

Кинетика и катализ, 2022, T. 63, № 5, стр. 614-627
Роль процессов формирования–дезактивации катализатора и свидетельства нелинейного механизма в реакции Мицороки–Хека с арилхлоридами
А. А. Курохтина a, Е. В. Ларина a, Н. А. Лагода a, А. Ф. Шмидт a, *
a ФГБОУ ВО Иркутский государственный университет, химический факультет
664003 Иркутск, ул. К. Маркса, 1, Россия
* E-mail: aschmidt@chem.isu.ru
Поступила в редакцию 14.02.2022
После доработки 17.04.2022
Принята к публикации 25.04.2022
- EDN: ERNYZU
- DOI: 10.31857/S0453881122050070
Аннотация
В работе представлены результаты операндо кинетического исследования реакции Мицороки–Хека с малореакционноспособными арилхлоридами в условиях применения наиболее привлекательных с практической точки зрения “безлигандных” каталитических систем, включающего анализ скорости накопления и концентраций продуктов реакции одновременно с контролем количества ацидокомплекса палладия [PdBr4]2–, формирующегося в каталитической системе. Полученные данные указывают на определяющее влияние процессов превращения катализатора за пределами каталитического цикла на протекание основной каталитической реакции. Динамика изменений концентрации спектроскопически наблюдаемого комплекса [PdBr4]2– не согласуется с общепринятой линейной схемой механизма каталитического цикла реакции, предполагающей последовательную активацию сочетающихся арилгалогенида и алкена в одном каталитическом цикле. В то же время симулирование кинетических зависимостей с использованием моделей, базирующихся на гипотезе так называемого кооперативного механизма катализа, подразумевающего активацию двух сочетающихся субстратов каталитически активными соединениями палладия в двух сопряженных каталитических циклах, приводит к результатам, качественно соответствующим наблюдаемым экспериментальным данным. Представленные данные указывают на необходимость учета реализации кооперативного катализа реакции Мицороки–Хека при анализе результатов экспериментальных исследований и формулировке гипотез механизма.
ВВЕДЕНИЕ
Реакция Мицороки–Хека, представляющая собой арилирование алкенов арилгалогенидами под действием катализаторов на основе палладия (схема 1 ), является одним из наиболее интенсивно изучаемых в последние десятилетия каталитических процессов. Высокий интерес исследовательского сообщества обусловлен уникальными синтетическими возможностями этой реакции, которые в настоящее время уже реализованы в производстве многих продуктов современного тонкого органического синтеза, в первую очередь, фармацевтических и биологически активных препаратов [1–4]. Разработано огромное количество экспериментальных протоколов для получения продуктов реакции с использованием в качестве субстратов арилбромидов и арилиодидов, гомогенных или гетерогенных палладиевых катализаторов [1, 2] с проведением процессов как в традиционных реакторах периодического действия, так и в проточных реакторах [5–7]. Однако задача эффективного вовлечения в реакцию Мицороки–Хека наименее реакционноспособных, но при этом наиболее доступных в ряду арилгалогенидов арилхлоридов на сегодняшний день не имеет общего решения, и в подавляющем большинстве случаев связана с применением каталитических систем, содержащих в своем составе сложные, чувствительные к влаге и кислороду и часто токсичные органические лиганды (в первую очередь, фосфиновые, аминовые, карбеновые), либо с жесткими условиями реакции (см., например, [2], а также [8, 9]). На наш взгляд, резкое падение эффективности каталитических систем при использовании арилхлоридов в качестве субстратов реакции Мицороки–Хека вместо арилиодидов и арилбромидов может быть обусловлено не только возрастанием прочности связи C–Cl в арилхлоридах, но и с изменением фундаментальных особенностей ее механизма. Уже при переходе от арилиодидов, обладающих наиболее высокой реакционной способностью в ряду арилгалогенидов, к менее реакционноспособным арилбромидам в условиях применения наиболее привлекательных с практической точки зрения так называемых “безлигандных” каталитических систем (т.е. систем, не содержащих добавок фосфиновых, аминовых, карбеновых или любых иных сильных органических лигандов) было однозначно показано, что ключевое влияние на каталитическую активность оказывают процессы превращения катализатора за пределами основного каталитического цикла [10–12]. Снижение реакционной способности арилбромидов по сравнению с арилиодидами в процессе эндогенной регенерации каталитически активных молекулярных комплексов палладия путем растворения неактивных гетерогенных форм является основной причиной их меньшей реакционной способности в реакции Мицороки–Хека.

Схема 1 . Общепринятая схема механизма каталитического цикла образования продуктов реакции Мицороки–Хека и сопряженных с ним процессов превращения палладия (лиганды при палладии опущены).
Использование арилхлоридов вместо арилиодидов и арилбромидов в реакции Мицороки–Хека сопровождается еще более выраженным падением реакционной способности и снижением выходов целевых продуктов. В течение долгого времени причиной этого считалось замедление скоростьопределяющей стадии активации арилхлоридов, которой, согласно общепринятым представлениям, является их окислительное присоединение к комплексам Pd(0) (схема 1 , стадия А) вследствие значительного увеличения прочности связи C–Cl в сравнении со связями C–I и C–Br (см., например, [13–15]). Тем не менее, эта гипотеза не имела прямых экспериментальных подтверждений. С другой стороны, существуют данные квантово-химического моделирования, указывающие на невысокие значения активационных барьеров окислительного присоединения арилхлоридов к молекулярным комплексам и кластерам палладия [16]. Кроме того, на основании результатов сравнительного исследования закономерностей превращения палладия в реакциях кросс-сочетания при использовании арилиодидов, арилбромидов и арилхлоридов авторами [17] было выдвинуто обоснованное предположение о том, что затруднения в окислительном присоединении арилхлоридов не могут являться единственной причиной их низкой реакционной способности, и ключевое влияние на эффективность катализа оказывает стабильность молекулярных комплексов палладия, образующихся в результате окислительного присоединения. В качестве убедительного свидетельства того, что окислительное присоединение арилхлоридов к Pd(0) не определяет скорость каталитической реакции, можно рассматривать данные о существенной обратимости (вплоть до квазиравновесного режима протекания) стадии активации арилхлоридов в каталитических условиях реакций кросс-сочетания [18]. Таким образом, совокупность результатов, получаемых в модельных [16, 17] и реальных [18] каталитических условиях, указывает на то, что проблемы вовлечения арилхлоридов в реакции кросс-сочетания и, в частности, реакцию Мицороки–Хека связаны не только с падением эффективности функционирования основного каталитического цикла процесса, но могут быть в большой степени обусловлены влиянием процессов превращения катализатора за его пределами.
В настоящей работе представлены данные совместного кинетического исследования закономерностей функционирования “безлигандных” каталитических систем и превращений катализатора в реакции Мицороки–Хека с арилхлоридами, направленного на установление причин низкой реакционной способности последних. Учитывая сложный характер взаимного влияния процессов, протекающих внутри основного каталитического цикла образования продуктов реакции, и процессов превращения катализатора за его пределами, все представленные экспериментальные данные получены в условиях реального каталитического процесса с высокими соотношениями субстрат/катализатор.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Пробы реакционной смеси анализировали на газожидкостном хроматографе Кристалл 5000.2 (“Хроматэк”, Россия, ДИП, колонка HP-5 15 м) в режиме деления потока (1/10) с программированным нагревом от 110 до 250°С. Количественный состав проб вычисляли методом внутреннего стандарта (внутренний стандарт − нафталин) с применением калибровки по аутентичным образцам. Материальный баланс, оцениваемый для каждой из проб реакционных смесей с использованием концентраций реагирующих арилхлоридов и алкена и суммарной концентрации образующихся продуктов, отклонялся от теоретического значения не более чем на 5%.
УФ-спектры исследуемых растворов регистрировали на приборе СФ-2000 (ОКБ “Спектр”, Россия) в области 190–600 нм с применением кварцевых кювет с толщиной поглощающего слоя 0.01 см. Измерение концентрации комплекса [PdBr4]2– проводили, используя интенсивность поглощения при 350 нм. Калибровочный график для этой длины волны был предварительно построен на основе спектров стандартных растворов [PdBr4]2–, полученных посредством последовательного разбавления раствора, содержащего 0.08 ммоля PdBr2 и 6.4 ммоля NBu4Br в 10 мл ДМФА. При этом путем предварительного построения калибровочных графиков учитывали поглощение при 350 нм от продуктов сочетания н-бутилакрилата (БА) с хлорбензолом (ХБ) или 1,4-дихлорбензолом (ДХБ), количества которых в ходе реакции определяли с помощью ГЖХ.
Скорости расходования субстратов и накопления продуктов реакции оценивали по изменению концентрации вещества за промежуток времени между двумя последовательно отбираемыми пробами реакционной смеси. Математическую обработку кинетических данных и построение аппроксимирующих зависимостей (фазовых траекторий) проводили с помощью средств программы “Microsoft Excel 2007” [19]. Построение зависимостей в трехмерном пространстве координат концентраций веществ-участников реакции выполняли с помощью ПО “Origin” [20]. Симулирование экспериментально полученных кинетических зависимостей выполняли с использованием ПО COPASI [21].
Каталитические эксперименты
Неконкурентные эксперименты проводили, смешивая при комнатной температуре в смеси N,N-диметилформамид (ДМФА)−NBu4Br (2.5 мл/2.58 г, 0.08 моля) арилхлорид (5–20 ммоль) и нафталин (0.5 ммоля) в качестве внутреннего стандарта для хроматографии. Полученный раствор помещали в круглодонный стеклянный реактор, снабженный резиновой мембраной и магнитной мешалкой, содержащий БА (0.625–2.5 ммоля), PdCl2 в качестве предшественника катализатора (0.01 ммоля) и NaOAc (3.25 ммоля) в качестве основания. Реакцию начинали, помещая реактор в предварительно нагретую до 140°С масляную баню при перемешивании (480 об/мин) в течение 30–100 мин. Пробы реакционной смеси для хроматографического анализа (100 мкл) периодически отбирали из реактора с помощью шприца с металлической иглой, после чего разбавляли и экстрагировали толуолом (100 мкл смеси толуол : : вода = 1 : 1). Каждый эксперимент выполняли 3 раза для проверки воспроизводимости.
Конкурентные эксперименты проводили, смешивая при комнатной температуре в смеси N-метилпирролидон (NMP)−NBu4Br (2.5 мл/2.58 г, 0.08 моля) конкурирующие ДХБ и ХБ (по 5 ммолей каждого) и нафталин (0.5 ммоля) в качестве внутреннего стандарта для хроматографии. Полученный раствор помещали в круглодонный стеклянный реактор, снабженный резиновой мембраной и магнитной мешалкой, содержащий БА (0.625 ммоля), PdCl2 в качестве предшественника катализатора (0.01 ммоля) и NaOAc (3.25 ммоля) в качестве основания. Реакцию начинали, помещая реактор в предварительно нагретую до 140°С масляную баню при перемешивании (480 об/мин) в течение 30–100 мин. Пробы реакционной смеси для хроматографического анализа (100 мкл) периодически отбирали из реактора с помощью шприца с металлической иглой, после чего разбавляли и экстрагировали толуолом (100 мкл смеси толуол : вода = 1 : 1). Каждый эксперимент выполняли 3 раза для проверки воспроизводимости.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Ранее было неоднократно показано, что количество арилгалогенида и его отношение к концентрации загружаемого в систему палладия являются одними из основных факторов, определяющих каталитическую активность в реакции Мицороки–Хека, особенно с относительно малореакционноспособными субстратами [10, 12]. Это объяснялось ключевой ролью арилгалогенида в процессах превращения катализатора за пределами основного каталитического цикла реакции (схема 1 ): его участия в процессе растворения металлического палладия (схема 1 , Г), образующегося в результате дезактивации каталитически активных растворенных молекулярных комплексов Pd(0) путем агломерации (схема 1 , Е), а также предотвращения последней за счет успешной конкуренции с ней стадии окислительного присоединения арилгалогенида [10, 12] (схема 1 , А).
Нами были установлены величины частных порядков скорости образования продуктов реакции Мицороки–Хека, под которой здесь и далее мы будем понимать суммарную скорость накопления α- и β-региоизомерных продуктов сочетания арилгалогенида и алкена (схемы 1 и 2 ), по концентрации арилхлорида для предварительной оценки его влияния на превращения катализатора внутри и за пределами каталитического цикла. При этом, учитывая известную высокую чувствительность скорости реакции Мицороки–Хека к природе арильного заместителя в арилгалогениде (см., например, [22–24]), нами были использованы два различающихся по реакционной способности арилхлорида – активированный 4-хлорацетофенон (ХАФ) и неактивированный ХБ. Величина частного порядка скорости реакции с ХАФ по его концентрации в условиях так называемого “неконкурентного” сочетания с БА (схема 2 ) оказывалась больше единицы (рис. 1а), что не может реализовываться в случае традиционного линейного механизма каталитического цикла реакции (порядок должен находиться в диапазоне от 0 до 1). Величина кинетического порядка реакции по концентрации арилхлорида больше единицы может быть результатом его участия в реакции не только как реагента в стадии основного каталитического цикла, т.е. окислительном присоединении к Pd(0) (схема 1 , А), но и в процессе образования каталитически активных растворенных молекулярных комплексов из коллоидного металлического палладия (схема 1 , Г), способном увеличивать количество активного катализатора с ростом концентрации арилхлорида. Значительное влияние процесса растворения металлического палладия на концентрацию каталитически активных соединений и, следовательно, скорость каталитической реакции многократно продемонстрировано в литературе (см., например, [9, 10, 12, 17]). При использовании неактивированного ХБ скорость реакции значительно слабее зависела от его концентрации, и значение частного порядка было близко к единице (рис. 1б). Тем не менее, это также не противоречило участию арилхлорида в ключевых определяющих активность стадиях, которые могут находиться в том числе и за пределами основного каталитического цикла.
Рис. 1.
Логарифмическая зависимость суммарной скорости накопления региоизомерных α- и β-продуктов (lnr) реакции Мицороки–Хека (схема 2 ) от концентрации арилхлорида (lnC) в реакции между: а – БА и ХАФ ([БА] = 1 M); б – БА и ХБ ([БА] = 0.25 M).
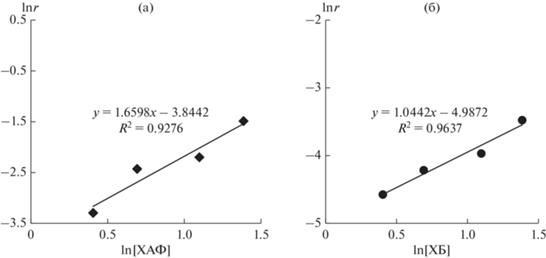

Схема 2 . Реакция Мицороки–Хека с арилхлоридами при использовании “безлигандной” каталитической системы на основе палладия.
На значительное влияние процессов за пределами каталитического цикла, определяющих скорости формирования и дезактивации активного катализатора указывал и характер интегральных кинетических зависимостей реакции. На кривых накопления продуктов реакции Мицороки–Хека всегда наблюдался период автоускорения, продолжительность которого практически не зависела от концентрации арилхлорида (от 1.5 до 3 мин). По окончании периода автоускорения максимальная скорость реакции была достаточно высокой (до 30 М/мин), но при этом период активной работы катализатора оказывался крайне непродолжительным, не превышая 5–10 мин, после чего реакция быстро тормозилась, не достигая полной конверсии реагирующих веществ в продукты (рис. 2).
Рис. 2.
Примеры начальных участков зависимостей суммарного выхода региоизомерных α- и β-продуктов от времени в реакции Мицороки–Хека (схема 2 ) между: а – БА и ХАФ ([БА] = 1 M); б – БА и ХБ ([БА] = 0.25 M) при различных начальных концентрациях арилхлоридов: 1.5 (1), 2 (2), 3 (3) и 4 M (4).

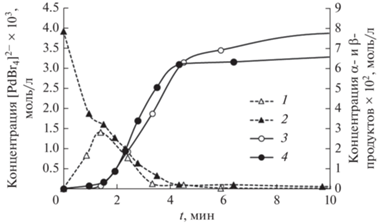
Базируясь на значительном экспериментальном материале, накопленном для реакции Мицороки–Хека с арилбромидами и арилиодидами, можно выделить два основных возможных маршрута дезактивации каталитически активных палладиевых соединений – образование гетерогенных (наноразмерных и более грубодисперсных) частиц металлического палладия (схема 1 , Е) и ацидокомплексов типа [PdX4]2– (где Х – галоген) (схема 1 , Д) [12, 25, 26]. При проведении реакции с арилхлоридами в качестве реакционной среды нами были использованы смеси ДМФА–NBu4Br или NMP–NBu4Br, благодаря чему в растворе всегда присутствовал значительный избыток бромид-ионов по отношению к палладию (800 экв.). Образования палладиевой черни в используемых нами условиях реакции не наблюдалось, но при этом вполне ожидаемым явилось детектирование в ходе реакции образующихся in situ ацидокомплексов [PdBr4]2–. Возможность спектроскопического детектирования в условиях каталитического процесса формы катализатора позволяет реализовать операндо кинетическое исследование, подразумевающее одновременную фиксацию изменений количеств такой формы катализатора и каталитической активности и/или селективности реакционной системы по продуктам каталитической реакции [27]. Типичные зависимости концентрации комплекса [PdBr4]2– от времени в ходе каталитической реакции приведены на рис. 3. В случае применения в качестве предшественника катализатора PdCl2 на начальном этапе реакции наблюдался рост концентрации комплекса [PdBr4]2–, которая после достижения максимума вновь падала до нуля. Такие временные изменения с определяемой нами точностью совпадали с периодом максимальной каталитической активности системы (рис. 3). Кроме того, на фиксируемых временных зависимостях с используемой нами точностью продолжительность периода автоускорения каталитической реакции совпадала по времени с моментом достижения максимальной концентрации [PdBr4]2– (рис. 3), что формально могло рассматриваться как свидетельство участия этого соединения либо в качестве каталитически активной частицы, либо в качестве соединения, непосредственно участвующего в образовании истинного катализатора. Однако при применении как предшественника катализатора заранее сформированного комплекса Na2PdBr4 в той же концентрации, что и ранее PdCl2 (4 × 10–3 М), концентрация [PdBr4]2– в ходе реакции уменьшалась практически до нуля одновременно с резким падением каталитической активности, при этом период автоускорения реакции также фиксировался. Зависимость концентрации палладиевого комплекса от времени в период активности каталитической системы практически совпадала с зафиксированной в эксперименте с PdCl2 как предшественником катализатора (рис. 3). С учетом высокой вероятности различного взаимного влияния процессов превращения катализатора внутри и за пределами каталитического цикла реакции Мицороки–Хека с арилбромидами, для которых [PdBr4]2– является неактивной формой катализатора, установление роли такого комплекса в реакции с арилхлоридами требовало дополнительных исследований.
Рис. 3.
Зависимости концентрации [PdBr4]2- (пунктирные линии) и суммарной концентрации региоизомерных α- и β-продуктов (сплошные линии) от времени в реакции между ХБ и БА (схема 2 ) при использовании PdCl2 (1, 3) и Na2PdBr4 (2, 4) в качестве предшественников катализатора ([ХБ] = 4 М, [БА] = 0.25 М).
В условиях конкуренции пары арилхлоридов (схема 3 ) закономерности изменения каталитической активности и концентрации [PdBr4]2– в реакции в целом демонстрировали аналогичный наблюдаемому в “неконкурентных” условиях характер (рис. 4). Рост концентрации палладиевого комплекса совпадал с периодом автоускорения, и после достижения наибольшей концентрации в момент, совпадающий с началом периода максимальной каталитической активности, которая в условиях конкуренции двух арилхлоридов оценивалась по расходованию общего для них реагента БА, концентрация комплекса далее падала до нуля. Интересно, что в данных экспериментах на концентрационных зависимостях [PdBr4]2– удалось зафиксировать этап увеличения скорости его образования, что указывало на то, что [PdBr4]2–, скорее всего, появляется не непосредственно из исходного PdCl2 путем обмена анионов галогена, а через некие промежуточные формы катализатора. Исчезновение [PdBr4]2– в конце реакции в большинстве случаев совпадало с моментом ее прекращения при полном расходовании алкена, находящегося в недостатке (рис. 4, кривые 1 и 4 с 800 экв. NBu4Br в расчете на палладий). Тем не менее, в отдельных экспериментах, характеризующихся низкими величинами каталитической активности, расходование алкена на образование продуктов из конкурирующих арилхлоридов наблюдалось в течение продолжительного времени после исчезновения [PdBr4]2– (в качестве примера на рис. 4 приведены зависимости для экспериментов с уменьшенным до 300 экв. в расчете на палладий количеством NBu4Br, кривые 2 и 5). Кроме того, при замене добавки NBu4Br на LiBr практически весь загружаемый в систему палладий переходил в форму [PdBr4]2–, и при этом реакция практически не протекала (рис. 4, кривые 3 и 6). Резкое падение активности в условиях применения LiBr при устойчиво высокой концентрации [PdBr4]2– противоречило гипотезе о возможной каталитической активности [PdBr4]2–.
Рис. 4.
Зависимости концентрации БА (сплошные линии) и концентрации [PdBr4]2– (пунктирные линии) от времени в конкурентной реакции с ХБ и ДХБ (схема 3 ) при использовании в качестве добавки (в расчете на загружаемый в систему палладий): 800 экв. NBu4Br (1, 4); 300 экв. NBu4Br (2, 5); 800 экв. LiBr (3, 6).
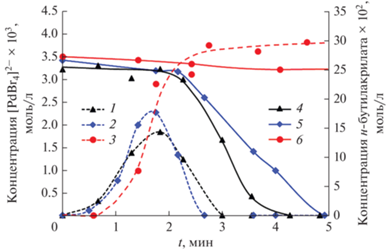
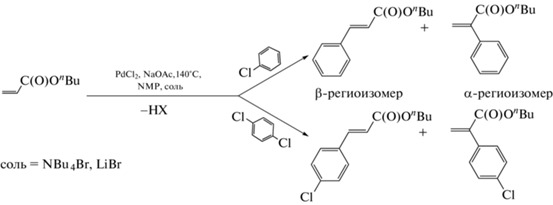
Схема 3 . Конкурентная реакция Мицороки–Хека с арилхлоридами при использовании “безлигандной” каталитической системы на основе палладия.
Для получения дополнительной информации о роли [PdBr4]2– в реакционной системе реакции Мицороки–Хека с арилхлоридами нами был применен метод фазовых траекторий конкурентных реакции [28]. Вид фазовых траекторий, представляющих собой зависимости неких изменяющихся во времени параметров реакционной системы друг от друга, по смыслу аналогичен “фазовому портрету” динамической системы, использующемуся в качественной теории дифференциальных уравнений [29] для анализа поведения таких систем. Ранее нами было показано, что при наличии экспериментальных данных о концентрации наблюдаемой формы катализатора в различные моменты времени дискриминация гипотез о роли этой формы в катализе может проводиться с помощью как традиционных зависимостей типа [количество формы катализатора]–[скорость накопления продукта], так и построенных с использованием интегральных кинетических данных фазовых траекторий реакции в координатах [количество формы катализатора]–[количество продукта реакции] [27]. При проведении конкурентной реакции с парой близких по свойствам конкурирующих субстратов (в нашем случае, пары арилхлоридов) фазовые траектории могут быть построены в координатах суммарных концентраций продуктов, образующихся из каждого из конкурирующих субстратов в различные моменты времени реакции, а также и концентраций наблюдаемых компонентов каталитической системы. В таком случае наклон касательной к любой точке фазовой траектории представляет собой отношение скоростей накопления/расходования веществ, которое количественно характеризует величину дифференциальной селективности реакции по соответствующим веществам [28]. Дифференциальная селективность по продуктам реакции, в отличие от каталитической активности, не зависит от количества активного катализатора и определяется только его природой, поэтому сравнение фазовых траекторий, построенных в координатах концентраций продуктов конкурирующих реакций, при варьировании условий проведения процесса может быть применено для выяснения вопроса о сохранении или изменении природы каталитически активных соединений.
Используя данные о суммарных концентрациях продуктов реакции Мицороки–Хека, образующихся из конкурирующих ХБ и ДХБ, а также концентрации комплекса [PdBr4]2–, фазовые траектории могут быть построены в трехмерном пространстве соответствующих координат (рис. 5). Проекции фазовых траекторий на координатные плоскости образуют на фазовых поверхностях соответствующих пар компонентов реакции фазовые траектории, получающиеся при варьировании параметров проведения реакции. Так, проекции на плоскостях типа [концентрация [PdBr4]2–]–[суммарный выход продуктов одной из конкурентных реакций] наглядно демонстрировали, что накопление продуктов превращения каждого из конкурирующих арилхлоридов продолжалось и после исчезновения [PdBr4]2– (рис. 5). При этом полное исчезновение [PdBr4]2– в ходе реакции после прохождения его концентрации через максимум никак не сказывалось на наклоне фазовой траектории, построенной в координатной плоскости концентраций продуктов конкурентных реакций, характеризующем дифференциальную селективность активного катализатора. Примерно постоянная величина дифференциальной селективности, в свою очередь, указывает на сохранение природы каталитически активных соединений на протяжении всей каталитической реакции, в том числе, и после исчезновения [PdBr4]2–. На наш взгляд, такой результат однозначно указывает на то, что образование продуктов реакции происходит с участием отличных от [PdBr4]2– соединений.
Рис. 5.
Фазовые траектории в пространстве координат: [концентрация [PdBr4]2– ]–[суммарные концентрации региоизомерных α- и β-продуктов, образующихся из конкурирующих ХБ и ДХБ в конкурентной реакции Мицороки–Хека] (схема 4 ) при использовании в качестве добавки (в расчете на загружаемый в систему палладий): 800 экв. NBu4Br (▲); 300 экв. NBu4Br (◼); 100 экв. NBu4Br (⚫). Проекции фазовых траекторий на различные плоскости: фазовые траектории в координатах [концентрация [PdBr4]2– ]–[суммарная концентрация региоизомерных α- и β-продуктов из ДХБ] (черные значки); фазовые траектории в координатах концентраций региоизомерных α- и β-продуктов, образующихся из конкурирующих ДХБ и ХБ (зеленые значки); фазовые траектории в координатах [концентрация [PdBr4]2– ]–[суммарная концентрация региоизомерных α- и β-продуктов из ХБ] (сиреневые значки).

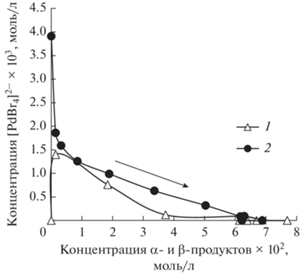
Вопрос о каталитической активности той или иной наблюдаемой формы катализатора может решаться и с помощью зависимостей другого типа. В случае каталитической активности [PdBr4]2– в образовании продуктов реакции Мицороки–Хека на закономерности изменения его концентрации с изменением скорости накопления продукта каталитической реакции должны наблюдаться достаточно протяженные участки возрастающей функции, соответствующие положительной зависимости концентрации каталитически активной специи от скорости каталитической реакции, и непродолжительные участки с убывающей функцией (отрицательная зависимость) [27]. Полученные нами закономерности изменения концентрации [PdBr4]2– и скорости накопления продукта реакции Мицороки–Хека в “неконкурентных” условиях (схема 2 ) показывали примерно одинаковые по протяженности положительные и отрицательные участки (рис. 6). Изменение концентрации [PdBr4]2– с ростом концентрации продукта реакции Мицороки–Хека (рис. 7) демонстрировало сложный характер, который также не соответствовал ни одному из вариантов, теоретически рассмотренных ранее в [27], что не позволяло сделать однозначного вывода о роли [PdBr4]2– в катализе реакции Мицороки–Хека с арилхлоридами.
Рис. 6.
Зависимость концентрации [PdBr4]2– от суммарной скорости накопления региоизомерных α- и β-продуктов в неконкурентной реакции БА с ХБ (схема 2 ) при использовании PdCl2 (1) или Na2PdBr4 (2) в качестве предшественника катализатора ([ХБ] = 4 М, [БА] = 0.25 М; стрелками показано временное развитие реакции).
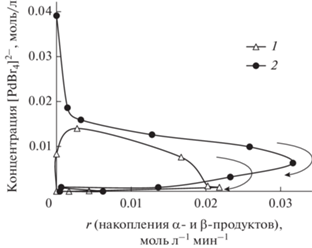
Рис. 7.
Зависимость концентрации [PdBr4]2– от суммарной концентрации региоизомерных α- и β-продуктов в неконкурентной реакции БА с ХБ (схема 2 ) при использовании PdCl2 (1) или Na2PdBr4 (2) в качестве предшественника катализатора ([ХБ] = 4 М, [БА] = 0.25 М; стрелкой показано временное развитие реакции).
Важно подчеркнуть, что приведенный выше анализ закономерностей изменения концентраций от времени (рис. 3 и 4), фазовых траекторий (рис. 5) и зависимостей концентрации [PdBr4]2– от скорости накопления или концентрации продукта каталитической реакции (рис. 6 и 7, соответственно) был проведен в рамках классического линейного механизма реакции Мицороки–Хека (схема 1 ). Однако ранее при проведении конкурентных экспериментов с использованием пары алкенов нами было показано, что на величину дифференциальной селективности реакции Мицороки–Хека по конкурирующим алкенам не влияет природа арильного заместителя и противоиона в арилгалогениде [30], что противоречило общепринятым представлениям о протекании катализа образования продуктов реакции в результате последовательной активации арилгалогенида и алкена внутри каталитического цикла, начинающегося с активного соединения Pd(0) (схема 1 ). Эти экспериментальные факты согласовывались с реализацией так называемого кооперативного механизма катализа [31, 32], предполагающего активацию сочетающихся арилгалогенида и алкена двумя различными активными палладиевыми соединениями, который является нелинейным с точки зрения химической кинетики. Учитывая эти результаты, нами было проведено симулирование экспериментально полученных зависимостей концентрации [PdBr4]2– от скорости накопления или концентрации продукта каталитической реакции в рамках гипотезы кооперативного механизма катализа с помощью ПО COPASI [21]. В использованной для симулирования простейшей модели кооперативного катализа (схема 4 а) и соответствующем ей наборе стехиометрических уравнений стадий (I) предполагалось, что один из сочетающихся субстратов (алкен) активируется неким соединением палладия (Pdact2), отличным от соединения, активирующего другой субстрат (Pdact1), реагирующий с арилгалогенидом (схема 4 а). Принимая во внимание, что активация арилгалогенида путем его окислительного присоединения к комплексам Pd(0) с образованием интермедиата типа Ar–Pd–X в настоящее время не вызывает сомнений [14, 17, 33–35], нами также был учтен известный побочный маршрут образования биарила в результате взаимодействия двух частиц типа Ar–Pd–X с формированием в качестве продуктов также Pd(0) (Pdact1) и Pd(II) (Pdact2) (схема 4 а) [12]. Симулированная на основании предложенной модели зависимость концентрации Pdact2 от скорости накопления продукта реакции (рис. 8) качественно была близка к полученной нами экспериментально (рис. 6), воспроизводя примерно одинаковые по протяженности положительные и отрицательные участки зависимости, что, как было отмечено выше, являлось нехарактерным для классического линейного механизма.
Рис. 8.
Зависимость концентрации катализатора Pdact2, активирующего один из сочетающихся субстратов, от скорости накопления продукта реакции Р1 при кооперативном механизме катализа (схема 4 а, набор стехиометрических уравнений стадий и начальных условий (I); стрелкой показано временное развитие реакции).
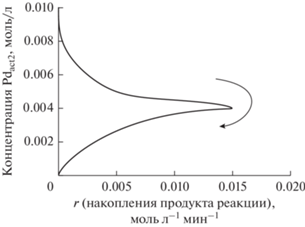
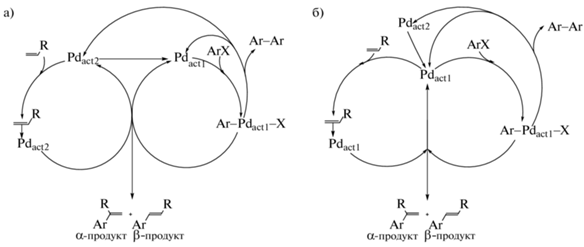
Схема 4 . Механизм образования продукта реакции Мицороки–Хека в предположении активации сочетающихся арилгалогенида и алкена в раздельных каталитических циклах двумя различными типами активных частиц (а) и одним типом активных частиц (б).
(I)
$\begin{gathered} {\text{P}}{{{\text{d}}}_{{{\text{act2}}}}}\xrightarrow{{k = \,\,1\,\,{\text{ми}}{{{\text{н}}}^{{ - 1}}}}}{\text{P}}{{{\text{d}}}_{{{\text{act1}}}}} \\ {\text{P}}{{{\text{d}}}_{{{\text{act1}}}}} + \,\,{\text{ArX}}\underset{{k = \,10\,\,{\text{ми}}{{{\text{н}}}^{{ - 1}}}}}{\overset{{k = \,10\,\,{\text{M/мин}}}}{\longleftrightarrow}}{\text{Ar}}{\kern 1pt} - {\kern 1pt} {\text{P}}{{{\text{d}}}_{{{\text{act1}}}}}{\kern 1pt} - {\kern 1pt} {\text{X}} \\ {\text{P}}{{{\text{d}}}_{{{\text{act2}}}}} + \,\,{\text{alkene}}\underset{{k = \,10\,\,{\text{ми}}{{{\text{н}}}^{{ - 1}}}}}{\overset{{k = \,10\,\,{\text{M/мин}}}}{\longleftrightarrow}}\,\,{\text{P}}{{{\text{d}}}_{{{\text{act2}}}}}{\kern 1pt} - {\kern 1pt} {\text{alkene}} \\ {\text{Ar}}{\kern 1pt} - {\kern 1pt} {\text{P}}{{{\text{d}}}_{{{\text{act1}}}}}{\kern 1pt} - {\kern 1pt} {\text{X}} + \,\,{\text{P}}{{{\text{d}}}_{{{\text{act2}}}}}{\kern 1pt} - {\kern 1pt} {\text{alkene}}\,\,\xrightarrow{{k = \,{{{10}}^{4}}\,\,{\text{M/мин}}}}\,\,(\alpha {\text{/}}\beta {\text{) - продукт + P}}{{{\text{d}}}_{{{\text{act1}}}}} + {\text{P}}{{{\text{d}}}_{{{\text{act2}}}}} \\ {\text{2Ar}}{\kern 1pt} - {\kern 1pt} {\text{P}}{{{\text{d}}}_{{{\text{act1}}}}}{\kern 1pt} - {\kern 1pt} {\text{X}}\xrightarrow{{k = \,{{{10}}^{4}}\,\,{\text{M/мин}}}}\,\,{\text{Ar}}{\kern 1pt} - {\kern 1pt} {\text{Ar + P}}{{{\text{d}}}_{{{\text{act1}}}}} + {\text{P}}{{{\text{d}}}_{{{\text{act2}}}}} \\ {{\left[ {{\text{ArX}}} \right]}_{0}} = {{\left[ {{\text{alkene}}} \right]}_{0}} = 1\,\,{\text{M,}}\,\,{{\left[ {{\text{P}}{{{\text{d}}}_{{{\text{act1}}}}}} \right]}_{0}} = 0,\,\,{{\left[ {{\text{P}}{{{\text{d}}}_{{{\text{act2}}}}}} \right]}_{0}} = 0.01\,\,{\text{M}} \\ \end{gathered} $Кооперативный механизм катализа может реализовываться и при раздельной (непоследовательной) активации двух сочетающихся субстратов, но одним и тем же типом активных частиц с образованием продукта реакции в нелинейной стадии взаимодействия двух палладийсодержащих интермедиатов (Pdact1, схема 4 б и соответствующий набор стехиометрических уравнений (II)). При этом в реакционной системе с большой вероятностью присутствует и каталитически неактивная форма палладия (Pdact2, схема 4 б), способная к взаимопревращениям с активной формой. В настоящий момент одной из вероятных гипотез относительно природы катализаторов, ответственных за активацию субстратов в рамках кооперативного механизма, может являться гипотеза активации и арилгалогенида, и алкена на одних и тех же активных соединениях Pd(0). Как было отмечено выше, точка зрения об активации арилгалогенидов в реакциях кросс-сочетания путем их окислительного присоединения к Pd(0) с образованием интермедиатов типа Ar–Pd–X на сегодняшний день является общепринятой [14, 17, 33–35]. При этом имеются литературные данные об образовании комплексов алкенов с соединениями Pd(0) в условиях реакций сочетания [36, 37], что позволяет предположить их в качестве интермедиатов одного из каталитических циклов в случае кооперативного механизма. Симулирование кинетических зависимостей для этого варианта механизма (схема 4 б) приводило к расчетным зависимостям типа [концентрация неактивной формы катализатора]–[скорость накопления продукта каталитической реакции] (рис. 9) и [концентрация неактивной формы катализатора]–[количество продукта каталитической реакции] (рис. 10), также качественно близким к экспериментальным зависимостям (рис. 6 и 7).
(II)
$\begin{gathered} {\text{P}}{{{\text{d}}}_{{{\text{act2}}}}}\xrightarrow{{k = \,\,1\,\,{\text{ми}}{{{\text{н}}}^{{ - 1}}}}}{\text{P}}{{{\text{d}}}_{{{\text{act1}}}}} \\ {\text{P}}{{{\text{d}}}_{{{\text{act1}}}}} + \,\,{\text{ArX}}\underset{{k = \,10\,\,{\text{ми}}{{{\text{н}}}^{{ - 1}}}}}{\overset{{k = \,10\,\,{\text{M/мин}}}}{\longleftrightarrow}}{\text{Ar}}{\kern 1pt} - {\kern 1pt} {\text{P}}{{{\text{d}}}_{{{\text{act1}}}}}{\kern 1pt} - {\kern 1pt} {\text{X}} \\ {\text{P}}{{{\text{d}}}_{{{\text{act1}}}}} + \,\,{\text{alkene}}\underset{{k = \,10\,\,{\text{ми}}{{{\text{н}}}^{{ - 1}}}}}{\overset{{k = \,10\,\,{\text{M/мин}}}}{\longleftrightarrow}}\,\,{\text{P}}{{{\text{d}}}_{{{\text{act1}}}}}{\kern 1pt} - {\kern 1pt} {\text{alkene}} \\ {\text{Ar}}{\kern 1pt} - {\kern 1pt} {\text{P}}{{{\text{d}}}_{{{\text{act1}}}}}{\kern 1pt} - {\kern 1pt} {\text{X}} + \,\,{\text{P}}{{{\text{d}}}_{{{\text{act1}}}}}{\kern 1pt} - {\kern 1pt} {\text{alkene}}\,\,\xrightarrow{{k = \,{{{10}}^{4}}\,\,{\text{M/мин}}}}\,\,(\alpha {\text{/}}\beta {\text{) - продукт + P}}{{{\text{d}}}_{{{\text{act1}}}}} + {\text{P}}{{{\text{d}}}_{{{\text{act2}}}}} \\ {\text{2Ar}}{\kern 1pt} - {\kern 1pt} {\text{P}}{{{\text{d}}}_{{{\text{act1}}}}}{\kern 1pt} - {\kern 1pt} {\text{X}}\xrightarrow{{k = \,{{{10}}^{4}}\,\,{\text{M/мин}}}}\,\,{\text{Ar}}{\kern 1pt} - {\kern 1pt} {\text{Ar + P}}{{{\text{d}}}_{{{\text{act1}}}}} + {\text{P}}{{{\text{d}}}_{{{\text{act2}}}}} \\ {{\left[ {{\text{ArX}}} \right]}_{0}} = {{\left[ {{\text{alkene}}} \right]}_{0}} = 1\,\,{\text{M,}}\,\,{{\left[ {{\text{P}}{{{\text{d}}}_{{{\text{act1}}}}}} \right]}_{0}} = 0,\,\,{{\left[ {{\text{P}}{{{\text{d}}}_{{{\text{act2}}}}}} \right]}_{0}} = 0.01\,\,{\text{M}} \\ \end{gathered} $Рис. 9.
Зависимость концентрации неактивной формы катализатора (Pdact2) от скорости накопления продукта реакции (P1) при кооперативном механизме катализа в случае активации сочетающих субстратов одним типом активных частиц (Pdact1, схема 4 б, набор стехиометрических уравнений стадий и начальных условий (II); стрелкой показано временное развитие реакции).
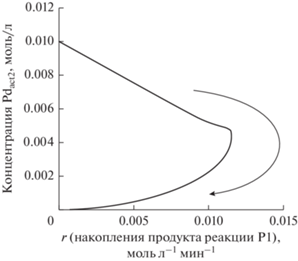
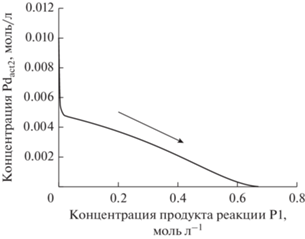
Рис. 10.
Зависимость концентрации неактивной формы катализатора (Pdact2) от концентрации продукта реакции (P1) при кооперативном механизме катализа в случае активации сочетающих субстратов одним типом активных частиц (Pdact1, схема 4 б, набор стехиометрических уравнений стадий и начальных условий (II); стрелкой показано временное развитие реакции).
Необходимо отметить, что вне зависимости от того, какой из вариантов активации алкенов и арилгалогенида, представленных на схеме 4 , реализуется, отношение скоростей накопления продуктов из двух конкурирующих алкенов будет определяться исключительно отношением констант скоростей их взаимодействия с общим катализатором, природа которого в любом из рассмотренных на схеме 4 случаев не зависит от природы арильного заместителя и противоиона арилирующего реагента. Поэтому обнаруженная нами ранее независимость дифференциальной селективности по конкурирующим алкенам от природы и концентрации арилирующего реагента [30] не противоречит обоим рассмотренным вариантам кооперативного механизма (схема 4 ). Кроме того, реализация любого из представленных на схеме 4 вариантов нелинейного механизма может приводить к тому, что величина порядка скорости реакции по концентрации палладиевого предшественника катализатора может находится в интервале от 1 до 2 в зависимости от чувствительности скорости реакции к скорости нелинейной стадии взаимодействия двух палладийсодержащих интермедиатов [32, 38], достигая значения 2 в предельном случае скоростьопределяющего характера нелинейной стадии. Действительно, экспериментально измеренные нами порядки по концентрации PdCl2, используемого в качестве предшественника, оказывались больше единицы: 1.5 для реакции сочетания н-бутилакрилата с ХАФ и 1.1 для реакции с ХБ (схема 2 ). На текущем этапе можно сделать вывод, что интерпретация полученных кинетических данных о закономерностях скоростей накопления и концентраций продуктов реакции Мицороки–Хека с арилхлоридами, а также концентрации ацидокомплексов [PdBr4]2– во времени в рамках линейного (общепринятого) механизма катализа (схема 1 ) приводит к противоречивым результатам. В то же время симулирование экспериментально обнаруженных зависимостей с применением моделей кооперативного катализа (схема 4 ) согласуется на качественном уровне с наблюдаемыми экспериментально зависимостями.
ЗАКЛЮЧЕНИЕ
Установленные кинетические закономерности накопления продуктов реакции Мицороки–Хека с малореакционноспособными арилхлоридами указывают на ключевой характер влияния процессов превращения катализатора за пределами основного каталитического цикла на эффективность функционирования “безлигандных” палладиевых каталитических систем. Проведенное с целью установления путей превращения катализатора в используемых условиях реакции операндо спектроскопическое исследование, включающее анализ кинетических закономерностей образования продукта реакции (концентрации, скорости и дифференциальной селективности) совместно с анализом кинетики изменения концентраций детектируемых спектроскопически соединений палладия показало, что в ходе реакции происходит формирование ацидокомплексов палладия [PdBr4]2–, и их концентрация драматически меняется в ходе каталитического процесса. Анализ экспериментальных зависимостей концентрации комплекса [PdBr4]2–, скорости накопления и концентрации продуктов реакции Мицороки–Хека в рамках общепринятой модели классического линейного механизма катализа приводит к противоречивым результатам. В то же время результаты симулирования таких зависимостей в приближении модели так называемого кооперативного механизма катализа, подразумевающего активацию сочетающихся арилгалогенида и алкена активными соединениями палладия в двух сопряженных каталитических циклах с нелинейной стадией взаимодействия двух палладийсодержащих интермедиатов, на качественном уровне оказываются адекватными экспериментальным данным. Таким образом, возможность образования продуктов реакции Мицороки–Хека по нелинейному (кооперативному) маршруту необходимо учитывать при анализе получаемых экспериментальных данных, а также дискриминации возможных гипотез механизмов функционирования каталитических систем.
Список литературы
Tabassum S., Zahoor A.F., Ahmad S., Noreen R., Khan S.G., Ahmad H. // Mol. Divers. 2021. № 0123456789.
Jagtap S. // Catalysts. 2017. V. 7. P 267.
Chong C., Zhang Q, Ke J., Zhang H., Yang X., Wang B., Ding W., Lu Z. // Angew. Chem. Int. Ed. 2021. V. 60. P. 13807.
Yedoyan J., Wurzer N., Klimczak U., Ertl T., Reiser O. // Angew. Chem. Int. Ed. 2019. V. 58. P. 3594.
Frost C.G., Mutton L. // Green Chem. 2010. V. 12. P. 1687.
Vásquez-Céspedes S., Betori R.C., Cismesia M.A., Kirsch J.K., Yang Q. // Org. Proc. Res. Dev. 2021. V. 25. P. 740.
Vucetic N., Virtanen P., Shchukarev A., Salmi T., Mikkola J.P. // Chem. Eng. J. 2022. V. 433. № 134432.
Hamasaka G., Ichii S., Uozumi Y. // Adv. Synth. Catal. 2018. V. 360. P. 1833.
Gnad C., Dachwald O., Raudaschl-Sieber G., Köhler K. // J. Catal. 2019. V. 375. P. 257.
de Vries A.H.M., Mulders J.M.C.A., Mommers J.H.M., Henderickx H.J.W., de Vries J.G. // Org. Lett. 2003. V. 5. P. 3285.
Schmidt A.F., Smirnov V.V. // J. Mol. Catal. A: Chem. 2003. V. 203. P. 75.
Schmidt A.F., Al Halaiqa A., Smirnov V.V. // Synlett. 2006. N. 18. P. 2861.
Consorti C.S., Ebeling G., Flores F.R., Rominger F., Dupont J. // Adv. Synth. Catal. 2004. V. 346. P. 617.
Jutand A. in The Mizoroki–Heck Reaction / Ed. Oestreich M. Munster: John Wiley & Sons Ltd. 2009. P. 1.
Schroeter F., Strassner T. // Inorg. Chem. 2018. V. 57. P. 5159.
Fernández E., Rivero-Crespo M.A., Domínguez I., Rubio-Marqués P., Oliver-Meseguer J., Liu L., Cabrero-Antonino M., Gavara R., Hernández-Garrido J.C., Boronat M., Leyva-Perez A., Corma A. // J. Am. Chem. Soc. 2019. V. 141. P. 1928.
Galushko A.S., Prima D.O., Burykina J.V., Ananikov V.P. // Inorg. Chem. Front. 2021. V. 8. P. 620.
Schmidt A.F., Kurokhtina A.A., Larina E.V., Vidyaeva E.V., Lagoda N.A. // J. Organomet. Chem. 2020. V. 929. № 121571.
Excel for Scientists and Engineers: Numerical Methods / E.J. Billo. John Wiley & Sons, 2007. 480 p.
Smith T.J., Stevenson K.J. // J. Am. Chem. Soc. 2003. V. 125. P. 3669.
Hoops S., Sahle S., Gauges R., Lee C., Pahle J., Simus N., Singhal M., Xu L., Mendes P., Kummer U. // Bioinformatics. 2006. V. 22. P. 3067.
Consorti C.S., Zanini M.L., Leal S., Ebeling G., Dupont J. // Org. Lett. 2003. V. 5. P. 983.
Böhm V.P.W., Herrmann W.A. // Chem. Eur. J. 2001. V. 7. P. 4191.
Шмидт А.Ф., Аль-Халайка А., Смирнов В.В. // Кинетика и катализ. 2007 Т. 48. С. 766. (Schmidt A.F., Al-Halaiqa A., Smirnov V.V. // Kinet. Catal. 2007. V. 48. P. 716.)
Reimann S., Stötzel J., Frahm R., Kleist W., Grunwaldt J.D., Baiker A. // J. Am. Chem. Soc. 2011. V. 133. P. 3921.
Шмидт А.Ф., Аль-Халайка А., Смирнов В.В., Курохтина А.А. // Кинетика и Катализ. 2008. Т. 49. С. 669. (Schmidt A.F., Al-Halaiqa A., Smirnov V.V., Kurokhtina A.A. // Kinet. Catal. 2008. V. 49. P. 638.)
Schmidt A.F., Kurokhtina A.A., Larina E.V. // Mendeleev Commun. 2017. V. 27. P. 213.
Шмидт А.Ф., Курохтина А.А., Ларина Е.В. // Кинетика и катализ. 2019. Т. 60. С. 555. (Schmidt A.F., Kurokhtina A.A., Larina E.V. // Kinet. Catal. 2019. V. 60. P. 551.)
Nonlinear Ordinary Differential Equations: An introduction for Scientists and Engineers / D.W. Jordan, P. Smith. New York: Oxford University Press Inc. 2007. 531 p.
Schmidt A.F., Kurokhtina A.A., Larina E.V., Vidyaeva E.V., Lagoda N.A., Schmidt E.Y. // ChemCatChem. 2020. V. 12. P. 5523.
Tan Y., Barrios-Landeros F., Hartwig J.F. // J. Am. Chem. Soc. 2012. V. 134. P. 3683.
Kim J., Hong S.H. // ACS Catal. 2017. V. 7. P. 3336.
de Vries A.H.M., Parlevliet F.J., Schmieder-van de Vondervoort L., Mommers J.H.M., Henderickx H.J.W., Walet M.A.M., de Vries J.G. // Adv. Synth. Catal. 2002. V. 344. P. 996.
Carrow B.P., Hartwig J.F. // J. Am. Chem. Soc. 2009. V. 132. P. 79.
Barrios-Landeros F., Carrow B.P., Hartwig J.F. // J. Am. Chem. Soc. 2009. V. 131. P. 8141.
Amatore C., Carré E., Jutand A., Medjour Y. // Organometallics. 2002. V. 21. P. 4540.
Amatore C., Bahsoun A.A., Jutand A., Mensah L., Meyer G., Ricard L. // Organometallics. 2005. V. 24. P. 1569.
Wang D., Izawa Y., Stahl S.S. // J. Am. Chem. Soc. 2014. V. 136. P. 9914.
Дополнительные материалы отсутствуют.
Инструменты
Кинетика и катализ