

Кинетика и катализ, 2022, T. 63, № 4, стр. 470-483
Синтез Фишера–Тропша на бифункциональных кобальтовых катализаторах с использованием иерархического цеолита HBeta
Р. Е. Яковенко a, *, В. Г. Бакун a, И. Н. Зубков a, О. П. Папета a, А. Н. Салиев a, М. Р. Аглиуллин a, А. П. Савостьянов a
a ФГБОУ ВО Южно-Российский государственный политехнический университет (НПИ) имени М.И. Платова
346128 Новочеркасск, ул. Просвещения, 132, Россия
* E-mail: jakovenko39@gmail.com
Поступила в редакцию 29.12.2021
После доработки 10.03.2022
Принята к публикации 12.03.2022
- EDN: XKKJBK
- DOI: 10.31857/S045388112204013X
Аннотация
Исследован процесс получения компонентов топлива в интегрированном синтезе Фишера–Тропша на бифункциональных кобальтовых катализаторах в форме смеси кобальтового катализатора Co–Al2O3/SiO2 и образцов иерархического цеолита HBeta со связующим. Определены условия приготовления образцов HBeta с развитой мезопористой структурой методом щелочного модифицирования с использованием раствора NaOH. Изучено влияние структуры и кислотных свойств HBeta на активность катализаторов в синтезе Фишера–Тропша, углеводородный и фракционный состав полученных продуктов. Исследования проведены в проточном реакторе со стационарным слоем катализатора при давлении 2 МПа, объемной скорости газа 1000 ч–1 и температуре 240°С. Показатели процесса для бифункциональных катализаторов и состав компонентов топлив коррелируют со структурой и состоянием поверхности образцов цеолита HBeta.
ВВЕДЕНИЕ
Традиционный синтез углеводородов из CO и H2 по методу Фишера–Тропша (ФТ) [1] является универсальным каталитическим процессом для производства различных углеводородов С5+, в том числе высокомолекулярных восков, с последующей переработкой полученных продуктов (гидрокрекинг или гидроочистка) [2, 3]. Современные процессы ФТ во многом ориентированы на развитие топливного направления, которое является известной “зеленой альтернативой” нефтяным топливам и перспективно как практически отработанная технология, особенно GTL [4, 5]. В последнее время активно изучаются и другие технологии XTL (X-to-liquid), предназначенные для переработки любого органического сырья (угля, биомассы, сланцевого газа и т.д.) [6]. Практически реализованы низкотемпературный (LTFT) и высокотемпературный (HTFT) варианты синтеза ФТ соответственно на кобальтовом или железном и железном катализаторах.
Новые потенциальные возможности открывает реализация экономически привлекательного интегрированного двухстадийного процесса ФТ для конверсии газообразных углеродсодержащих ресурсов и получения продуктов с добавленной стоимостью [7]. В присутствии полифункциональной каталитической системы в виде комбинации катализатора или активного металла ФТ и кислотного катализатора первичные продукты синтеза, образующиеся на металлических активных центрах, подвергаются вторичным превращениям на кислотных центрах катализатора (изомеризация, гидрокрекинг, олигомеризация, ароматизация и гидрирование углеводородов и др.) [7–9]. В этом процессе особая роль принадлежит полифункциональному катализатору, обеспечивающему синтез и последующее превращение углеводородов в топливные продукты.
Рассматриваются различные способы приготовления и составы катализаторов, в том числе промотированных [5, 10–14], однако одним из условий их эффективного функционирования остается обеспечение тесного контакта металлического и кислотного активных центров катализаторов, а также формирование оптимальной пористой структуры для транспортировки исходных реагентов и продуктов синтеза, даже при наличии диффузионных ограничений [11, 13, 15]. Большой интерес представляет создание катализаторов ФТ для непосредственного превращения синтез-газа в углеводороды с определенным числом атомов углерода и разветвленной структурой [16]. Тем не менее, разработка новых полифункциональных катализаторов с высокой селективностью в отношении образования продуктов топливных фракций остается сложной задачей [16, 17], решению которой способствует развитие мезопористости катализаторов [5, 12].
Как указывают авторы [12, 18–22], сравнивая подходы к созданию иерархических структур для адсорбции и катализа, в том числе синтеза ФТ, формирование мезопористой пористой структуры цеолитов может осуществляться во время синтеза (подход снизу вверх) или методами постсинтеза (подход сверху вниз) [17, 23, 24] при извлечении кремния или алюминия из кристаллического каркаса цеолитов. Среди постсинтетических стратегий создания внутрикристаллических мезопор [19] процесс извлечения кремния: i) относится к числу наиболее эффективных и экономичных [20] и включает селективное удаление Т-атомов каркаса цеолита с помощью гидротермальной [16, 24, 25], кислотной [17, 26] и/или щелочной обработки [12, 19, 21, 23, 27, 28]; ii) сопровождается увеличением числа различных кислотных центров Бренстеда и Льюиса и изменением их локализации и соотношения [20]; iii) ведет, как показано на примере катализаторов с ZSM-5 [21, 23, 29], к предотвращению дезактивации (за счет улучшения молекулярной диффузии в системе пор и ограничения блокирования микропор углеродсодержащими отложениями). Для катализаторов на основе цеолитов со структурой MFI, особенно ZSM-5 с соотношением Si/Al около 25–50 как коммерчески востребованного продукта, и было проведено большинство фундаментальных исследований по извлечению атомов кремния [22], рассматривающих среди других не только проблему изменения условий молекулярной диффузии, но и повышения доступности активных центров катализаторов на основе цеолитов. В то же время при использовании цеолитов как составляющих катализаторов для получения топлив нет однозначных представлений о том, как структура и топологические характеристики цеолитов влияют на синтез компонентов топлив с различными свойствами (например, октанового числа изоалканов), которое растет с увеличением числа ответвлений у разветвленных алканов в составе фракций топлив ФТ). Кроме того, наряду с составом и особенностями строения исследуемого цеолита, при осуществлении щелочного модифицирования следует учитывать, что оптимальные условия проведения обработки во многом зависят от свойств конкретной партии используемого цеолита [22].
Целью настоящей работы явилось изучение влияния щелочного модифицирования на параметры пористой структуры и кислотность цеолита HBeta, свойства и каталитические показатели бифункциональных кобальтовых катализаторов синтеза ФТ на основе мезопористых образцов цеолита и углеводородный состав синтезируемых фракций топлива.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Исходный цеолит Beta с мольным соотношением SiО2/Al2О3 = 40.5 (“Zeolyst International”) прокаливали 6 ч при температуре 550°С. При приготовлении модифицированных образцов навеску цеолита Beta в течение 1 ч при температуре 70°С обрабатывали раствором NaOH концентрацией 0.1, 0.15, 0.25, 0.3 и 0.5 M. По окончании обработки при комнатной температуре промывали дистиллированной водой, после чего 8 ч сушили при 100–120°С и прокаливали 3 ч при 300°С. Для получения цеолита в H-форме образцы обрабатывали раствором NH4NO3 концентрацией 1 M в течение 1 ч при 70°С, после чего 8 ч сушили при 100–120°С и прокаливали 6 ч при 550°С. Образцы цеолита HBeta обозначали: исходный – Нβ и модифицированные – xНβ, где x – концентрация раствора NaOH при обработке: 0.1, 0.15, 0.25, 0.3 и 0.5 M.
Образцы бифункциональных кобальтовых катализаторов синтеза ФТ c цеолитом Beta в H-форме и связующим бемитом получали в форме смеси компонентов. В качестве компонентов использовали кобальтовый катализатор Co–Al2O3/SiO2 для селективного синтеза длинноцепочечных углеводородов [30–32] с силикагелевым носителем КСКГ (ООО “Салаватский катализаторный завод”), исходный и модифицированные образцы цеолита HBeta и связующее бемит Al(OH)O (“Sasol”, TH 80).
Образцы готовили смешением порошков (фракция <0.1 мм), мас. %: катализатор Со‒Al2O3/SiO2 – 35, цеолит HBeta – 30, бемит – 35 [33, 34]. Для пластификации связующего бемита применяли водно-спиртовой раствор триэтиленгликоля и азотной кислоты (раствор азотной кислоты получали внесением 1–2 мл азотной кислоты концентрацией 65 мас. % в 90–100 мл дистиллированной воды на 100 г смеси порошков, триэтиленгликоль вводили, исходя из объемного соотношения азотная кислота : триэтиленгликоль в смеси равного 1 : 3). Гранулы катализатора формовали методом экструзии. Сушили 24 ч при комнатной температуре, 4–6 ч при 80–100°С, 2–4 ч при 100–150°С и прокаливали 5 ч при 400°С. Катализаторы обозначали: исходный – Нβ+, модифицированные – xНβ+, где x – концентрация раствора NaOH: 0.1, 0.15, 0.25, 0.3 и 0.5 М.
Определение содержания кобальта в катализаторах проводили методом рентгено-флуоресцентного анализа на рентгеновском энергодисперсионном спектрометре ARL QUANT’X (“Thermo Scientific”, США), при следующих условиях: среда – воздух, тефлоновая подложка, эффективная площадь облучения – 48.9 мм2.
Изучение параметров пористой структуры образцов цеолита и катализаторов осуществляли методом адсорбции–десорбции азота с использованием сорбометра Nova 1200e (“Quantachrome”, США). Величину удельной поверхности методом БЭТ (Брунауэра–Эммета–Теллера) вычисляли при относительном парциальном давлении газа P/P0 = 0.20. Объем пор определяли методом BJH (Баррета–Джойнера–Халенды) при относительном парциальном давлении P/P0 = 0.95, распределение пор по размерам рассчитывали по кривой десорбции BJH, объем микропор в присутствии мезопор был измерен с помощью t-метода (де Бур и Липпенс). Предварительно образцы подвергали вакуумной обработке в течение 5 ч при температуре 350°С.
Рентгенофазовый анализ (РФА) проводили на дифрактометре Thermo Scientific ARLX’TRA Powder Diffractometer (“Thermo Fisher Scientific”, Швейцария) с монохроматизированным CuKα-излучением методом сканирования по точкам (шаг – 0.01°, время накопления в точке – 2 с) в интервале 2θ от 5° до 90°. Определение качественного фазового состава образцов HBeta и катализаторов осуществляли с помощью PDF-2 [35] в программном комплексе Crystallographica.
Обработку рентгенограмм выполняли в программе FullProf, расчет среднего размера частиц Co3O4 (d(Co3O4), нм) для характеристической линии со значением 2θ равным 36.8° – по уравнению Шеррера [36]:
(1)
$d({\text{C}}{{{\text{o}}}_{{\text{3}}}}{{{\text{O}}}_{{\text{4}}}}) = \frac{{K{{\lambda }}}}{{{{\beta }}{\kern 1pt} \cos {\kern 1pt} {{\theta }}}},$Расчет среднего размера частиц кобальта d(Co0) выполняли с использованием формулы [37]:
(2)
$d({\text{C}}{{{\text{o}}}^{0}}) = d({\text{C}}{{{\text{o}}}_{{\text{3}}}}{{{\text{O}}}_{{\text{4}}}}) \times 0.75.$Дисперсность металлического компонента (D, %) определяли по формуле [38]:
Исследования катализаторов методом температурно-программированного восстановления водородом (ТПВ-Н2) проводили на анализаторе ChemiSorb 2750 (“Micromeritics”, США) с детектором по теплопроводности (ДТП). Перед ТПВ-Н2 катализаторы выдерживали в токе гелия (20 мл/мин) в течение 1 ч при температуре 200°С. Затем охлаждали до комнатной температуры и подавали смесь 10% водорода и 90% азота (20 мл/мин). Исследования выполняли в интервале температур 20–800°С со скоростью нагрева 20°С/мин.
Оценку кислотных свойств осуществляли на анализаторе Autosorb (“Quantachrome”, США). Перед адсорбцией образцы цеолита и катализаторы дегазировали в токе гелия при 600°С в течение 5 ч и продували для удаления физически адсорбированного аммиака. Адсорбцию аммиака проводили при температуре 100°C в токе аммиачно-гелиевой смеси (10% NH3 по объему) в течение 2 ч.
Десорбцию аммиака (ТПД NH3) выполняли в интервале температур 100–600°С при линейном нагреве со скоростью 10°С/мин, газ-носитель – гелий.
Изучение каталитических свойств в синтезе ФТ вели в изотермическом реакторе диаметром 16 мм со стационарным слоем катализатора. В реактор загружали 5–10 см3 катализатора (фракция 1–2 мм) в смеси с 15–30 см3 кварца. Катализатор восстанавливали водородом в течение 1 ч при температуре 400°С и объемной скорости газа 3000 ч–1. Активацию образцов синтез-газом с соотношением Н2/СО = 2 и каталитические испытания проводили при давлении 2.0 МПа и объемной скорости газа 1000 ч–1, поднимая температуру от 180 до 230–250°С со скоростью 2.5°С/ч. Балансовые опыты осуществляли в течение 70–80 ч, каждые 2 ч анализируя состав и количество газа на выходе установки. Об активности катализаторов судили по конверсии СО, селективности и производительности катализаторов, фракционному и углеводородному составу продуктов синтеза.
Состав исходного газа и газообразных продуктов синтеза анализировали с помощью газового хроматографа Кристалл 5000 (“Хроматэк”, Россия), оснащенного детектором по теплопроводности и двумя колонками (активная фаза Haysep R и молекулярные сита NaX). Режим анализа – температурно-программируемый со скоростью нагрева 8°С/мин.
Конденсированные продукты синтеза разделяли дистилляцией при атмосферном давлении, выделяя топливные фракции с температурой кипения: бензиновая – до 180°С, дизельная – 180–330°С, кубовый остаток – выше 330°С. Состав углеводородов С5+ определяли с помощью хромато-масс-спектрометра 7890A (“Agilent Techno1ogies”, США), снабженного детектором MSD 5975С (“Agilent”, США) и капиллярной колонкой HP-5MS.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
В качестве кислотного компонента бифункциональных кобальтовых катализаторов синтеза ФТ использовали исходный и модифицированные образцы алюмосиликатного высококремнеземного цеолита Beta типа BEA [39] в H-форме. Мольное соотношение Si/Al Beta варьируется в пределах 10–100 и выше [40], для исследованного образца оно равно 40.5. Внутрикристаллическая структура цеолитов зависит от условий синтеза, соотношения Si/Al, типа катионов и др. [41, 42] и описывается топологией каркаса, геометрией и размерами полостей и каналов. Размер элементарной ячейки цеолита Beta составляет 1.3 × 1.3 × × 2.6 нм [39, 43], структура трехмерна и образована системой взаимно пересекающихся 12-членных прямых кольцевых каналов (3D, 12 MR), сформированных сочетанием политипов тетрагональной и моноклинной симметрии с отверстиями 0.68 и 0.73 нм или 0.60 и 0.73 нм соответственно (эллиптическое сечение). Диаметр входного окна для каждого из политипов равен 0.55 × 0.55 нм и 0.64 × 0.75 нм по данным [44–46] или 0.56 × × 0.56 нм и 0.66 × 0.67 нм по данным [42].
Рентгенофазовый анализ согласно [35] подтвердил фазовый состав образцов HBeta (рис. 1) и обнаружил изменения в структуре цеолита, связанные с обработкой раствором NaOH концентрацией 0.1–0.5 М. При использовании раствора NaOH, который является сильным модификатором [28] (концентрация наиболее сильно влияет на развитие мезопористости HBeta [47]), кремниевый каркас образцов частично растворяется. Вымывание атомов кремния из каркаса происходит преимущественно с внешней стороны кристаллов, вызывает разрушение структуры цеолита и снижение соотношения Si/Al [23]. Основное воздействие, судя по уширению и уменьшению интенсивности основных характеристических линий образцов, оказывает использование раствора щелочи концентрацией выше 0.25 М [28]. Результаты изучения механизма модифицирования свидетельствуют об особом влиянии алюминия (защищает атомы кремния от извлечения) на кинетику этого процесса. По данным [20, 24] частичная потеря кристалличности и аморфизация структуры цеолита при высоких концентрациях раствора NaOH может быть связана с меньшей стабильностью каркасного алюминия в структуре Beta, чем в структурах типа MFI и MOR [48], и наличием внутренних дефектов упаковки [21, 49]. Кроме того, увеличение концентрации алюминия в структуре модифицированного HBeta не только играет преобладающую роль в регулировании извлечения атомов кремния, но и способствует образованию значительной мезопористости с регулярным распределением пор по размерам [20].
Рис. 1.
Дифрактограммы образцов цеолита HBeta: 1 – Нβ; 2 – 0.1Нβ; 3 – 0.15Нβ; 4 – 0.25Нβ; 5 – 0.3Нβ; 6 – 0.5Нβ.
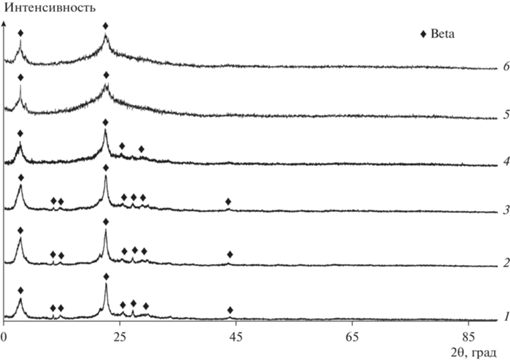
Пористое строение цеолита Beta зависит от топологии каркаса: размер канала или раскрытие поры определяются размером кольца – числом Т-атомов (центральный атом оксида тетраэдра ТО4) в минимальном замкнутом цикле связей. Диаметр пор находится в пределах 0.59–0.67 нм [43], удельная поверхность достигает 420–1050 м2/г [50]. Исходный образец HBeta обладает развитой поверхностью и системой пор (табл. 1). Доля микропор в величине удельной поверхности (БЭТ) составляет порядка 55%, в измеренном объеме пор – 45%. Определяя избирательность цеолитов, в присутствии больших молекул в процессах, аналогичных синтезу ФТ, микропоры обусловливают появление диффузионных ограничений, вызывая быструю дезактивацию катализаторов [21, 29]. При повышении концентрации раствора модификатора, вследствие трансформации каркаса цеолита, количество микропор (и, соответственно, поверхность и объем микропор) образцов постепенно снижается (максимально в 4.5 раза и до 10% объема у образца 0.5Нβ), вторичных мезопор – увеличивается. Основное развитие мезопористости, несмотря на появление межкристаллических мезопор, определяется изменениями, происходящими в объеме кристаллов (удаление T-атомов кристаллов) [20]. Степень трансформации пористой структуры образцов цеолита HBeta в ходе модифицирования иллюстрирует рис. 2.
Таблица 1.
Параметры пористой структуры образцов цеолита HBeta
| Цеолит HBeta | Удельная поверхность, м2/г | Объем пор, см3/г | ||
|---|---|---|---|---|
| БЭТ | микропоры | микропоры | суммарный | |
| Нβ | 576 | 325 | 0.17 | 0.37 |
| 0.25Нβ | 373 | 160 | 0.08 | 0.34 |
| 0.5Нβ | 331 | 72 | 0.04 | 0.41 |
Рис. 2.
Изотермы адсорбции–десорбции азота и распределение пор по размерам для образцов цеолита HBeta: (а) – Нβ; (б) – 0.25Нβ; (в) – 0.5Нβ.
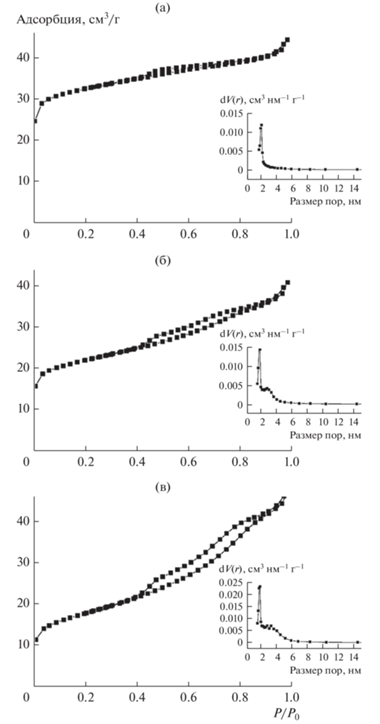
Представленные результаты показывают, что выбранный метод постсинтеза эффективен для создания иерархической мезопористой структуры цеолита HBeta. Изотермы адсорбции–десорбции азота исходного HBeta близки к изотермам типа I, характерным для микропористых цеолитсодержащих материалов (рис. 2а). Изотермы модифицированных образцов 0.25Нβ и 0.5Нβ (рис. 2б и 2в) типичны для материалов, содержащих микро- и мезопоры, и могут быть отнесены к типу IV. Нарастание гистерезиса между изотермами адсорбции и десорбции отражает динамику образования мезопор и процесса капиллярной конденсации в мезопорах (петля гистерезиса типа H4 встречается у материалов с щелевидной формой пор). По данным BJH распределение пор исходного HBeta по размерам имеет узкий максимум в микропористой области, соответствующей размеру пор порядка 1.5–2 нм. Увеличение концентрации раствора NaOH ведет к росту размера мезопор и появлению второго максимума на кривой распределения, соответствующего для образца 0.25Нβ мезопорам размером 2–4 нм, для образца 0.5Нβ – 2–7 нм. Подобные закономерности в изменении структуры и изотерм адсорбции–десорбции цеолитов Beta и ZSM-5 при щелочном модифицировании отмечались в ряде работ, например, в [13, 21, 29].
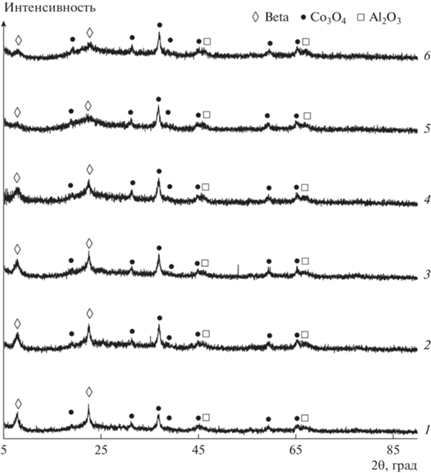
Концентрация кобальта в исследуемых бифункциональных катализаторах составляет 7.4–7.5 мас. % и определяется равным количеством введенного кобальтсодержащего компонента – катализатора Co–Al2O3/SiO2. В структуре Co‒Al2O3/SiO2 согласно [30, 31] и катализаторов по данным РФА (рис. 3) кобальт содержится в виде оксида-предшественника Co3O4 с кубической структурой шпинели (Fd3m) [35]. Размер кристаллитов металлического кобальта, рассчитанный по уравнению Шеррера, при среднем размере частиц Co3O4 равном 13.0–17.5 нм, составляет 9.8–13.0 нм. SiO2 рентгеноаморфен. Рефлексы цеолитного компонента катализаторов аналогичны рефлексам на рис. 1. Частичная потеря кристалличности образцами цеолита делает более доступным определение фазы Co3O4 катализаторов 0.25Нβ+, 0.3Нβ+ и 0.5Нβ+ – интенсивность основных линий на дифрактограммах увеличивается. Оксид алюминия представлен рефлексами при 2θ ≈ 47°–70°. Слабая интенсивность рефлексов Al2O3 указывает на низкую степень кристалличности фазы оксида, формирующегося в процессе термообработки связующего компонента бемита.
Рис. 3.
Дифрактограммы катализаторов: 1 – Нβ+; 2 – 0.1Нβ+; 3 – 0.15Нβ+; 4 – 0.25Нβ+; 5 – 0.3Нβ+; 6 – 0.5Нβ+.
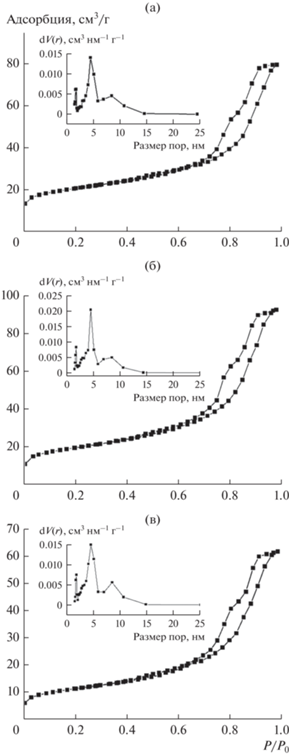
Параметры пористой структуры гранул катализаторов приведены в табл. 2. Удельная поверхность (БЭТ) катализатора Co–Al2O3/SiO2 равна 246 м2/г, бемита – 260 м2/г. Таким образом, уменьшение удельной поверхности и объема микропор катализаторов обусловлено снижением этих параметров у модифицированных образцов цеолита HBeta со значительной степенью трансформации пористой структуры. Так, доля микропор в величине удельной поверхности (БЭТ) образца с исходным HBeta составляет 28%, в измеренном объеме пор – 8%, а для образца 0.5Нβ+ – 7 и 2% соответственно. Изотермы адсорбции–десорбции азота катализаторов относятся к типу IV (рис. 4) с формой петли гистерезиса H4. По данным BJH образцы отличает широкое бидисперсное распределение пор по размерам в интервале 2–15 нм c двумя максимами около 5 и 8 нм.
Таблица 2.
Параметры пористой структуры катализаторов
| Катализатор | Удельная поверхность, м2/г | Объем пор, см3/г | ||
|---|---|---|---|---|
| БЭТ | микропоры | микропоры | суммарный | |
| Нβ+ | 311 | 87 | 0.04 | 0.55 |
| 0.25Нβ+ | 261 | 36 | 0.02 | 0.55 |
| 0.5Нβ+ | 222 | 17 | 0.01 | 0.54 |
Рис. 4.
Изотермы адсорбции–десорбции азота и распределение пор по размерам для катализаторов: (а) – Нβ+; (б) – 0.25Нβ+; (в) – 0.5Нβ+.
Спектры ТПВ-Н2 катализаторов идентичны (рис. 5). Восстановление оксида Co3O4 катализаторов идет поэтапно: Co3O4 → CoО и CoО → Со0 [51]. Отношение площадей пиков поглощения водорода, необходимого для осуществления ступенчатого перехода Co3+ → Co0, отличается от 3 – теоретически ожидаемого значения в соответствии со стехиометрией реакций восстановления Co3O4 (табл. 3). Снижение температуры максимумов и более пологая форма пиков, относимых к этапу восстановления CoО катализаторов на основе образцов HBeta, полученных обработкой раствором NaOH концентрацией 0.15–0.3 М, вероятно, могут быть связаны как с наименьшим размером частиц Co3O4 (13.0–14.1 нм) или Co0, так и развитием мезопористости; повышение температуры восстановления катализатора 0.5Нβ+ – с их укрупнением, и, по-видимому, более заметным изменением соотношения Si/Al, увеличением количества и влияния алюминия в структуре образцов на состояние кобальта. Отсутствие пиков поглощения Н2 в высокотемпературной области показывает, что основная часть кобальта, зафиксированная в оксидной форме в составе катализатора Co‒Al2O3/SiO2, не вступает в значимое взаимодействие с цеолитом или оксидом алюминия связующего с образованием трудновостанавливаемых соединений [51].
Рис. 5.
Профили ТПВ-Н2 катализаторов: 1 – Нβ+; 2 – 0.1Нβ+; 3 – 0.15Нβ+; 4 – 0.25Нβ+; 5 – 0.3Нβ+; 6 – 0.5Нβ+.
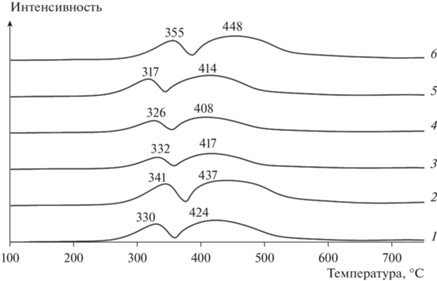
Таблица 3.
Данные ТПВ-Н2 катализаторов
| Катализатор | Пик 1 | Пик 2 | S2/S1 | ||
|---|---|---|---|---|---|
| Тmax, °С | площадь S1, % | Тmax, °С | площадь S2, % | ||
| Нβ+ | 330 | 29.2 | 424 | 70.8 | 2.4 |
| 0.1Нβ+ | 341 | 28.4 | 437 | 71.6 | 2.5 |
| 0.15Нβ+ | 332 | 26.9 | 417 | 73.1 | 2.7 |
| 0.25Нβ+ | 326 | 27.5 | 408 | 72.6 | 2.6 |
| 0.3Нβ+ | 317 | 27.3 | 414 | 72.7 | 2.7 |
| 0.5Нβ+ | 355 | 25.9 | 448 | 74.1 | 2.9 |
Поскольку содержание алюминия определяет и количество кислотных центров на внутренней и внешней поверхности кристаллов цеолита, во многом обусловливая каталитические свойства, можно полагать, что рост концентрации алюминия в структуре образцов HBeta будет вести к увеличению числа активных центров Бренстеда (B) и Льюиса (L) [21]. В работе [47] было обнаружено, что у образцов Beta, обработанных раствором NaOH концентрацией менее 0.4 М, количество кислотных центров L сохраняется, сильных центров B – увеличивается, а при концентрации выше 0.4 М уменьшается. В то же время по данным [13], если вначале цикла модифицирования кислотность B поверхности образцов снижается, то дальнейший ионный обмен мезопористого Beta с ${\text{NH}}_{4}^{ + }$, последующее прокаливание и образование H-формы может ее восстановить.
Кислотность поверхности исследуемых образцов HBeta по результатам ТПД NH3 представлена центрами трех типов (табл. 4). По мере увеличения степени модифицирования HBeta наблюдается перераспределение состава кислотных центров: суммарно количество кислотных центров, обработанного раствором NaOH 0.25 М, снижается не более чем на 10%; при повышении концентрации раствора до 0.5 М – падает в 2 и более раз, при этом воздействию подвергаются слабые и сильные кислотные центры. Количество слабых и средних кислотных центров бифункциональных катализаторов суммарно находится в пределах 87–169 мкмоль/г, сильные центры не обнаружены.
Таблица 4.
Кислотные свойства образцов цеолита HBeta и катализаторов
| Цеолит, катализатор+ | Кислотные центры, мкмоль/г* | ||
|---|---|---|---|
| слабые (100–250°С) |
средние (250–400°С) |
сильные (400–550°С) |
|
| Нβ | 229 | 174 | 114 |
| 0.25Hβ | 160 | 206 | 106 |
| 0.5Нβ | 53 | 84 | 8 |
| Нβ+ | 117 | 52 | – |
| 0.1Нβ+ | 106 | 48 | – |
| 0.15Нβ+ | 74 | 33 | – |
| 0.25Нβ+ | 60 | 27 | – |
| 0.3Нβ+ | 67 | 29 | – |
| 0.5Нβ+ | 83 | 36 | – |
Полученные бифункциональные катализаторы проявляют высокую активность в процессе синтеза ФТ при температуре 240°С (табл. 5). Максимальные показатели по конверсии CO и производительности в отношении углеводородов C5+ имеют катализаторы на основе образцов HBeta со значительной степенью трансформации кристаллического каркаса и иерархической мезопористой структурой цеолита, полученной методом постсинтеза при обработке раствором NaOH концентрацией 0.25–0.5 М. Эти катализаторы отличаются развитой удельной поверхностью и объемом пор, сформированным мезопорами 2–15 нм с бидисперсным распределением пор по размерам, и активной поверхностью, представленной слабыми и средними кислотными центрами [10, 52].
Таблица 5.
Каталитические показатели синтеза ФТ
| Катализатор | Конверсия CO, % | Селективность, % | Производитель-ность по C5+, ${\text{кг м}}_{{{\text{кат}}}}^{{ - 3}}{\text{ }}{{{\text{ч}}}^{{ - 1}}}$ | |||
|---|---|---|---|---|---|---|
| СН4 | С2–С4 | С5+ | СО2 | |||
| Нβ+ | 81.7 | 16.0 | 8.2 | 73.2 | 2.6 | 135.6 |
| 0.1Нβ+ | 67.9 | 21.4 | 12.6 | 62.9 | 3.1 | 92.8 |
| 0.15Нβ+ | 76.3 | 19.5 | 10.6 | 67.5 | 2,5 | 110.1 |
| 0.25Нβ+ | 81,7 | 19.2 | 9.6 | 68.6 | 2,7 | 117.3 |
| 0.3Нβ+ | 80.7 | 18.2 | 9.8 | 69.1 | 3.0 | 120.2 |
| 0.5Нβ+ | 83.1 | 15.1 | 9.0 | 73.0 | 2.5 | 126.7 |
Синтезированные углеводороды С5+ являются алканами и алкенами нормального и изостроения фракций С5–С10, С11–С18 и С19+ (табл. 6). Нафтеновые и ароматические углеводороды не обнаружены.
Таблица 6.
Углеводородный состав продуктов синтеза ФТ
| Катализатор | Углеводороды | Групповой состав углеводородов С5+, мас. % | Всего | изо/н** | о/п*** | ||
|---|---|---|---|---|---|---|---|
| C5–C10 | C11–С18 | С19+ | |||||
| Нβ+ | н-Алканы | 20.4 | 29.2 | 9.6 | 59.2 | 0.4 | 0.3 |
| изо-Алканы | 5.1 | 9.0 | 2.0 | 16.1 | |||
| Алкены | 9.1 | 2.1 | – | 11.2 | |||
| Алкены* | 12.6 | 0.9 | – | 13.5 | |||
| Итого | 47.2 | 41.2 | 11.6 | 100.00 | |||
| 0.1Нβ+ | н-Алканы | 25.9 | 23.6 | 7.2 | 56.7 | 0.4 | 0.4 |
| изо-Алканы | 4.6 | 9.6 | 2.4 | 16.6 | |||
| Алкены | 8.9 | 3.7 | 0.6 | 13.2 | |||
| Алкены* | 10.8 | 2.7 | – | 13.5 | |||
| Итого | 50.2 | 39.6 | 10.2 | 100.00 | |||
| 0.15Нβ+ | н-Алканы | 25.0 | 19.4 | 12.6 | 57.0 | 0.5 | 0.3 |
| изо-Алканы | 5.4 | 8.5 | 5.6 | 19.5 | |||
| Алкены | 8.2 | 2.7 | 0.6 | 11.5 | |||
| Алкены* | 10.2 | 1.5 | 0.3 | 12.0 | |||
| Итого | 48.8 | 32.1 | 19.1 | 100.00 | |||
| 0.25Нβ+ | н-Алканы | 25.1 | 23.9 | 8.0 | 57.0 | 0.5 | 0.3 |
| изо-Алканы | 4.9 | 11.4 | 3.5 | 19.8 | |||
| Алкены | 7.3 | 5.1 | 0.6 | 13.0 | |||
| Алкены* | 8.2 | 2.0 | – | 10.2 | |||
| Итого | 45.5 | 42.4 | 12.1 | 100.00 | |||
| 0.3Нβ+ | н-Алканы | 29.9 | 22.7 | 9.8 | 62.4 | 0.4 | 0.3 |
| изо-Алканы | 3.3 | 9.4 | 3.2 | 15.9 | |||
| Алкены | 5.7 | 4.0 | 0.4 | 10.1 | |||
| Алкены* | 9.1 | 2.5 | – | 11.6 | |||
| Итого | 48.0 | 38.6 | 13.4 | 100.0 | |||
| 0.5Нβ+ | н-Алканы | 25.5 | 29.6 | 8.3 | 63.4 | 0.3 | 0.2 |
| изо-Алканы | 2.9 | 11.0 | 3.9 | 17.8 | |||
| Алкены | 2.3 | 1.7 | – | 4.0 | |||
| Алкены* | 11.2 | 3.6 | – | 14.8 | |||
| Итого | 41.9 | 45.9 | 12.2 | 100.0 | |||
Изменения кристаллической структуры, пористого строения и состояния поверхности кислотного компонента катализаторов оказывают влияние на состав полученных групп углеводородов. Мезопористость и кислотность HBeta, по-видимому, способствуют селективному гидрокрекингу первичных высокомолекулярных углеводородов, образующихся на металлических активных центрах катализаторов, в углеводороды топливных фракций [13, 17, 25]. Количество н-алканов, синтезированных на катализаторах 0.1Нβ+–0.3Нβ+, в сравнении с катализатором Нβ+ на 5–10% повышается в составе бензиновой фракции C5–C10 и уменьшается в составе углеводородов дизельной фракции C11–C18. Противоположная картина наблюдается для алкенов. Изомеризация, следующая за гидрокрекингом длинноцепочечных углеводородов [25], способствует увеличению содержания углеводородов изостроения в продуктах синтеза. В свою очередь мезопористые каналы и структура 12MR HBeta обеспечивают близость реагирующих молекул, усиливая интенсивность процесса изомеризации. Кроме того, цеолит Beta, в сравнении с другими цеолитами, отличается бóльшим количеством кислотных центров Льюиса [11], на которых легко образуются ионы карбения, являющиеся промежуточным звеном для синтеза разветвленных углеводородов и их селективного превращения в изопарафины. Как следствие, в составе фракций C11–C18 и C19+ всех катализаторов с модифицированным кислотным компонентом интенсивно образуются углеводороды изостроения – изоалканы и разветвленные алкены, в особенности в присутствии образца 0.5Нβ+. Суммарное количество углеводородов C5+ изостроения меняется в пределах 28–33%. Максимальное количество изоалканов – порядка 20% – фиксируется в продуктах синтеза для катализаторов 0.15Нβ+ и 0.25Нβ+, характеризующихся наибольшим показателем изо/н. По мере нарастания изменений свойств образцов при использовании растворов щелочи концентрацией 0.25–0.5 М баланс между крекирующей и изомеризующей функциями катализаторов меняется: количество н-алканов, образующихся на кобальтсодержащем компоненте и участвующих во вторичных реакциях на кислотных центрах цеолита, снижается. Близкие результаты были получены при изучении влияния цеолитного компонента на свойства композитного катализатора в работе [13].
Молекулярно-массовое распределение (ММР) углеводородов С5+ (рис. 6) иллюстрирует процесс изменения состава синтезируемых продуктов. В определенной мере такой тип ММР может быть следствием наличия на поверхности катализаторов различных центров роста цепи и метанирования, центров, активных в процессах вторичных превращениях синтезируемых углеводородов, а также проявлением возникающих диффузионных торможений. Основой для их формирования, вероятно, является пористое строение образцов HBeta, а также вторичная пористость катализаторов, появляющаяся в процессе их приготовления. По мере развития мезопористости и расширения диапазона размеров образующихся пор образцов цеолита, а затем и катализаторов, сопровождаемых трансформацией кислотных свойств и возможным частичным преодолением диффузионных торможений, характер ММР становится ближе к мономодальному (рис. 6в).

ЗАКЛЮЧЕНИЕ
Проведено сравнение каталитических показателей бифункциональных кобальтовых катализаторов синтеза ФТ, приготовленных на основе образцов иерархического цеолита HBeta. Определено, что катализаторы обладают высокой активностью и селективностью в отношении образования углеводородов С5+. Производительность катализаторов при температуре синтеза 240°С находится в пределах 92.8–135.6 ${\text{кг м}}_{{{\text{кат}}}}^{{ - 3}}{\text{ }}{{{\text{ч}}}^{{ - 1}}}$.
Изучены особенности развития мезопор и формирования оптимальной иерархической пористой структуры цеолита HBeta в процессе щелочного модифицирования. Показано, что синергетическое взаимодействие регулярной мезопористой структуры и относительно умеренной кислотности цеолита – это основа для селективного синтеза углеводородов топливных фракций в присутствии бифункциональных катализаторов. Установлено, что в качестве кислотного компонента катализаторов предпочтительно использовать цеолит HBeta с модифицированной кристаллической структурой, полученной при обработке раствором NaOH концентрацией 0.15–0.25 М. Основными параметрами, определяющими образование углеводородов бензиновой С5–С10 и дизельной С11–С18 фракций с высокой степенью изомеризации, являются объем, геометрия и характер распределения пор цеолита HBeta.
Список литературы
Khodakov A.Y., Chu W., Fongarland P. // Chem. Rev. 2007. V. 107. P. 1692.
Dry M.E. // Catal. Today. 2002. V. 71. P. 227.
Klerk A. Fischer-Tropsch refining. John Wiley & Sons, 2012. 620 p.
Tso W.W., Niziolek A.M., Onel O., Demirhan C.D., Floudas C.A., Pistikopoulos E.N. // Comp. Chem. Eng. 2018. V. 113. P. 222.
Klerk A., Li Y.W., Zennaro R. / Greener Fischer–Tropsch Processes for Fuels and Feedstocks. 2013. P. 53.
Sartipi S., Alberts M., Santos V.P., Nasalevich M., Gascon J., Kapteijn F. // ChemCatChem. 2014. V. 6. P. 142.
Adeleke A.A., Liu X., Lu X., Moyo M., Hildebrandt D. // Rev. Chem. Eng. 2020. V. 36. № 4. P. 437.
Sartipi S., Parashar K., Valero-Romero M., Santos V., Linden B., Makkee M., Kapteijn F., Gascon J. // J. Catal. 2013. V. 305. P. 179.
Sartipi S., Makkee M., Kapteijn F., Gascon J. // Catal. Sci.Technol. 2014. V. 4. P. 893.
Subramanian V., Zholobenko V.L., Cheng K., Lancelot C., Heyte S., Thuriot J., Paul S., Ordomsky V.V., Khodakov A.Y. // ChemCatChem. 2016. V. 8. № 2. P. 380.
Xing C., Li M., Zhang G., Noreen A., Fu Y., Yao M., Lu C., Gao X., Yang R., Cederick C.A. // Fuel. 2021. V. 285. P. 119233.
Min Ji-E., Kim S., Kwak G., Kim Y.T., Han S.J., Lee Y., Jun K.-W., Kim S.K. // Catal. Sci. Technol. 2018. V. 8. № 24. P. 6346.
Cheng K., Kang J., Huang S., You Z., Zhang Q., Ding J., Hua W., Lou Y., Deng W., Wang Y. // ACS Catalysis. 2012. V. 2. I. 3. P. 441.
Adipah S., Takase M. // Кинетика и катализ. 2020. Т. 61. №. 3. С. 382. (Adipah S., Takase M. // Kinet. Catal. 2020. V. 61. №. 3. P. 405.)
Мурзин Д.Ю. // Кинетика и катализ. 2020. Т. 61. №. 1. С. 85. (Murzin D.Y. // Kinet. Catal. 2020. V. 61. № 1. P. 80.)
Li H., Hou B., Wang J., Qin C., Zhong M., Huang X., Jia L., Li D. // Mol. Catal. 2018. V. 459. P. 106.
Sadek R., Chalupka K.A., Mierczynski P., Rynkowski J., Millot Y., Laetitia V., Casale S., Dzwigaj S. // Catal. Today. 2020. V. 354. P. 109.
Fernandez S., Ostraat M.L., Zhang K. // AIChE J. 2020. V. 66. № 9. P. 16943.
Liu Q., Fan W. // Chem. J. Chin Univ. 2021. V. 42. №. 1. P. 60.
Jin Y., Zhang L., Liu J., Zhang S., Sun S., Asaoka S., Fujimoto K. // Micropor. Mesopor. Mater. 2017. V. 248. P. 7.
Tarach K., Góra-Marek K., Tekla J., Brylewska K., Datka J., Mlekodaj K., Makowski W., Igualada López M.C., Martínez Triguero J., Rey F. // J. Catal. 2014. V. 312. P. 46.
Verboekend D., Pérez-Ramirez J. // Catal. Sci. Technol. 2011. V. 1. P. 879.
Sun H., Wang A., Sun K., Jiang J., Wang F., Gu Z. // J. Porous Mater. 2019. V. 26. P. 961.
Zhang K., Fernandez S., Converse E.S., Kobaslija S. // Catal. Sci. Technol. 2020. V. 10. № 14. P. 4602.
Tomasek S., Lonyi F., Valyon J., Wollmann A., Hancsók J. // ACS Omega. 2020. V. 5. № 41. P. 26413.
Chalupka K.A., Sadek R., Szkudlarek L., Mierczynski P., Maniukiewicz W., Rynkowski J., Gurgul J., Casale S., Brouri D., Dzwigaj S. // Res. Chem. Intermed. 2021. V. 47. P. 397
Verboekend D., Vilé G., Pérez-Ramírez J. // Crystal Growth and Design. 2012. № 12. P. 3123.
Guo X., Guo L., Zeng Y., Kosol R., Gao X., Yoneyama Y., Yang G., Tsubaki N. // Catal. Today. 2021. V. 368. P. 196.
Wang F., Zheng Y., Huang Y., Yang X., Xu G., Kang J., Liu C., Zheng Z. // J. Anal. Appl. Pyrolysis. 2017. V. 126. P. 180.
Савостьянов А.П., Яковенко Р.Е., Нарочный Г.Б., Бакун В.Г., Сулима С.И., Якуба Э.С., Митченко С.А. // Кинетика и катализ. 2017. Т. 58. №. 1. С. 1. (Savost’yanov A.P., Yakovenko R.E., Narochnyi G.B., Bakun V.G., Sulima S.I., Yakuba E.S., Mitchenko S.A. // Kinet. Catal. 2017. V. 58. № 1. P. 81.)
Сулима С.И., Бакун В.Г., Яковенко Р.Е., Шабельская Н.П., Салиев А.Н., Нарочный Г.Б., Савостьянов А.П. // Кинетика и катализ. 2018. Т. 59. № 2. С. 240. (Sulima S.I., Bakun V.G., Yakovenko R.E., Shabel’skaya N.P., Saliev A.N., Narochnyi G.B., Savost’yanov A.P. // Kinet. Catal. 2018. V. 59. № 2. P. 218.)
Нарочный Г.Б., Яковенко Р.Е., Савостьянов А.П., Бакун В.Г. // Катализ в промышленности. 2016. Т. 16. №. 1. С. 37. (Narochnyi G.B., Yakovenko R.E., Savost’yanov A.P., Bakun V.G. // Catal. Indust. 2016. V. 8. P. 139.)
Yakovenko R.E., Savost’yanov A.P., Narochniy G.B., Soromotin V.N., Zubkov I.N., Papeta O.P., Svetogorov R.D., Mitchenko S.A. // Catal. Sci. Technol. 2020. V. 10. P. 7613.
Савостьянов А.П., Нарочный Г.Б., Яковенко Р.Е., Салиев А.Н., Сулима С.И., Зубков И.Н., Некроенко С.В., Митченко С.А. // Нефтехимия. 2017. Т. 57. № 6. Р. 809. Savost’yanov A.P., Narochnyi G.B., Yakovenko R.E., Saliev A.N., Sulima S.I., Zubkov I.N., Nekroenko S.V., Mitchenko S.A. // Petrol. Chem. 2017. V. 57. № 12. Р. 1186.)
PDF-2. The powder diffraction file TM. International Center for Diffraction Data (ICDD). PDF-2 Release 2012. web site: www.icdd.com. 2014.
Young R.A. The Rietveld Method. Oxford University Press, 1995. 298 p.
Schanke D., Vada S., Blekkan E.A., Hilmen A.M., Hoff A., Holmen A. // J. Catal. 1995. V. 156. № 1. P. 85.
Xu D., Li W., Duan H., Ge Q., Xu H. // Catal. Lett. 2005. V. 102. № 3–4. P. 229.
Baerlocher C., McCusker L.B., Olson D.H. Atlas of zeolite framework types. Elsevier, 2007. 308 p.
Caullet P., Hazm J., Guth J.L., Joly J.F., Lynch J., Raatz F. // Zeolites. 1992. V. 12. № 3. P. 240.
Peng G., Nana Y., Lei W., Xiaodong Z. // Crystal Growth and Design. 2017. V. 17. № 12. P. 6821.
Akhmedov V.M., Al-Khowaiter S.H. // Catal. Rev. 2007. V. 49. P. 33.
Database of Zeolite Structures URL: http://www.iza-structure.org/databases/ (дата обращения 14.09.2021).
Newsam J., Treacy M., Koetsier W., Gruyter C.B. // Proceedings of The Royal Society A: Mathematical, Physical and Engineering Sciences. 1988. V. 420. P. 375.
Camblor M., Mifsud A., Pérez-Pariente J. // Zeolites. 1991. V. 11. P. 792.
Xie Z., Qingling C., Chengfang Z., Jiaqing B., Yuhua C. // J. Phys. Chem B. 2000. V. 104. № 13. P. 2853.
de Oliveira A.A., da Silva V.L., de Aguiar P.D., Almeida D.F., Silva D.S.A., Ferreira J.M.M., Santos R.C., Urquieta-Gonzalez E.A., Pontes L.A.M. // React. Kinet. Mechanisms Catal. 2021. V. 132. P. 401.
Groen J.C., Peffer L.A.A., Moulijn J.A., Pérez-Ramirez J. // Micropor. Mesopor. Mater. 2004. V. 69. № 1–2. P. 29.
Mathieu R., Vieillard P. // Micropor. Mesopor. Mater. 2010. V. 132. № 3. P. 335.
Sakthivel A., Iida A., Komura K., Sugi Y. // J. Nanosci. Nanotechnol. 2009. V. 9. P. 475.
Bulavchenko O.A., Cherepanova S.V., Tsybulya S.V. // Eleventh European Powder Diffraction Conference. 2015. V. 9. P. 329.
Lonyi F., Valyon J. // Micropor. Mesopor. Mater. 2001. V. 47. P. 293.
Дополнительные материалы отсутствуют.
Инструменты
Кинетика и катализ