

Кинетика и катализ, 2022, T. 63, № 5, стр. 628-642
Жидкофазное пероксидное окисление метана в присутствии катализатора Cu-ZSM-5: влияние модифицирования палладием
С. А. Яшник a, *, В. В. Болтенков a, Д. Э. Бабушкин a, Т. А. Суровцова a, В. Н. Пармон a
a ФИЦ Институт катализа им. Г.К. Борескова СО РАН
630090 Новосибирск, просп. Акад. Лаврентьева, 5, Россия
* E-mail: yashnik@catalysis.ru
Поступила в редакцию 30.03.2022
После доработки 17.05.2022
Принята к публикации 24.05.2022
- EDN: HTQESC
- DOI: 10.31857/S0453881122050197
Аннотация
В реакции жидкофазного пероксидного окисления метана исследованы свойства катализаторов Cu-ZSM-5, модифицированных Pd. Катализаторы готовили методом поликонденсации ионов Cu2+ и Pd2+ в поровом пространстве цеолита. В качестве щелочного агента, промотирующего поликонденсацию гидратированных ионов металлов, использованы растворы аммиака или аммиакатных комплексов одного из вводимых металлов. Исследованы электронное состояние ионов Cu2+ и Pd2+, кислотные и окислительно-восстановительные свойства, а также текстурные характеристики катализаторов. Показано, что при поликонденсации в порах формируются ассоциированные ионы Cu2+ и полиядерные гидроксо/оксо комплексы ионов Cu2+/Pd2+, расположенные в каналах и на поверхности кристаллитов цеолита. Установлено, что добавление Pd воздействует на окислительно-восстановительные, кислотные и каталитические свойства Cu-ZSM-5, но это влияние не всегда положительно сказывается на селективности процесса относительно жидких оксигенатов: метанола и муравьиной кислоты. Среди биметаллических катализаторов наиболее активным и селективным по жидким продуктам окисления был образец Cu/Pd-ZSM-5, синтезированный введением ионов [Cu(NH3)4]2+ после ионов Pd2+. Предполагается, что его каталитические характеристики связаны с меньшей силой Cu2+–ЛКЦ и с большей устойчивостью изолированных ионов Cu2+ и полиядерных PdO-подобных кластеров к восстановлению по сравнению с образцом, полученным одновременным введением Cu2+ и Pd2+.
ВВЕДЕНИЕ
Низкотемпературная функционализация метана, моделирующая действие природных ферментов метаноксигеназ, по-прежнему остается одной из наиболее актуальных научных, а в будущем, мы надеемся, – и прикладных задач.
Известно, что основную роль в активации и окислении метана природными метанмонооксигеназами выполняют простетические группы, содержащие би- [1, 2] и трехъядерные [3] Fe- [1] или Cu- [2, 3] структуры. Поэтому большинство исследований по окислительной функционализации метана в мягких условиях сфокусированы на катализаторах, в состав которых входят би- [5–8] и трехъядерные [8–10] комплексы ионов Fe3+/Fe2+ [4, 5] и/или Cu2+ [4, 6–10] с мостиковыми О2– и ОН–-группами. Активность Cu2+-структур с мостиковым кислородом, локализованных в каналах медьсодержащих цеолитов, и их селективность в газофазном окислении метана молекулярным кислородом изучены достаточно детально [6–10]. В то же время, число работ по исследованию Cu-цеолитов в жидкофазном окислении метана пероксидом водорода в целом невелико [11–17] и, к тому же, проводится силами всего нескольких научных групп.
Причина низкого интереса к Cu-цеолитам в качестве катализаторов пероксидного окисления метана, вероятно, связана с отведенной катионам Cu(II) ролью ловушки OH• радикалов [12], образующихся при разложении пероксида водорода. Благодаря этому, как полагают Hammond с соавт. [12], окисление метана, катализируемое двухъядерными комплексами ионов Fe3+, протекает до метанола через метилгидропероксид на Cu,Fe-содержащих цеолитах; маршрут дальнейшего окисления метанола в муравьиную кислоту и диоксид углерода блокируется. С другой стороны, роль кислородных соединений Cu(II) в пероксидном окислении метана и С2-алканов была выявлена на примере нанесенного гидроксида меди [18], который образует в щелочной среде реакционноспособные пероксокомплексы [19, 20].
Проведенные нами исследования подтвердили, что реакционную способность Cu-центров в пероксидном окислении метана можно контролировать путем варьирования структуры активных центров Cu(II) как на стадии синтеза Cu-ZSM-5 [17], так и в ходе активации катализатора в реакционной среде. К примеру, плоско-квадратные биядерные оксо/гидроксо комплексы Cu2+ повышают активность Cu,Fe-ZSM-5 с низким (0.09 мас. %) содержанием ионов Fe3+ [17]. Это свидетельствует о важной роли Cu-центров, связанных с внекаркасным кислородом, в пероксидном окислении метана в водной среде.
Установлено, что в цеолитах, содержащих Cu2+ [8], наиболее устойчивыми к восстановлению водородом и СО являются изолированные ионы Cu2+ [21], тогда как Cu-структуры с внекаркасным кислородом – биядерные комплексы [CuOCu]2+ и [CuO2Cu]2+, плоско-квадратные CuO-подобные кластеры – легко восстанавливаются до Cu+ [21–23]. Внекаркасный кислород, встраиваемый в Cu-структуры при синтезе катализаторов или при их активации в O2 и N2O, является реакционноспособным в окислении оксида азота [21–24] и аммиака [23], а также в газофазном окислении метана в метанол [8, 10]. Структура Cu-центра определяет окислительно-восстановительные и каталитические свойства Cu-содержащего цеолита в целом, поэтому можно полагать, что именно Cu-структуры с внекаркасным кислородом представляют интерес для жидкофазного окисления метана в оксигенаты. Это предположение подтверждено в работе [17], в которой были проварьированы состояния Cu(II) от изолированных ионов до разнообразных Cu-структур с внекаркасным кислородом и показана высокая активность Cu-структур с внекаркасным кислородом.
Положительное влияние на окислительно-восстановительные свойства медьсодержащих катализаторов оказывает их модифицирование добавками благородного металла. К примеру, небольшие количества PdO снижают температуру восстановления наночастиц CuО и CuO-подобных кластеров, нанесенных на SiO2 [25, 26]. Вдобавок монометаллические Pd-катализаторы считаются наиболее активными системами для синтеза пероксида водорода из молекулярного водорода и кислорода [27–30] и разложения Н2О2 [27, 30], что также может влиять на каталитические свойства биметаллического Pd,Cu-содержащего цеолита.
Ранее уже отмечалась активность Pd и PdAu, нанесенных на TiO2 [31, 32] или цеолит H-ZSM-5 [33, 34], в генерировании Н2О2 и в пероксидном окислении метана. Так, в работе [34] показано, что состав продуктов окисления метана пероксидом водорода при 50–95°С слабо зависел от содержания Pd (от 0.01 до 2 мас. %) в катализаторе Pd/ZSM-5, в продуктах регистрировались в основном муравьиная кислота и следовые количества метилпероксида, метанола и СО2. На основании данных EXAFS и ПЭМ Huang и др. сделали вывод [34] о высокой активности в пероксидном окислении метана одиночных Pd2+-центров, закрепленных на внутренней поверхности микропор цеолита ZSM-5, и об отсутствии активности у наночастиц PdO, расположенных на его внешней поверхности.
В некоторых исследованиях отмечается положительное влияние добавок со-катализатора на основе соединений меди [32, 34, 35], ванадия [35] и железа [36] на поведение Pd-катализаторов в пероксидом окислении метана. К примеру, добавление 2.0 мас. % CuO в качестве со-катализатора изменяло селективность превращения метана: основным продуктом реакции становился метанол вместо муравьиной кислоты [34]. После добавления растворимых соединений меди или ванадия, Pd-катализатор на углеродном носителе проявлял активность в окислении СН4 до оксигенатов в присутствии H2 и O2 [35]; гидропероксидные комплексы Cu2+ и пероксомонованадат были предложены авторами в качестве активных частиц для активации связи C–H в СН4. Введение меди в состав биметаллического образца Au–Pd/TiO2 [32] приводило к повышению скорости окисления СН4 пероксидом водорода и селективности образования метанола.
Настоящая работа посвящена изучению влияния добавки Pd и последовательности ее введения в Cu-ZSM-5 на способность этих катализаторов к пероксидному окислению метана. Для выяснения роли, которую играют катионы Pd2+ и Cu2+, а также протонные центры цеолита, образцы охарактеризованы методами ТПВ-Н2 и ТПД NH3. Для наиболее активного биметаллического катализатора были оптимизированы условия проявления его каталитической активности и селективности.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Приготовление катализаторов Pd–Cu-ZSM-5
Исходные реактивы: палладий(II) нитрат – раствор с массовой долей Pd не менее 25%, чистый (ОАО “Аурат”, Россия); медь(II) азотнокислая 3-водная, ч. д. а. (АО “ЛенРеактив”, Россия); аммиак водный – ос. ч.-25-5 (ООО “АО Реахим”, Россия); газообразный кислород – технический, сорт I, чистота не менее 99.7%, ГОСТ 5583-78 (ООО “ПромГаз”, Россия). Для приготовления катализаторов использован коммерческий цеолит со структурой ZSM-5 и атомным соотношением Si/Al = 17, предоставленный ПАО “НЗХК” (Новосибирск, Россия).
Каталитически активные компоненты вводили методом поликонденсации в порах, варьируя последовательность нанесения катионов палладия и меди.
Для приготовления катализатора 1 (далее Pd–Cu/ZSM-5) поры цеолита H-ZSM-5 заполняли смешанным раствором нитратов палладия(II) и меди(II) (методом пропитывания по влагоемкости), концентрация нитратов Pd(II) и Cu(II) составляла 0.06 и 0.21 М соответственно. Пропитанный образец выдерживали 30 мин. Затем его обрабатывали водным раствором аммиака с концентрацией 1.7 М и объемом, соответствующим влагоемкости образца цеолита H-ZSM-5.
Для получения катализатора 2 (далее Cu/Pd-ZSM-5) поры цеолита H-ZSM-5 заполняли 0.06 М раствором нитрата палладия (методом пропитывания по влагоемкости), выдерживали 30 мин и затем проводили поликонденсацию водно-аммиачным раствором, содержащим аммиакатный комплекс меди(II). Раствор аммиакатного комплекса меди(II) готовили путем растворения нитрата меди(II) в дистиллированной воде с последующим добавлением водного аммиака до мольного соотношения NH3/(Cu(II) + Pd(II)) = 6; концентрация Cu(NO3)2 в растворе – 0.21 М.
Для приготовления катализатора 3 (далее Pd/Cu-ZSM-5) поры цеолита H-ZSM-5 заполняли 0.21 М раствором нитрата меди (методом пропитывания по влагоемкости), выдерживали 30 мин и затем осуществляли поликонденсацию водно-аммиачным раствором, содержащим аммиакатные комплексы палладия(II) с концентрацией Pd 0.06 М. Раствор тетрааммин палладия(II) нитрата готовили путем добавления водного аммиака в раствор нитрата палладия(II) до мольного соотношения NH3/(Cu(II) + Pd(II)) = 6.
Монометаллический образец Cu-ZSM-5 получали путем заполнения пор пропитыванием по влагоемкости цеолита H-ZSM-5 раствором нитрата меди(II) с концентрацией 0.21 М. Пропитанный образец выдерживали 30 мин. Затем его обрабатывали водным раствором аммиака с концентрацией 1.25 М и объемом, соответствующим влагоемкости цеолита H-ZSM-5.
Содержание меди и палладия в образцах составляло 1 и 0.5 мас. % соответственно. Содержание Cu(II), равное 1 мас. %, выбрано на основании результатов работы [17], в которой указанного количества Cu(II) было достаточно для обеспечения заметной активности и селективности в пероксидном окислении метана. Мольное соотношение NH4OH/(Cu + Pd) = 6 выбрано для стабилизации ионов Cu2+ в составе полиядерных гидроксокомплексов в непрокаленных образцах и в виде полиядерных оксокомплексов в прокаленных образцах [37]. Термическую обработку проводили при 450°С в течение 3 ч.
Исследование физико-химических свойств катализаторов
Химический анализ образцов проводили рентгеноспектральным флуоресцентным методом (РФлА) на анализаторе ARL PERFORM’X (“Thermo Scientific”, США) c Rh-анодом.
Текстурные свойства образцов – удельную поверхность (SБЭТ, м2/г), внешнюю поверхность кристаллитов (St, м2/г) и объем микропор (Vm, см3/г) – были исследованы методом низкотемпературной адсорбции азота при 77 К на установке ASAP 2400 (“Micrometrics”, США) после дегазации образцов под вакуумом при 250°С в течение 6 ч. Экспериментальные изотермы адсорбции азота обрабатывали по методу БЭТ для нахождения удельной поверхности катализатора и экстраполяцией линейной части t-графиков (t-метод Хэлси) для определения внешней поверхности кристаллитов и объема микропор цеолита.
Электронные спектры диффузного отражения (ЭСДО) порошков катализаторов сняты на спектрофотометре UV-2501 PC (“Shimadzu”, Япония) с приставкой диффузного отражения ISR-240A, относительно стандарта отражения (BaSO4) в диапазоне 11 000–54 000 см–1. Зарегистрированные ЭСДО представлены в координатах функции Кубелки–Мунка–волное число.
Кислотные свойства образцов изучали методом термопрограммируемой десорбции аммиака (ТПД-NH3) при выбранных ранее условиях [38]: адсорбция NH3 при 75°С/1 ч из смеси 0.35 об. % NH3/He, удаление слабо адсорбированного NH3 при 75°С/1 ч в потоке He, последующий нагрев в потоке гелия (30 см3/мин) со скоростью 10°/мин от 50 до 600°С. Регистрацию десорбирующегося NH3 проводили с помощью катарометра. Перед адсорбцией аммиака образец (100 мг) подвергали обработке в газообразном кислороде, продуваемом через слой катализатора со скоростью 30 см3/мин при 450°С/2 ч для удаления адсорбированной воды. Общее количество кислотных центров в образце определяли по количеству выделившегося NH3. Разложением профиля ТПД-NH3 на отдельные компоненты находили концентрацию центров определенной природы.
Окислительно-восстановительные свойства катализаторов изучали с помощью термопрограммируемого восстановления водородом (ТПВ-H2) на установке, оборудованной проточным реактором и детектором по теплопроводности. Восстановление осуществляли в интервале температур от 0 до 950°C со скоростью подъема температуры 10°C/мин, пропуская смесь 10% H2 в Ar со скоростью 30 см3/мин через навеску образца. Перед проведением эксперимента образец подвергали тренировке в потоке газообразного кислорода, продуваемом через реактор со скоростью 30 см3/мин при 450°С в течение 30 мин. Навеска образца составляла 100 мг, размер частиц – 250–500 мкм, для снятия экзотермических эффектов образец смешивали со 100 мг кварца с аналогичным размером частиц. Воду, образующуюся в ходе восстановления, удаляли из газовой смеси вымораживанием в ловушке при температуре –70°С. Количество поглощенного водорода калибровали относительно H2, потребляемого на восстановление CuO в аналогичных условиях, предполагая полное восстановление CuO до Cu. Обработку экспериментов ТПВ-Н2 выполняли следующим образом: аппроксимировали профиль ТПВ-Н2 отдельными пиками, максимум которых выбирали в соответствии с известной информацией о температурных интервалах восстановления отдельных структур активного компонента; находили количество поглощенного водорода (моль/г) интегрированием площадей отдельных пиков; рассчитывали соотношение количества Н2, поглощенного в каждом температурном интервале, к общему содержанию Pd или Cu (моль/г) в катализаторе; с учетом стехиометрии восстановления конкретного состояния Pd и Cu оценивали его долю (%) в катализаторе. Пример разложения профиля ТПВ-Н2 приведен на рис. 1г для Cu-ZSM-5 с содержанием Cu 1 мас. %.
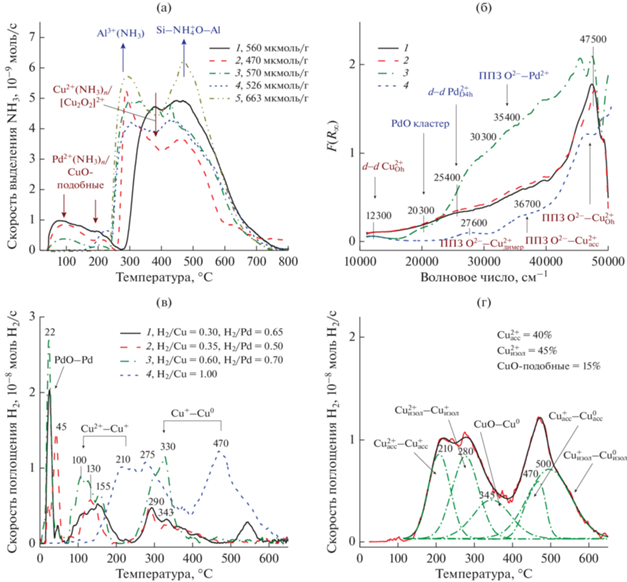
Рис. 1.
Профили ТПД-NH3 (а), спектры ЭСДО (б) и профили ТПВ-Н2 (в, г) образцов: 1 – Cu–Pd/ZSM-5; 2 – Cu/Pd-ZSM-5; 3 – Pd/Cu-ZSM-5; 4 – Cu-ZSM-5; 5 – H-ZSM-5. а – Для каждого образца приведена концентрация кислотных центров; в – для каждого образца приведено соотношение поглощенного водорода к введенному активному компоненту; г – разложение профиля ТПВ-Н2 образца Cu-ZSM-5 на отдельные компоненты, их отнесение к отдельным состояниям Cu2+ и их количество (%) относительно общего содержания меди в катализаторе (1 мас.% Cu, принят за 100%).
Исследование каталитических свойств
Катализаторы были испытаны в жидкофазном пероксидном окислении метана. Реакцию вели в реакторе высокого давления (автоклав “Amar Equipment Pvt. Ltd.”, Индия). В реактор объемом 0.45 л помещали 80 мл 1 М раствора H2O2, герметизировали его и 5 раз продували метаном. Автоклав нагревали до необходимой температуры реакции (50°C), после чего подавали в него 0.216 г катализатора (2.7 г/л) с помощью шарового клапана системы загрузки. В системе фиксировали рабочее давление метана (30 бар), запускали перемешивание (1500 об/мин) и выдерживали необходимое время. Кинетику окисления СН4 изучали проведением отдельных экспериментов с варьированием времени реакции от 5 до 90 мин без отбора проб на анализ в процессе эксперимента. После окончания реакции газовую фазу отбирали с помощью системы газового пробоотбора и анализировали методом ГХ. Реакционный раствор фильтровали на ацетатцеллюлозном фильтре с порами размером 0.45 мкм и анализировали методами ВЭЖХ и Н1 ЯМР.
Предварительный эксперимент, выполненный в описанных выше условиях, но без катализатора, показал, что окисления метана в отсутствие катализатора не наблюдается.
Для наиболее активного катализатора (образец 2) варьировали: температуру в интервале 40–60°С, давление метана – 20–40 бар, концентрацию катализатора – 1.35–4.05 г/л, концентрацию Н2О2 – 0.1–1 М, рН – 3, 5 и 10.
Для исключения гомогенного протекания реакции с участием акватированных катионов металлов, перешедших с катализатора в реакционный раствор, были проведены дополнительные эксперименты с реакционными растворами, отфильтрованными от гетерогенного катализатора (так называемый “личинг”-тест (leaching test) в англоязычной литературе). Для этого раствор после реакции фильтровали, используя ацетатцеллюлозный шприцевой фильтр с диаметром пор 0.45 мкм. Определяли концентрацию пероксида водорода в растворе и добавляли H2O2 до концентрации 1 M. Реакцию вели в автоклаве при условиях, указанных выше для реакции пероксидного окисления метана в присутствии катализатора. На примере катализатора Cu-ZSM-5 было показано, что гомогенные ионы Cu2+ не обладали активностью в превращении СН4 в оксигенаты [17].
Конверсию пероксида водорода, Х(Н2О2), в пероксидном окислении метана определяли по отношению количества молей прореагировавшего пероксида водорода, νпрореаг(Н2О2), к количеству его молей в начальный момент времени, ν0(Н2О2):
Конверсию метана, Х(СН4), в реакции пероксидного окисления определяли как отношение суммарного количества молей всех образовавшихся продуктов реакции (CO, CO2, CH3OOH, CH3OH, CH2O, и HCOOH), Σν(продукты), к начальной концентрации метана, ν0(СН4), в реакторе по формуле:
Селективность, S, образования индивидуальных продуктов рассчитывали по формуле:
Катализаторы сравнивали между собой и с описанными в литературе [32, 34, 35] по величине числа оборотов реакции (TOF(CH4)):
Состав газовой фазы анализировали на газовом хроматографе Кристалл 2000М (“Хроматэк”, Россия), оснащенном пламенно-ионизационным детектором и метанатором. Колонка 2 м × 2 мм заполнена Chromosorb 102, газ-носитель – аргон. Концентрацию муравьиной кислоты определяли методом высокоэффективной жидкостной хроматографии (ВЭЖХ) на жидкостном хроматографе Миллихром A02 (ООО ИХ “ЭкоНова”, Россия), оснащенном спектрофотометрическим детектором (λ = 210 нм) и ионообменной колонкой Диасфер-250ПА, 2 × 75 мм. Элюенты: 0.04 M водный раствор LiClO4 и H2O; скорость потока – 0.2 мл/мин.
1Н ЯМР- и 13C ЯМР-спектры жидких продуктов регистрировали с помощью спектрометра AVANCE-400 (“Bruker”, Германия) в ЯМР ампулах (диаметр – 5 мм) без разбавления D2O при 400.13 MHz (1H) и 100.61 MHz (13C). Подробно методика приведена в [17].
Концентрацию Н2О2 в реакционном растворе определяли спектрофотометрическим методом по образованию комплекса с титанил сульфатом на спектрофотометре Cary 60 UV-Vis (“Agilent Technologies”, США) в соответствии с процедурой, описанной в [39].
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Текстурные свойства катализаторов
Изучение текстурных свойств моно- и биметаллических катализаторов показало, что их удельная поверхность (табл. 1, SБЭТ) примерно на 8% ниже, чем у исходного цеолита H-ZSM-5. Внешняя поверхность кристаллитов (St) в биметаллическом катализаторе увеличивается относительно исходного цеолита Н-ZSM-5, однако она превосходит таковую в монометаллическом катализаторе только в случае последовательного введения катионов [Cu(NH3)4]2+ после Pd2+. Внешняя поверхность кристаллитов изменяется в ряду: H-ZSM-5 (26 м2/г) < Pd/Cu-ZSM-5 (29 м2/г) < Cu–Pd/ZSM-5 (31 м2/г) < Cu-ZSM-5 (32 м2/г) < < Cu/Pd-ZSM-5 (38 м2/г). Бóльшие значения St у катализаторов по сравнению с цеолитом H-ZSM-5 указывают на локализацию части активного компонента на поверхности мезопор кристаллитов цеолита.
Таблица 1.
Физико-химические свойства носителя и катализаторов
| № | Катализатор | Текстурные характеристики | Количество кислотных центров, 10–6 моль/г* | ||||||
|---|---|---|---|---|---|---|---|---|---|
| SБЭТ, м2/г | St, м2/г | Vm, см3/г | <250 °C |
270–330 °C |
355–415 °C | 470–510 °C | общее | ||
| 1 | H-ZSM-5 | 385 | 26 | 0.118 | – | 158 | – | 527 | 685 ± 40 |
| 2 | Cu-ZSM-5 | 350 | 32 | 0.109 | 7 | 26(108) | 168 | 243 | 526 ± 30 |
| 3 | Cu–Pd/ZSM-5 | 354 | 31 | 0.119 | 56 | 71 | 120 | 310 | 560 ± 30 |
| 4 | Cu/Pd-ZSM-5 | 354 | 38 | 0.114 | 40 | 30(48) | 119 | 228 | 470 ± 40 |
| 5 | Pd/Cu-ZSM-5 | 349 | 29 | 0.117 | 16 | 43(114) | 83 | 324 | 580 ± 50 |
Объем микропор цеолита после модифицирования цеолита катионами Cu2+ и Pd2+ находится в пределах 0.109–0.119 см3/г. Минимальные значения зарегистрированы для катализаторов Cu-ZSM-5 (0.109 см3/г) и Cu/Pd-ZSM-5 (0.114 см3/г), и они ниже, чем у исходного цеолита H-ZSM-5 (0.118 см3/г). Наблюдаемое снижение объема микропор у одних образцов и его сохранение у других может быть обусловлено несколькими возможными причинами. С одной стороны, это заполнение каналов цеолита полиядерными гидроксо/оксо структурами катионов Cu2+ и Pd2+, которое максимально у катализаторов Cu-ZSM-5 и Cu/Pd-ZSM-5, однако внутриканальная локализация структур активного компонента не должна приводить к росту внешней поверхности кристаллитов цеолита. С другой стороны, расположение полиядерных гидроксо/оксо структур ионов Cu2+ и Pd2+ или CuO/PdO-подобных кластеров на внешней поверхности мезопор кристаллитов цеолита может частично блокировать вход в канал цеолита для молекулярного азота, используемого для измерения текстурных свойств катализаторов. Кинетический диаметр N2 всего в 1.5 раза меньше, чем диаметр канала цеолита: 0.364 нм [40] против 0.54–0.56 нм [41]. В этом случае, чем выше дисперсность частиц активного компонента, тем больше их поверхность и значительнее вероятность недоступности каналов для адсорбата. Возможность блокировки каналов ZSM-5 была доказана ранее на примере катализатора Cu(1 мас. %)-ZSM-5 [42].
Электронное состояние активных металлов в катализаторах
Результаты ТДП-NH3 (рис. 1а) дают информацию о кислотных центрах цеолита H-ZSM-5 и катализаторов, полученных на его основе. Исходный цеолит содержит два типа кислотных центров, различающихся силой: слабые и сильные центры, с которых аммиак удаляется при температурах 295 и 470°С соответственно при выбранных нами условиях [38]. К первому типу центров относят Льюисовские кислотные центры (ЛКЦ) из внекаркасных катионов Al3+ и терминальные SiOH-группы цеолита. Второй представлен Бренстедовскими кислотными центрами (БКЦ) решетки цеолита Si(OH)Al. Количество слабых кислотных центров и БКЦ уменьшается после введения Pd2+ и Cu2+ в цеолит H-ZSM-5, одновременно появляются новые ЛКЦ. Концентрация БКЦ (табл. 1, 475–510 °С) зависит от способа введения модифицирующих ионов и увеличивается в ряду: Cu/Pd-ZSM-5 < Cu-ZSM-5 < Cu–Pd/ZSM-5 < < Pd/Cu-ZSM-5 < H-ZSM-5. Новые ЛКЦ соответствуют комплексам [Cu(NH3)n]2+ (350–400°С) и CuO–NH3 (200°С) [38], а также комплексам [Pd(NH3)n]2+ (275–285°С) и PdO–NH3 (100°С). Концентрация слабых ЛКЦ, с которых аммиак удаляется при температурах до 200°С, возрастает в ряду: Cu-ZSM-5 < Pd/Cu-ZSM-5 < Cu/Pd-ZSM-5 < < Cu–Pd/ZSM-5. Количество более сильных Cu2+–ЛКЦ, десорбирующих аммиак при 350–400°С, изменяется в ряду: Pd/Cu-ZSM-5 < < Cu/Pd-ZSM-5 ~ Cu–Pd/ZSM-5 < Cu-ZSM-5.
ЭСДО (рис. 1б) прокаленных биметаллических Cu–Pd-катализаторов отчетливо демонстрируют присутствие в образцах изолированных ионов Cu2+ и Pd2+. Спектры образцов Cu–Pd/ZSM-5 и Cu/Pd-ZSM-5 полностью идентичны. Они содержат полосы поглощения (п. п.) с энергией d–d-перехода (12 300 см–1) и полосы переноса заряда лиганд–металл (ППЗ L–M, 47 500 см–1) от изолированных ионов [Cu(H2O)6-n(OH)n]2+ в тетрагонально-искаженном октаэдрическом окружении (${\text{Cu}}_{{{\text{Oh}}}}^{{2 + }}$) кислородсодержащих лигандов [43]. В биметаллических образцах не исключено присутствие ионов Cu2+ в составе димеров (ППЗ L–M 27 600 см–1 от Cu2+–O–Cu2+ [43]) и ассоциатов (ППЗ L–M 36 700 см–1 от Cu2+ [37]), а также CuO-подобных кластеров (поднятие фона). Характеристические ППЗ L–M от димеров Cu2+–O–Cu2+ и ассоциатов ионов Cu2+ отчетливо проявляются в спектре монометаллического катализатора Cu-ZSM-5. В спектрах биметаллических образцов наблюдаются также полосы, энергия которых соответствует d–d-переходу (25 400 см–1) и ППЗ L–M (36 700 см–1) от изолированных ионов Pd2+ в псевдо-октаэдрическом окружении (${\text{Pd}}_{{{\text{D4h}}}}^{{2 + }}$) ОН- или Н2О-лигандов [44]. Значительное уширение п. п. 25 400 см–1, вероятно, связано с образованием полиядерных форм ионов Pd2+. Кроме того, не исключено присутствие ионов Pd2+ в составе ассоциатов (ППЗ L–M 35 400 см–1) и PdO-подобных кластеров (плечо 20 300 см–1), но их количество невелико.
В отличие от описанных выше, образец Pd/Cu-ZSM-5-17 помимо изолированных ионов ${\text{Cu}}_{{{\text{Oh}}}}^{{2 + }}$ и ${\text{Pd}}_{{{\text{D4h}}}}^{{2 + }}$ содержит PdO-подобные кластеры, на которые указывает четкая полоса 20300 см–1 [45] в электронном спектре Pd/Cu-ZSM-5.
Наличие изолированных ионов Cu2+ в исследуемых образцах подтверждается данными ТПВ-Н2. Профили поглощения водорода в ТПВ-Н2 (рис. 1в) содержат три пика с максимумами при 22–45, 100–155 и 290–330°С. Два последних пика соответствуют, по-видимому, 2-стадийному восстановлению изолированных и/или ассоциированных ионов Cu2+. Отметим, что как 1-я, так и 2‑я стадии восстановления упомянутых ионов Cu2+ в биметаллических образцах протекают при температурах, которые на 90–120°С ниже, чем для монометаллического катализатора 1%Cu-ZSM-5 (рис. 1г) [22, 37], указывая на промотирующее действие палладия на восстановительную способность катионов Cu2+. Pd-содержащий активный компонент биметаллических катализаторов Cu,Pd-ZSM-5 восстанавливается при температуре 22–45°С, что в соответствии с [46] демонстрирует наличие в них высокодисперсных наночастиц или кластеров PdO. Благодаря хемосорбции и диссоциации Н2 на образовавшемся при восстановлении металлическом Pd с последующей диффузией водорода к ионам Cu2+, ускоряется восстановление ионов Cu2+, находящихся в каналах и на поверхности цеолита. Улучшение способности CuO к восстановлению в присутствии PdO хорошо известно и наблюдалось ранее даже для систем с сильным взаимодействием CuO и PdO с носителем SiO2 [26]. Кроме того, в ТПВ-Н2 профиле наших образцов присутствует плечо слабой интенсивности в интервале температур 300–450°С. Это плечо может быть связано с PdO-подобными кластерами в каналах цеолита и изолированными ионами Pd2+, взаимодействующими с водородом при 300 и 450°С соответственно [47, 48].
С учетом количества водорода, поглощенного образцами при 0–50°С, и стехиометрии восстановления PdO до Pd (теоретическое значение H2/Pd = 1), можно полагать, что в биметаллических катализаторах соотношение (в %) количества PdO, регистрируемого методом ТПВ-Н2 в этой температурной области, к общему содержанию палладия в образцах (0.5 мас. % или 4.72 × × 10–5 моль/г) возрастает в ряду: Cu/Pd-ZSM-5 (до 50%) < Cu–Pd/ZSM-5 (около 65%) < < Pd/Cu-ZSM-5 (около 70%). Если связать все количество водорода, поглощенного образцом в интервале 80–400°С, с катионами Cu2+, то доля ионов Cu2+ (суммарно в виде изолированных, димерных и ассоциированных ионов) от общего содержания меди в катализаторах (1 мас. % или 1.56 × 10–4 моль/г) увеличивается в аналогичной последовательности: Cu/Pd-ZSM-5 (до 30%) < Cu–Pd/ZSM-5 (35%) < < Pd/Cu-ZSM-5 (60%).
В отличие от монометаллического образца Cu-ZSM-5, суммарное количество поглощенного катионами Cu2+ и Pd2+ водорода ниже, чем требуется для полного восстановления введенных ионов до металлического состояния. Различие между теоретическим и экспериментальным значениями соотношения H2/(Cu + Pd) можно объяснить несколькими причинами. Во-первых, формированием металлических частиц палладия и меди в ходе синтеза катализаторов в результате восстановления ионов Pd2+ и Cu2+ аммиаком и их последующим сплавлением. Частицы металлического Pd регистрировали после окислительного прокаливания Pd-содержащих цеолитов, если ионы Pd2+ вводили в цеолит из аммиакатных комплексов [49–51]. Отметим, что отрицательный пик в ТПВ-Н2, который типичен для металлического Pd и связан с разложением гидрида Pd [52, 53], не характерен для сплавных частиц PdCu (рис. 1в) в силу ингибирования образования гидрида Pd вторым металлом [53–55]. Во-вторых, стабилизацией ионов Cu+ в ходе синтеза, но это предположение противоречит данным ЭСДО (рис. 1б). Следовательно, часть активных металлов в образцах Cu,Pd-ZSM-5 может находиться в металлическом высокодисперсном состоянии, несмотря на окислительные условия термической обработки.
Пероксидное окисление метана в присутствии катализаторов Cu,Pd-ZSM-5
Методом Н1 ЯМР определен состав жидких продуктов пероксидного окисления метана, получаемых с участием Cu,Pd-ZSM-5 катализаторов. Ими оказались метилгидропероксид, метанол, метиленгликоль и муравьиная кислота. В меньших количествах обнаружены метилформиат и надмуравьиная кислота. Основным газообразным продуктом согласно данным ГХ был СО2. В следовых количествах зафиксирован СО. Указанные жидкие и газообразные продукты регистрировали и в случае использования монометаллического катализатора 1%Cu-ZSM-5 [17]. Таким образом, модифицирование Cu-ZSM-5 палладием в количестве 0.5 мас. % не влияет на состав продуктов реакции. Некоторые из перечисленных жидких (CH3OH, HCOOH, CH3OOH) и газообразных (CO2) продуктов детектировали в большей части работ по пероксидному окислению метана [11–15, 31–34].
Каталитические характеристики образцов H-ZSM-5, Cu-ZSM-5 и Cu,Pd-ZSM-5 суммированы в табл. 2. Их сопоставление показывает, что катионы Cu2+ в составе Cu-ZSM-5 действительно тормозят превращение метана в муравьиную кислоту: селективность образования НСООН понижается (с 62 до 49%), тогда как селективность по метанолу повышается практически вдвое в сравнении с H-ZSM-5. В образце H-ZSM-5 каталитически активными являются катионы Fe2+/Fe3+, содержащиеся в нем в следовых количествах (0.09 мас. % Fe). [16, 17]. С одной стороны, влияние ионов Cu2+ на поведение H-ZSM-5 согласуется с результатами работы [12]. Рост селективности по СО2 (с 5 до 9%) и двукратное увеличение конверсии Н2О2, напротив, демонстрируют активность ионов Cu2+ в разложении Н2О2 и окислении образовавшейся муравьиной кислоты до СО2.
Таблица 2.
Пероксидное окисление метана на биметаллических Cu- и Pd-содержащих цеолитах типа ZSM-5*
| № | Катализатор | H2O2 | CH4 | TOF(H2O2)/ TOF(CH4) | Селективность, % | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Х(Н2О2), % | TOF, ч–1 | Х(СН4), % | TOF, ч–1 | CH3OOH | CH3OH | CH2(OH)2 | HCOOH+ + HCOОOH | газы | |||
| 1 | H-ZSM-5 | 14.6 | 2200 | 0.56 | 447 | 4.9 | 8 | 14 | 8 | 62 | 5 |
| 2 | Cu-ZSM-5 | 30.1 | 430 | 0.73 | 54 | 8.0 | 2 | 31 | 6 | 49 | 9 |
| 3 | Cu–Pd/ZSM-5 | 80.1 | 900 | 0.52 | 30 | 30.0 | 3 | 23 | 7 | 45 | 20 |
| 4 | Cu/Pd-ZSM-5 | 47.5 | 534 | 0.82 | 47 | 11.6 | 3 | 19 | 7 | 59 | 10 |
| 5 | Pd/Cu-ZSM-5 | 54.0 | 600 | 0.79 | 46 | 13.0 | 3 | 21 | 7 | 56 | 13 |
Рис. 2 иллюстрирует кинетику накопления основных продуктов реакции и изменения селективности пероксидного окисления метана на примере образца Cu/Pd-ZSM-5. Приведенные на рис. 2 данные свидетельствуют, что метилгидропероксид и формальдегид накапливаются в незначительном количестве при изученных временах реакции. Согласно результатам Hammond с соавт. [11] и нашим предыдущим работам [16, 17] CH3OOH – весьма реакционноспособное вещество, и поэтому его концентрация может резко снижаться уже за первые 2–5 мин реакции. В большем количестве на начальном этапе реакции регистрируется метанол; его количество продолжает расти на протяжении всего эксперимента, однако после 30 мин скорость накопления СН3ОН замедляется. Через 30 мин реакции начинается активное накопление муравьиной кислоты в продуктах реакции и увеличение выхода углекислого газа, причем к концу эксперимента НСООН уже преобладает в продуктах реакции. Схема последовательного превращения метана в метанол и муравьиную кислоту, предполагаемая на основании обнаруженных промежуточных и конечных продуктов реакции и кинетики их накопления, приведена ниже (схема 1 ).
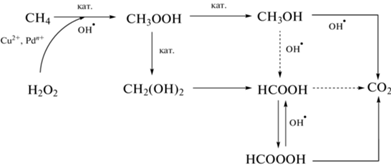
Схема 1 . Схема окислительного превращения метана в присутствии Cu,Pd-ZSM-5 и Н2О2.
Рис. 2.
Профили накопления (а) и изменения селективности (б) основных продуктов реакции пероксидного окисления метана на катализаторе Cu/Pd-ZSM-5. Условия: 50°C, 30 бар СН4, 1 M H2O2, 2.7 г/л катализатора.
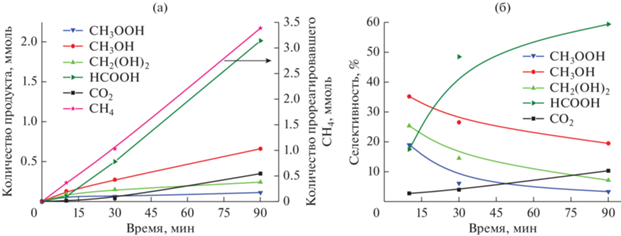
Схожие закономерности накопления продуктов реакции характерны и для образцов Pd/Cu-ZSM-5 и Cu–Pd/ZSM-5. Впрочем, для катализатора Cu–Pd/ZSM-5, в сравнении с образцами с последовательным нанесением металлов, отмечается минимальный период индукции накопления муравьиной кислоты (1 мин вместо 3–5 мин), но меньшая скорость ее накопления в продуктах реакции и более низкие ее концентрации за время эксперимента (0.97 против 1.85–2.00 ммоль), в то время как выделение СО2 в 1.5–2 раза выше и начинает регистрироваться при меньшем времени эксперимента (1 мин против 9–15 мин). Образование СО2 в значительных количествах указывает на бóльшую склонность первичных продуктов реакции к окислению в присутствии катализатора Cu–Pd/ZSM-5, чем при участии Cu/Pd-ZSM-5 и Pd/Cu-ZSM-5. Таким образом, модифицирование Cu-ZSM-5 палладием и последовательность его введения оказывают воздействие на концентрацию продуктов реакции пероксидного окисления метана (табл. 2), вероятно, из-за изменения способности системы Cu,Pd-ZSM-5/Н2О2 в целом к формированию активных частиц окислителя (из Н2О2) и активации метана.
Сопоставление каталитических характеристик Cu-ZSM-5 и Cu,Pd-ZSM-5 (табл. 2) показывает, что добавление палладия и последовательность его введения в Cu-ZSM-5 влияют на достигаемые конверсии H2O2 и CН4. Так, совместное нанесение катионов Pd(II) и Cu(II) приводит даже к уменьшению активности катализатора: конверсия СН4 составляет 0.52% для Cu–Pd/ZSM-5 против 0.73% для Cu-ZSM-5. Одновременно значительно (c 30 до 80%) возрастает конверсия пероксида водорода, что свидетельствует о неселективном расходовании окислителя. Двукратное увеличение селективности по газовым продуктам реакции (с 9 до 20%) также подтверждает разложение Н2О2 с образованием существенного количества радикалов ОН·, так как считается, что ОН· способствует глубокому окислению метана [11, 14].
При последовательном нанесении катионов металлов активность биметаллических образцов Cu/Pd-ZSM-5 и Pd/Cu-ZSM-5, оцениваемая по конверсии метана, выше, чем у монометаллического образца Cu-ZSM-5 (0.82 и 0.79% против 0.73%) и цеолита H-ZSM-5 (только 0.56%). Неселективное расходование Н2О2 на катализаторах, полученных последовательным нанесением Pd(II) и Cu(II), снижается по сравнению с образцом Cu–Pd/ZSM-5, но остается выше, чем в присутствии монометаллического катализатора Cu-ZSM-5 и цеолита Н-ZSM-5. Упомянутые экспериментальные результаты свидетельствуют о способности одного или нескольких состояний палладия промотировать пероксидное окисление метана, катализируемое Фентон-подобной системой из ионов Cu(II)/Cu(I) в составе Cu-ZSM-5 и H2O2 [17], и одновременно проводить разложение пероксида водорода. То, что металлический Pd может ускорять разложение Н2О2, доказано в [28, 30]. В единичных работах рассматривались каталитические свойства Pd-ZSM-5 и Pd/TiO2 в пероксидном окислении метана [31, 33]. Добавление в качестве сокатализатора соединений меди (CuO и других [34]) к Pd-ZSM-5 (с изолированными ионами Pd2+ [34]) приводило к замедлению превращения CН4 в оксигенаты, в том числе, и в муравьиную кислоту [34], т.е. по существу CuO участвовал в разложении избытка H2O2 и ингибировал образование радикалов OH·. Добавление Pd(II) в наши образцы Cu-ZSM-5, в отличие от результатов работы [34], наоборот, способствует увеличению селективности по продуктом более глубокого окисления, таким как муравьиная кислота (на 7–10% для Cu/Pd-ZSM-5 и Pd/Cu-ZSM-5) и СО2 (на 1–4%). Это указывает на вклад палладия в окисление СН4 в НСООН, возможно, за счет образования активных частиц окислителя при разложении Н2О2 с участием одного из состояний палладия (изолированных ионов Pd(II), PdO или Pd), выявленных в биметаллических катализаторах.
Таким образом, среди изученных биметаллических катализаторов наиболее активным и селективным в окислении метана оказался образец Cu/Pd-ZSM-5, в который Cu(II) вводили после Pd(II). Одновременно с этим он был менее активным в разложении пероксида водорода (47.5%). Сопоставление с литературными данными [17, 32, 34] показывает, что катализаторы Cu,Pd-ZSM-5 проявляют сравнимую [17], а в ряде случаев даже бóльшую [32, 34] активность в пероксидном окислении метана. Однако по селективности образования метанола они уступают описанным в литературе образцам на основе Pd и Cu [32, 34], обеспечивая высокую селективность по муравьиной кислоте (табл. 3). Различие в поведении каталитических систем Cu/Pd-ZSM-5 (настоящая работа) и (2% CuO + Pd-ZSM-5) [34] может быть связано с разным электронным и структурным состоянием каталитически активных катионов.
Таблица 3.
Сопоставление результатов пероксидного окисления метана с литературными данными*
| № | Катализатор | TOF(H2O2), ч–1 | TOF(CH4), ч–1 | S(СН3ОН), % | S(НСООН), % | Источник |
|---|---|---|---|---|---|---|
| 1 | Cu-ZSM-5-17 | 410 | 75 | 32 | 31 | [17] |
| 2 | Cu–Pd/ZSM-5-17 | 920 | 57 | 26 | 33 | Настоящая работа |
| 3 | Cu/Pd-ZSM-5-17 | 650 | 45 | 26 | 49 | |
| 4 | Pd/Cu-ZSM-5-17 | 730 | 67 | 28 | 47 | |
| 5 | 2.5% Pd + 2.5% Cu/TiO2 | 513 | 0.59 | 18 | 0 | [32] |
| 6 | 2.5% Au +2.5% Pd/ 1.0% Cu/TiO2 |
832 | 1.40 | 83 | 0 | [32] |
| 7 | 0.01 мас. % Pd/ZSM-5 + + 2 мас. % CuO | – | 15 | 78 | 6 | [34] |
Кинетика пероксидного окисления метана с участием Cu/Pd-ZSM-5
Наиболее активный катализатор Cu/Pd-ZSM-5 испытан при варьировании параметров процесса для определения основных маршрутов пероксидного окисления метана и оптимизации процесса по продуктам реакции. Рис. 3 демонстрирует влияние температуры, давления, концентрации катализатора и начальной концентрации окислителя на количество образующихся продуктов, общую селективность по жидким продуктам (оксигенатам) и селективности образования метанола и муравьиной кислоты.
Рис. 3.
Влияние температуры (а), давления (б), концентрации катализатора (в) и концентрации H2O2 (г) на общее количество образующихся продуктов и на селективность по метанолу, муравьиной кислоте и суммарно по жидким оксигенатам, получаемым в реакции пероксидного окисления метана с участием Cu/Pd-ZSM-5 за 90 мин. В каждом эксперименте варьировали только один параметр, остальные соответствовали условиям, при которых проводилось сравнение катализаторов: 50°C, 30 бар СН4, 1 M H2O2, 2.7 г/л катализатора.
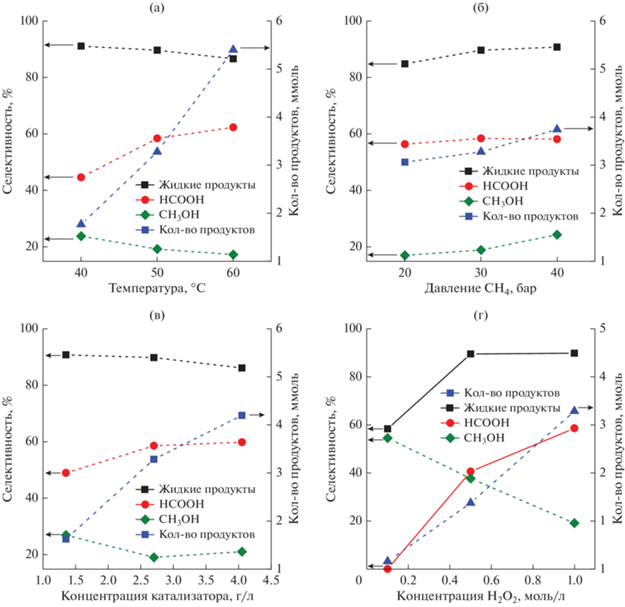
При повышении температуры от 40 до 60°С (рис. 3а) втрое (с 1.8 до 5.4 ммоль) возрастает общее количество образующихся жидких продуктов окисления метана и увеличивается (от 45 до 62%) селективность по муравьиной кислоте, между тем общая селективность по жидким продуктам незначительно (на 2%) снижается, в основном за счет уменьшения селективности по метанолу (с 24 до 17%). Следовательно, при выбранных условиях образующиеся жидкие продукты – преимущественно СН3ОН и НСООН – достаточно стабильны в интервале температур 40–60°С, их дальнейшее окисление до СО2 изменяется очень слабо. Одновременно с этим при подъеме температуры всего на 10°С (от 50 до 60°С) в 2 раза увеличивается конверсия пероксида водорода, которая достигает 99% при 60°С за 90 мин эксперимента.
Давление метана (рис. 3б) оказывает слабое влияние на селективность и выход продуктов реакции. Так, при повышении давления СН4 в 2 раза (с 20 до 40 бар) количество жидких продуктов реакции возрастает всего на 16% (с 3.1 до 3.7 ммоль), селективность по общему количеству жидких продуктов – с 85 до 91%, селективность по СН3ОН – с 17 до 24.5%, а по муравьиной кислоте находится в пределах 56–58%. Конверсия пероксида водорода снижается с 49 до 45%. Полученные экспериментальные данные указывают, что в выбранном интервале давлений метана реакция его окисления пероксидом водорода имеет нулевой порядок по CH4. В этом случае наблюдаемое незначительное увеличение количества образующихся оксигинатов объясняется изменением растворимости СН4 в воде и возрастанием количества метана, адсорбированного на поверхности катализатора. Так, растворимость метана в воде при 40°С повышается в 1.5 раза при изменении давления от 20 до 40 бар [56]. Некоторое падение селективности по СО2 с ростом давления СН4 обусловлено конкурентным ингибированием метаном адсорбции метанола. Это согласуется с наблюдаемым в [11] понижением стабильности СН3ОН, адсорбированного на поверхности ZSM-5(Si/Al = = 15), в среде СН4 в сравнении с гелием.
Содержание катализатора (рис. 3в) в реакционном растворе влияет на количество оксигенатов, образующихся в реакции при 50°С и давлении СН4 в 30 бар. Так, при 3-кратном (от 1.35 до 4.05 г/л) увеличении концентрации катализатора количество продуктов реакция возрастает в 2.5 раза. Это свидетельствует о протекании реакции в кинетическом режиме в выбранных нами условиях проведения реакции. Вместе с тем, при повышении концентрации катализатора от 2.7 до 4.05 г/л селективности по СО2, СН3ОН и НСООН практически не изменяются.
Общее количество образующихся продуктов реакции пероксидного окисления метана прямолинейно возрастает с начальной концентрацией Н2О2 (рис. 3г). Ранее аналогичный результат был получен для ZSM-5(Si/Al = 15) [11], на основании чего авторы предположили участие гидропероксидной (–ООН) частицы в инициировании процесса окисления СН4 в первичный продукт – СН3ООН.
Полученные экспериментальные результаты позволяют сделать предположение, что пероксидное окисление метана на биметаллических Cu,Pd-содержащих цеолитах и монометаллическом Cu-ZSM-5 протекает по схожим маршрутам. Первоначально образуется метилгидропероксид, который далее окисляется до СН3ОН и муравьиной кислоты, а они затем окисляются до углекислого газа (схема 1 ). На металлическом палладии идет процесс неселективного разложения пероксида водорода с появлением дополнительного количества радикалов ОН·, что способствует образованию продуктов реакции глубокого окисления, в частности СО2. Следовательно, для достижения высокой селективности по СН3ОН и НСООН необходимо исключить факторы, способствующие образованию свободных радикалов ОН·: подъем температуры (>50°С) и высокие концентрации биметаллического катализатора Cu/Pd-ZSM-5, активного в разложении Н2О2.
Взаимосвязь между электронным и структурным состояниями активного компонента и каталитическими характеристиками Cu,Pd-ZSM-5
Сопоставление электронного и структурного состояний каталитически активных металлов в моно- и биметаллических образцах с обеспечиваемыми ими каталитическими характеристиками (конверсия метана и селективность по продуктам) в пероксидном окислении метана позволяет сделать несколько предположений.
Во-первых, прослеживается тенденция к увеличению достигаемых конверсий Н2О2 (от 47.5 до 80%) с ростом силы слабых Cu2+–ЛКЦ и Pd2+–ЛКЦ/Al3+–ЛКЦ, идентифицируемых по ТПД-NH3 в виде пиков десорбции при 309–350 и 275–285°С в образцах Cu,Pd-ZSM-5. Сила упомянутых центров в катализаторах изменяются в ряду: Cu/Pd-ZSM-5 (285/309°С) < Pd/Cu-ZSM-5 (275/318°С) < Cu–Pd/ZSM-5 (–/345°С). Их относительное количество изменяется слабо и находится в пределах 10–14% от всего количества центров, регистрируемых по десорбции NH3. Однако в последовательность изменения силы слабых Cu2+–ЛКЦ, обсуждаемую выше для биметаллических катализаторов, плохо вписывается монометаллический образец Cu-ZSM-5, который обеспечивает конверсию Н2О2 на уровне 30% против 47.5% на Cu/Pd-ZSM-5 при близкой силе слабых Cu2+–ЛКЦ (275/311°С).
Пик десорбции аммиака при 270–295°С в профиле ТПД-NH3 катализатора Cu-ZSM-5 связывают обычно с несколькими структурами, среди которых основными считаются слабые кислотные центры (SiOH) и внекаркасные катионы Al3+ [38]. С другой стороны, при разложении профиля ТПД-NH3 образца Cu-ZSM-5 всегда выделяют еще одну компоненту в области 320–400°С, которую приписывают разложению комплексов [Cu(NH3)4]2+ [38]. Смещение максимума в область меньших температур связывают [38] с изменением электронного состояния Cu2+ от изолированного иона до би/полиядерных структур с внекаркасным кислородом. Это предположение о структуре Cu2+–ЛКЦ в Cu-ZSM-5 можно перенести и на биметаллические катализаторы Cu,Pd-ZSM-5.
В таком случае природа пиков с максимумом при 311°С в профиле ТПД-NH3 Cu-ZSM-5 и 309–350°С в профиле Cu,Pd-ZSM-5 описывается структурами [Cu2O2]2+. Присутствие Pd2+, по-видимому, увеличивает силу Cu2+–ЛКЦ в составе [Cu2O2]2+ по сравнению с аналогичными центрами в монометаллическом катализаторе Cu-ZSM-5, но степень этого изменения зависит от способа введения активного компонента.
Вторая корреляция прослеживается между силой Cu2+–ЛКЦ, природа которых соответствует изолированным ионам Cu2+, и конверсией Н2О2. В биметаллических катализаторах вышеуказанные центры идентифицируются по пику десорбции NH3 в области от 355 до 415°С, то есть их сила больше, чем у структур [Cu2O2]2+ [38]. Судя по температурам десорбции NH3, сила центра возрастает в том же ряду катализаторов: Cu/Pd-ZSM-5 (355°С) < < Pd/Cu-ZSM-5 (395°С) < Cu–Pd/ZSM-5 (415°С).
Прямых корреляцией между силой БКЦ (SiOHAl), их количеством и каталитическими характеристиками биметаллических образцов в разложении Н2О2 и окислении СН4 в оксигенаты не обнаружено.
Таким образом, можно предположить, что чем больше сила Cu2+–ЛКЦ (как в составе структур [Cu2O2]2+, так и изолированных ионов Cu2+), тем легче он катализирует разложение Н2О2 (Cu‑ZSM-5 (${{Х}_{{{{{\text{Н}}}_{{\text{2}}}}{{{\text{О}}}_{{\text{2}}}}}}}$ = 30%) < Cu/Pd-ZSM-5 (47.5%) < < Pd/Cu-ZSM-5 (54%) < Cu–Pd/ZSM-5 (80%)) и генерацию радикалов ОН·, приводя к окислению первичных продуктов реакции – СН3ОН и НСООН – в СО2 (Cu-ZSM-5 (9% СО2) < Cu/Pd-ZSM-5 (10% СО2) < Pd/Cu-ZSM-5 (13% СО2) < Cu–Pd/ZSM-5 (20% СО2).
С учетом большей способности ионов Cu+ к взаимодействию с H2O2 в сравнении с Cu2+ [57], а также различия в активностях металлического Pd и PdO [28, 30], необходимо сопоставить окислительно-восстановительные свойства катализаторов. Как видно из рис. 1в, наиболее легко ионы Cu+ формируются при восстановлении водородом в катализаторе Cu–Pd/ZSM-5 – уже при 100°С. Для двух других биметаллических катализаторов соответствующие температуры несколько выше (130–150°С), но эти значения существенно ниже, чем для образца Cu-ZSM-5 (210°С). Отношения H2/Pd, определенные в экспериментах ТПВ-Н2 с катализаторами Cu,Pd-ZSM-5, свидетельствуют, что часть Pd в биметаллических катализаторах находится в металлическом состоянии. Таким образом, высокая склонность биметаллических катализаторов к формированию Cu+ и металлического Pd может также объяснять их высокую активность в разложении Н2О2 с формированием свободного ОН· радикала, активно участвующего в окислении первичных продуктов реакции пероксидного окисления метана.
ЗАКЛЮЧЕНИЕ
Установлено, что при одинаковом химическом составе биметаллического катализатора изменение последовательности введения активного компонента (катионов Cu2+ и Pd2+) методом поликонденсации в порах цеолита H-ZSM-5 позволяет контролировать кислотные и окислительно-восстановительные свойства образца и тем самым воздействовать на его каталитические характеристики в пероксидном окислении метана в оксигенаты.
Обнаружено, что последовательность введения каталитически активных металлов не влияет на формирующие при синтезе электронные состояния металлов. По данным ТПВ-Н2 и ЭСДО основными состояниями каталитически активного компонента являются ассоциаты изолированных ионов Cu2+/Pd2+ и полиядерные гидроксо/оксо кластеры ионов Cu2+/Pd2+, расположенные в каналах и на поверхности кристаллитов цеолита. Предполагается также присутствие Pd в металлическом состоянии. Соотношение между количеством изолированных ионов Cu2+/Pd2+ и количеством полиядерных структур ионов Cu2+/Pd2+ зависит от последовательности введения активных металлов в цеолит.
Выявлено, что катализатор Cu/Pd-ZSM-5, в который ионы Cu2+ вводили после Pd2+, способен превращать метан в метанол и муравьиную кислоту с большей эффективностью и селективностью, чем биметаллический образец Cu–Pd/ZSM-5, полученный введением Cu2+ и Pd2+ из совместного раствора. Образец Cu/Pd-ZSM-5 менее активен в разложении пероксида водорода, что позволяет минимизировать нежелательное окисление первичных продуктов окисления метана в СО2. Предполагается, что перечисленные различия в каталитических свойствах биметаллических образцов связаны с меньшей силой Cu2+–ЛКЦ и с большей устойчивостью ионов Cu2+ и полиядерных PdO-подобных кластеров к восстановлению в образце Cu/Pd-ZSM-5, чем в катализаторе Cu–Pd/ZSM-5.
В сравнении с монометаллическим катализатором Cu-ZSM-5, ассоциированные ионы Cu2+ легче восстанавливаются до Cu+ (при 100–155°С вместо 210–275°С) в биметаллических катализаторах Cu,Pd-ZSM-5. Сила Cu2+–ЛКЦ (в составе структур [Cu2O2]2+ и изолированных ионов Cu2+) оказалась выше в биметаллических катализаторах. Оба фактора влияют на образование свободных радикалов ОН· при разложении Н2О2, приводя к нежелательному окислению продуктов реакции – СН3ОН и НСООН – в СО2 в присутствии биметаллических катализаторов.
Список литературы
Rosenzweig A.C., Nordlund P., Takahara P.M., Frederick C.A., Lippard S.J. // Chem. Biol. 1995. V. 2. P. 409.
Lieberman R.L., Rosenzweig A.C. // Nature. 2005. V. 434. P. 177.
Chen K.H.-C., Chen C.-L., Tseng C.-F., Yu S.S.-F., Ke S.-S., Lee J.-F., Nguyen H.-T., Elliott S.J., Alben J.O., Chan S.I. // J. Chin. Chem. Soc. 2004. V. 51. P. 1081.
Smeets P.J., Woertink J.S., Sels B.F., Solomon E.I., Schoonheydt R.A. // Inorg. Chem. 2010. V. 49. P. 3573.
Olivos-Suarez A.I., Szecsenyi A., Hensen E.J.M., Ruiz-Martinez J., Pidko E.A., Gascon J. // ACS Catal. 2016. V. 6. P. 2965.
Smeets P.J., Groothaert M.H., Schoonheydt R.A. // Catal. Today. 2005. V. 110. P. 303.
Vanelderen P., Hadt R.G., Smeets P.J., Solomon E.I., Schoonheydt R.A., Sels B.F. // J. Catal. 2011. V. 284. P. 157.
Kulkarni A.R., Zhao Z., Siahrostami S., Norskov J.K., Studt F. // Catal. Sci. Technol. 2018. V. 8. P. 114.
Grundner S., Markovits M.A.C., Li G., Tromp M., Pidko E.A., Hensen E.J.M., Jentys A., Sanchez-Sanchez M., Lercher J.A. // Nature Commun. 2015. V. 6. P. 1.
Gabrienko A.A., Kolganov A.A., Arzumanov S.S., Yashnik S.A., Kriventsov V.V., Freude D., Stepanov A.G. // J. Phys. Chem. C. 2021. V. 125. P. 2182.
Hammond C., Jenkins R.L., Dimitratos N., Lopez-Sanchez J.A., Ab Rahim M.H., Forde M.M., Thetford A., Murphy D.M., Hagen H., Stangland E.E., Moulijn J.M., Taylor S.H., Willock D.J., Hutchings G.J. // Chem. A. Eur. J. 2012. V. 18. P. 15735.
Hammond C., Forde M.M., Ab Rahim M.H., Thetford A., He Q., Jenkins R.L., Dimitratos N., Lopez-Sanchez J.A., Dummer N.F., Murphy D.M., Carley A.F., Taylor S.H., Willock D.J., Stangland E.E., Kang J., Hagen H., Kiely C.J., Hutchings G.J. // Angew. Chem. Int. Ed. 2012. V. 51. P. 5129.
Hammond C., Dimitratos N., Lopez-Sanchez J.A., Jenkins R.L., Whiting G., Kondrat S.A., Ab Rahim M.H., Forde M.M., Thetford A., Hagen H., Stangland E.E., Moulijn J.M., Taylor S.H., Willock D.J., Hutchings G.J. // ACS Catalysis. 2013. V. 3. P. 1835.
Hammond C., Dimitratos N., Jenkins R.L., Lopez-Sanchez J.A., Kondrat S.A., Ab Rahim M.H., Forde M.M., Thetford A., Taylor S.H., Hagen H., Stangland E.E., Kang J.H., Moulijn J.M., Willock D.J., Hutchings G.J. // ACS Catal. 2013. V. 3. P. 689.
Hammond C., Hermans I., Dimitratos N. // ChemCatChem. 2015. V. 7. P. 434.
Taran O.P., Yashnik S.A., Boltenkov V.V., Parkhomchuk E.V., Sashkina K.A., Ayusheev A.B., Babushkin D.E., Parmon V.N. // Top. Catal. 2019. V. 62. № 5–6. P. 491.
Yashnik S.A., Boltenkov V.V., Babushkin D.E., Taran O.P., Parmon V.N. // Top. Catal. 2020. V. 63. № 1. P. 203.
Kuzmin A.O., Elizarova G.L., Matvienko L.G., Savinova E.R., Parmon V.N. // Mendeleev Comm. 1998. № 6. P. 210.
Elizarova G.L., Matvienko L.G., Kuzmin A.O., Savinova E.R., Parmon V.N. // Mendeleev Comm. 2001. V. 11. P. 15.
Елизарова Г.Л., Одегова Г.В., Матвиенко Л.Г., Талзи Е.П., Коломейчук В.Н., Пармон В.Н. // Кинетика и катализ. 2003. Т. 44. № 2. С. 227.
Яшник С.А., Исмагилов З.Р. // Кинетика и катализа. 2016. Т. 57. № 6. С. 777. (Yashnik S.A., Ismagilov Z.R. // Kinet. Catal. 2016. V. 57. № 6. P. 776.)
Yashnik S., Ismagilov Z. // Appl. Catal. B: Env. 2015. V. 170–171. P. 241.
Yashnik S., Ismagilov Z. // Appl. Catal. A: Gen. 2021. V. 615. P. 118054.
Яшник С.А., Ануфриенко В.Ф., Сазонов В.А., Исмагилов З.Р., Пармон В.Н. // Кинетика и катализ. 2012. Т. 53. № 3. С. 377. (Yashnik S.A., Anufrienko V.F., Sazonov V.A., Ismagilov Z.R., Parmon V.N. // Kinet. Catal. 2012. V. 53. № 3. P. 363.)
Jiang X., Wang X., Nie X., Koizumi N., Guo X., Song C. // Catal. Today. 2018. V. 316. P. 62.
Yang J., Fan Y., Li Z.-L., Peng Z., Yang J.-H., Liu B., Liu Z. // Mol. Catal. 2020. V. 492. P. 110992.
Menegazzo F., Signoretto M., Ghedini E., Strukul G. // Catalysts. 2019. V. 9. P. 251.
Gaikwad A.G., Sansare S.D., Choudhary V.R. // J. Mol. Catal. A: Chem. 2002. V. 181. P. 143.
Choudhary V.R., Samanta C., Jana P. // Appl. Catal. A: Gen. 2007. V. 332. P. 70.
Wang F., Xia C., De Visser S.P., Wang Y. // J. Am. Chem. Soc. 2019. V. 141. P. 901.
Ab Rahim M.H., Forde M.M., Jenkins R.L., Hammond C., He Q., Dimitratos N., Lopez-Sanchez J.A., Carley A.F., Taylor S.H., Willock D.J., Murphy D.M., Kiely C.J., Hutchings, G.J. // Angew. Chem. Int. Ed. 2012. V. 52. № 4. P. 1280.
Ab Rahim M.H., Armstrong R.D., Hammond C., Dimitratos N., Freakley S.J., Forde M.M., Morgan D.J., Lalev G., Jenkins R.L., Lopez-Sanchez J.A., Taylor S.H., Hutchings G.J. // Catal. Sci. Technol. 2016. V. 6. P. 3410.
Lewis R.J., Bara-Estaun A., Agarwal N., Freakley S.J., Morgan D.J., Hutchings G.J. // Catal. Lett. 2019. V. 149. № 11. P. 3066.
Huang W., Zhang S., Tang Y., Li Y., Nguyen L., Li Y., Shan J., Xiao D., Gagne R., Frenkel A.I., Tao F.F. // Angew. Chem. Int. Ed. 2016. V. 55. P. 13441.
Park E.D., Hwang Y.-S., Lee C.W., Lee J.S. // Appl. Catal. A: Gen. 2003. V. 247. P. 269.
Kang J., Puthiaraj P., Ahn W., Park E.D. // Appl. Catal. A: Gen. 2020. V. 602. P. 117711.
Yashnik S.A., Salnikov A.V., Vasenin N.T., Anufrienko V.F., Ismagilov Z.R. // Catal. Today. 2012. V. 197. P. 214.
Yashnik S.A., Ismagilov Z.R. // Top. Catal. 2019. V. 62. P. 179.
Sendel E. Colorimetric Determination of Traces of Metals. New York: Interscience Publishers, Inc., 1959.
Брек Д. Цеолитовае молекулярные сита. Москва: Мир, 1976. 781 с.
Baerlocher Ch., Meier W.M., Olson D.H. Atlas of zeolite framework types, 5th revised edn. Amsterdam: Elsevier, 2001. 132 p.
Krivoruchko O.P., Larina T.V., Shutilov R.A., Gavrilov V.Yu., Yashnik S.A., Sazonov V.A., Molina I.Yu., Ismagilov Z.R. // Appl. Catal. B: Env. 2011. V. 103. P. 1.
Yashnik S.A., Anufrienko V.F., Ismagilov Z.R. // Catal. Today. 2005. V. 110. P. 320.
Gaspar A.B., Dieguez L.C. // Appl. Catal. A: Gen. 2000. V. 201. P. 241.
Lomot D., Juszczyk W., Pielaszek J., Kaszkur Z., Bakuleva T.N., Karpinski Z. // New J. Chem. 1995. V. 19. P. 263.
Adelman B.J., Sachtler W.M.H. // Appl. Catal. B: Env. 1997. V. 14. P. 1.
Pieterse J.A.Z., Booneveld S. // Appl. Catal. B: Env. 2007. V. 73. P. 327.
Gu Y., Zelinsky R.P., Chen Y.-R., Epling W.S. // Appl. Catal. B: Env. 2019. V. 258. P. 118032.
Homeyer S.T., Sachtler W.M.H. // J. Catal. 1989. V. 117. P. 91.
Reifsnyder S.N., Otten M.M., Lamb H.H. // Catal. Today. 1998. V. 39. 317.
Yashnik S.A., Urzhuntsev G.A., Stadnichenko A.I., Svintsitskiy D.A., Ishchenko A.V., Boronin A.I., Ismagilov Z.R. // Catal. Today. 2019. V. 323. P. 257.
Yashnik S.A., Ismagilov Z.R. // Top. Catal. 2012. V. 55. P. 818.
Яшник С.А., Суровцова Т.А., Ищенко А.В., Каичев В.В., Исмагилов З.Р. // Кинетика и катализа. 2016. Т. 57. № 4. С. 535. (Yashnik S.A., Surovtsova T.A., Ishchenko A.V., Kaichev V.V., Ismagilov Z.R. // Kinet. Catal. 2016. V. 57. № 4. P. 528.)
Lesiak M., Binczarski M., Karski S., Maniukiewicz W., Rogowski J., Szubiakiewicz E., Berlowska J., Dziugan P., Witonska I. // J. Mol. Catal. A: Chem. 2014. V. 395. P. 337.
Liu Y., He Y., Zhou D., Feng J., Li D. // Catal. Sci. Technol. 2016. V. 6. P. 3027.
Намиот А.Ю. Растворимость газов в воде. Справочное пособие. Москва: Изд. Недра, 1991. 167 с.
Masarwa M., Cohen H., Meyerstein D., Hickman D.L., Bakac A., Espenson J.H. // J. Am. Chem. Soc. 1988. V. 110. P. 4293.
Дополнительные материалы отсутствуют.
Инструменты
Кинетика и катализ