 2019. V. 579. P. 151.
Kantzer E., Dobber D., Kiessling D., Wendt G. // Stud. Surf. Sci. Catal. 2002. V. 143. P. 489.
Дополнительные материалы отсутствуют.
Инструменты
Кинетика и катализ