

Кинетика и катализ, 2022, T. 63, № 5, стр. 592-601
Двухкомпонентная Ni–Mg–O/V–Mg–O каталитическая система. I. Синтез, физико-химические и каталитические свойства в окислительном дегидрировании этана
Г. Б. Веселов a, *, Е. В. Ильина a, М. В. Тренихин b, А. А. Ведягин a
a ФГБУН Институт катализа им. Г.К. Борескова СО РАН
630090 Новосибирск, просп. Акад. Лаврентьева, 5, Россия
b Центр новых химических технологий Института катализа им. Г.К. Борескова СО РАН
644040 Омск, ул. Нефтезаводская, 54, Россия
* E-mail: g.veselov@catalysis.ru
Поступила в редакцию 20.04.2022
После доработки 22.05.2022
Принята к публикации 24.05.2022
- EDN: UPLMUP
- DOI: 10.31857/S045388112205015X
Аннотация
В настоящей работе оксидная система Ni–Mg–O исследована в составе совместной каталитической системы Ni–Mg–O/V–Mg–O в реакции окислительного дегидрирования этана. Для синтеза образцов использован золь–гель-метод, что позволило достичь высоких текстурных характеристик системы и равномерного распределения частиц NiO в матрице оксида магния. Продемонстрировано, что добавление Ni–Mg–O-компоненты приводит к существенному росту выхода этилена при температурах 600–650°C. При этом значительного снижения селективности не происходит. Окислительно-восстановительные свойства Ni–Mg–O изучены в циклах температурно-программируемого восстановления в атмосфере водорода или монооксида углерода и реокисления кислородом воздуха. Показано, что система Ni–Mg–O способна воспроизводимо поглощать водород и конвертировать CO, что позволяет рассматривать ее в качестве перспективного агента для химических циклов. Наблюдаемое увеличение выхода этилена обусловлено поглощением водорода, выделяемого на стадии дегидрирования этана, и смещением термодинамического равновесия.
ВВЕДЕНИЕ
На сегодняшний день дегидрирование углеводородов по-прежнему является одним из важнейших промышленных процессов. Реакция дегидрирования этана эндотермическая (ΔH298 = = 136 кДж/моль) и требует высоких температур для достижения приемлемых значений конверсии [1]. В каталитическом дегидрировании углеводородов наиболее распространены нанесенные катализаторы на основе оксида хрома или платины с небольшими добавками таких металлов как Sn, Zn, Ga и т.д. [2–6]. В ходе реакции катализаторы дезактивируются в основном за счет образования кокса, поэтому требуется проводить регенерацию катализатора кислородом воздуха. Это может приводить к достаточно большим выбросам оксидов углерода в атмосферу, что обусловливает дополнительные затраты на очистку отходящих газов [7, 8].
Альтернативой является окислительное дегидрирование, когда в реактор совместно с углеводородом подается кислород. Углеродные отложения в этом случае не образуются, однако для таких процессов характерна низкая селективность, связанная с побочными реакциями окисления углеводородов до CO и CO2. Катализаторами для процессов окислительного дегидрирования в основном служат оксиды переходных металлов, таких как ванадий, молибден, кобальт и т.д. [9–13]. Также вызывает опасения безопасность этого процесса, поскольку смеси углеводородов и кислорода способны легко самовоспламеняться. Избежать прямого контакта кислорода и реакционной смеси позволяет технология химических циклов, где в качестве окислителя используется не кислород воздуха, а твердофазные переносчики кислорода [12, 14–17]. После израсходования решеточного кислорода эти материалы подвергаются регенерации в параллельном реакторе. В последние годы такой подход привлекает все большее внимание исследователей, в том числе для получения этилена из этана, ввиду его высокой экологичности [18–24]. Оксид магния представляется перспективным носителем для переносчиков кислорода в химических циклах, что связано с его термической стабильностью и отсутствием фазовых переходов [12, 16]. Известно также, что MgO широко используется для улавливания CO2 в других технологиях химических циклов за счет обратимого образования карбонатов [25].
Ранее было показано, что конверсия алканов в алкены может быть значительно повышена благодаря применению мембранных реакторов [26–28]. Селективные мембраны, проницаемые только для водорода, позволяют удалять водород из реактора и тем самым смещать термодинамическое равновесие реакции в сторону образования продуктов. Совмещение данного процесса с окислением водорода на внешней стороне мембраны дополнительно способствует увеличению выхода этилена.
Некоторыми авторами был предложен альтернативный подход, который основан на протекании процессов дегидрирования углеводорода и окисления на разных активных центрах одного и того же катализатора. Таким образом, удается совместить благоприятные термодинамические условия процесса окислительного дегидрирования с достаточно высокой селективностью процесса неокислительного дегидрирования. Так, в работе [29] были испытаны нанесенные катализаторы на основе оксида никеля и цеолитов. Такие системы содержат два типа форм никеля – изолированные ионы Ni2+, обладающие высокой кислотностью по Льюису и активностью в дегидрировании этана, и кластеры NiO, способные к поглощению водорода. Энергия связи кислорода в NiO достаточно высока, что препятствует протеканию избыточного окисления этилена, в результате чего удается достичь практически 100% селективности по целевому продукту.
В настоящей работе предложен и испытан комбинированный подход, заключающийся в применении для окислительного дегидрирования этана в этилен механической смеси катализатора и сокатализатора, выполняющего функцию акцептора водорода. В качестве катализатора дегидрирования был использован образец V–Mg–O, ранее хорошо зарекомендовавший себя в окислительном дегидрировании пропана [13, 30, 31]. Несмотря на большую распространенность платиновых и хром-оксидных катализаторов в неокислительном дегидрировании, некоторые авторы отмечают высокую термическую стабильность ванадий-оксидных катализаторов и их пониженную склонность к образованию кокса [32–34]. В качестве акцептора водорода была использована оксидная система Ni–Mg–O, которая, как было показано ранее, демонстрирует стабильное поведение в окислительных и восстановительных средах при повышенных температурах. Синтезированные образцы катализатора V–Mg–O и сокатализатора Ni–Mg–O были охарактеризованы методами низкотемпературной адсорбции азота, электронной спектроскопии диффузного отражения (ЭСДО), рентгенофазового анализа (РФА), просвечивающей электронной микроскопии (ПЭМ), ТПВ-H2 и ТПВ-CO и испытаны в модельной реакции окислительного дегидрирования этана.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Приготовление образцов
Синтез чистого оксида магния проводили с помощью золь–гель-метода. Сначала 2 г магния растворяли в 86 мл метанола при температуре 45°C и интенсивном перемешивании. К полученному раствору метоксида магния добавляли 86 мл толуола и далее проводили гидролиз, прикапывая 6.7 мл дистиллированной воды. Полученный гель перемешивали в течение 1 ч, а затем высушивали в течение 2 ч при комнатной температуре. После этого образец подвергали термической обработке. Для удаления большей части органических примесей образец сначала нагревали до 200°C со скоростью 2°C/мин, после чего повышали температуру до 500°C со скоростью 1°C/мин и выдержали при ней 1 ч. Полученный таким образом образец служил для разбавления катализатора и сокатализатора с целью обеспечения постоянства объема образца, загружаемого в каталитический реактор.
Синтез оксидной системы Ni–Mg–O осуществляли по схожей методике. В этом случае гидролиз проводили не водой, а предварительно приготовленным раствором нитрата никеля в воде. Для этого 2.28 г Ni(NO3)2‧6H2O растворяли в 5.82 г дистиллированной воды. Сушку и прокаливание выполняли по вышеописанной процедуре. Содержание NiO в полученной оксидной системе Ni–Mg–O составляет 15 мас. %.
В качестве образца сравнения использовали массивный NiO, синтезированный прокаливанием соли Ni(NO3)2·6H2O при 500°C в течение 1 ч. Скорость нагрева до указанной температуры составляла 1°C/мин.
Образец катализатора V–Mg–O синтезировали следующим способом: 2.4 г магниевой ленты растворяли в 90 мл метанола. Смесь грели 1 ч при постоянном перемешивании, после чего добавляли толуол (100 мл). Далее проводили медленный гидролиз добавлением 3.6 мл дистиллированной воды. Через час после образования геля гидроксида магния приливали раствор предшественника активного компонента (1.04 г ацетилацетоната ванадила в смеси 20 мл метанола, 10 мл толуола и 1.8 мл H2O). На следующий день полученный совместный гель помещали в автоклав и нагревали со скоростью 1.3°C/мин, что обеспечивало удаление растворителя при сверхкритических условиях. Конечное давление в автоклаве составляло 80 атм, а температура – 265°C. Затем ванадий-магниевый аэрогель помещали в муфельную печь и нагревали со скоростью 1°C/мин до 500°C и выдерживали при этой температуре 1 ч.
Исследование образцов физико-химическими методами
Изотермы адсорбции азота были записаны при 77 К с помощью прибора ASAP-2400 (“Micromeritics”, США). Из полученных изотерм были рассчитаны значения удельной поверхности с помощью метода Брунауэра–Эметта–Теллера (БЭТ). Значения объема пор и среднего радиуса пор были определены методом Баррета–Джойнера–Халенды.
Спектры диффузного отражения были зарегистрированы в интервале длин волн 190–800 нм с использованием спектрофотометра Varian Cary 300 UV/VIS Bio (“Agilent”, США) с интегрирующей сферой DRA-CA-3300 (“Labsphere”, США). Полученные спектры были преобразованы в функцию Кубелки–Мунка F(R) = (1 − R)2/2R, где R – экспериментально измеренные значения отражения образца. Ширину запрещенной зоны Eg определяли по методике Тауца для прямых разрешенных переходов по спектрам диффузного отражения.
Рентгенофазовый анализ (РФА) образцов проводили на X’TRA (“Thermo ARL”, Швейцария) с излучением CuKα (Ni-фильтр) при длине волны 1.54178 Å. Для определения фазового состава выполняли сканирование в диапазоне углов 2θ от 15° до 85° с шагом 0.05°.
Снимки просвечивающей электронной микроскопии были получены на приборе JEM 2100 (“JEOL”, Япония) при ускоряющем напряжении 200 кВ, разрешение – 0.14 нм.
В экспериментах по температурно-программированному восстановлению в атмосфере водорода (ТПВ-H2) образец помещали в кварцевый реактор, подавали реакционную смесь (10% H2, 90% N2) с объемной скоростью 57.8 мл/мин, после чего реактор нагревали от 30 до 700°C со скоростью 10°C/мин и выдерживали 15 мин при 700°C. Концентрацию водорода (мол. %) определяли с помощью газоанализатора ГАММА-100 (“Аналитприбор”, Россия) с детектором по теплопроводности. Далее реактор охлаждали до комнатной температуры. Для реокисления образца в реактор подавали поток воздуха. Реактор нагревали со скоростью 20°C/мин до 500°C, после чего выдерживали при этой температуре 30 мин. Циклы восстановления/окисления повторяли 5 раз в случае массивного NiO и 9 раз для системы Ni–Mg–O.
Циклические эксперименты по температурно-программированному восстановлению в атмосфере монооксида углерода (ТПВ-CO) проводили схожим образом. Реакционная смесь содержала 10% CO в азоте, объемная скорость ее подачи составляла 57.8 мл/мин. Концентрацию оксидов углерода CO и CO2 (мол. %) определяли с помощью газоанализатора ГАММА-100 (“Аналитприбор”, Россия) с двумя оптико-акустическими каналами. Для системы Ni–Mg–O было проведено семь циклов восстановления/окисления, для массивного NiO – пять циклов.
Тестирование образцов в модельной реакции окислительного дегидрирования этана
Каталитическая система Ni–Mg–O/V–Mg–O и образцы сравнения (MgO; Ni–Mg–O/MgO; V–Mg–O/MgO) были испытаны в модельной реакции окислительного дегидрирования этана. В каждом эксперименте в кварцевый реактор загружали 100 мг катализатора и 100 мг сокатализатора либо чистого оксида магния. Образцы загружали в реактор в виде гранул размером 0.25–0.5 мм. В каждом эксперименте образцы нагревали в потоке гелия (50 мл/мин) до 450°C со скоростью 10°C/мин, далее в реактор подавали реакционную смесь (5% этана в гелии; 100 мл/мин). Активность образцов исследовали в температурном интервале 450–700°C с шагом 25°C. Состав газовой смеси на выходе из реактора анализировали с помощью хроматографа Кристалл 2000М (“Хроматэк”, Россия). При каждой температуре производили 5 отборов проб реакционной смеси с интервалом 8 мин. Значения площадей пиков усредняли.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Исследование образцов физико-химическими методами
В настоящее время хорошо известно, что текстурные характеристики носителя оказывают решающее влияние на эффективность работы катализатора. Носитель должен обладать максимальной удельной поверхностью, а также оптимальной пористой структурой, чтобы обеспечить эффективный транспорт реагента к частицам активного компонента. Наилучших текстурных характеристик оксида магния удается добиться при использовании для его синтеза золь–гель-подхода.
Известно, что pH на стадии гидролиза метоксида магния оказывает важнейшее влияние на свойства конечного материала. Ранее было установлено, что лучшие текстурные характеристики в случае раствора нитрата никеля могут быть получены при pH 5 [8]. Так как система Ni–Mg–O должна обеспечить функцию переносчика кислорода в химических циклах, необходимо, чтобы материал содержал наибольшее количество доступного кислорода. Таким образом, чем выше концентрация NiO, тем более эффективной должна быть система. С другой стороны, излишне высокая концентрация оксида никеля существенно ухудшает текстурные характеристики оксида магния. Кроме того, значительно снижается дисперсность оксида никеля, что приводит к неэффективному использованию активного компонента при применении систем в качестве катализаторов. Как было показано ранее, оптимальная концентрация в случае систем Ni–Mg–O составляет 15 мас. % [35]. Удельная поверхность образца, определенная методом БЭТ, равна 137 м2/г, а объем пор – 1.40 см3/г. К преимуществам золь–гель-метода синтеза двухкомпонентных систем следует отнести равномерность распределения оксида никеля по объему образца. Наиболее наглядно это видно на микроскопических снимкам (рис. 1).

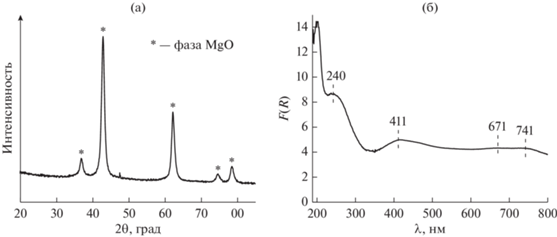
Следует отметить, что взаимодействие NiO с MgO с образованием твердых растворов замещения NixMg1–xO позволяет предотвратить спекание частиц оксида [8, 36]. В случае системы Ni–Mg–O формирование твердых растворов может быть доказано методами рентгенофазового анализа и электронной спектроскопии диффузного отражения. Как видно из РФА-дифрактограммы (рис. 2а), несмотря на достаточно высокую концентрацию NiO (15 мас. %), для образца Ni–Mg–O наблюдаются только рефлексы MgO. Рассчитанное значение параметра решетки составляет 4.220 нм. Спектр диффузного отражения образца в области 200–800 нм представлен на рис. 2б. Полосы при 411, 671 и 741 нм относят d–d-переходам Ni2+ в октаэдрическом положении. Полоса поглощения при ~240 нм характеризует перенос заряда O2−(2p) → Ni2+(3d) и определяет ширину запрещенной зоны частиц NixMg1-xO. Рассчитанное значение (3.97 эВ) согласно литературным данным [37] соответствует частицам состава Ni0.85Mg0.15O.
Рис. 2.
Дифрактограмма РФА образца Ni–Mg–O (а); спектр диффузного отражения в области 200–800 нм для образца Ni–Mg–O (б).
Микроскопические снимки для образца Ni–Mg–O, демонстрирующие морфологию материала, представлены на рис. 3. Видно, что образец состоит из кристаллитов неопределенной формы с размером 5–10 нм (рис. 3а). Эти кристаллиты формируют пористую структуру образца. Средний радиус пор равен 41 нм. Частицы твердого раствора NixMg1–xO хорошо различимы на рис. 3б и 3в. Средний размер таких частиц оценочно составляет 7 нм.

В качестве катализатора дегидрирования в настоящей работе выбрана система V–Mg–O, приготовленная по аэрогельной методике. Системы такого типа показали высокую активность в реакции окислительного дегидрирования пропана [30, 31]. Использованный образец характеризуется высокими значениями удельной поверхности (351 м2/г) и объема пор (3.05 см3/г).
Тестирование образцов в модельной реакции окислительного дегидрирования этана
Кривые зависимости конверсии этана от температуры приведены на рис. 4. Первый эксперимент проведен в отсутствие катализатора – в реактор было загружено 200 мг чистого оксида магния. Как видно из соответствующей кривой на рис. 4а, реакция начинается при 650°C, а максимальное значение конверсии (4.3%) достигается при 700°C. Известно, что при высоких температурах реакция способна протекать без катализатора в газовой фазе. На катализаторе V–Mg–O конверсия этана фиксируется уже при 550°C. При повышении температуры этот показатель также увеличивается. Для системы Ni–Mg–O наблюдается схожая картина. Этот образец демонстрирует меньшую активность по сравнению с V–Mg–O при Т < 675°C, но более активен при высоких температурах. Активность Ni–Mg–O в реакции дегидрирования обусловлена, предположительно, протеканием процесса окислительного дегидрирования с участием решеточного кислорода NiO и процесса дегидрирования на металлическом никеле, образующемся в результате восстановления оксида.
Рис. 4.
Кривые зависимости конверсии этана (а), выхода этилена (б), селективности по этилену (в) и селективности по метану (г) от температуры.

Картина значительно меняется при загрузке в реактор механической смеси V–Mg–O и Ni–Mg–O. При температурах 600–650°C отмечается существенный рост конверсии этана, которая достигает максимального значения 12.8% при 625°C. По мере расходования решеточного кислорода NiO и восстановления оксида до металлического никеля предположительно происходит закоксовывание частиц никеля. При 675–700°C значения конверсии этана практически совпадают с таковыми для катализатора V–Mg–O. Стоит отметить, что наблюдаемое повышение конверсии этана нельзя объяснить простым “сложением” активностей двух систем. В противном случае максимальный выход этилена при температуре 625°C должен был бы составить 8.7% (рис. 4б).
На рис. 4в и 4г представлены кривые зависимости селективности образования этилена и метана от температуры. Остановимся на них подробнее. Для чистого MgO селективность по этилену максимальна за счет низкой конверсии этана, а образования метана практически не происходит. В случае катализатора V–Mg–O значения селективности образования этилена ниже, что может быть связано как с отложением углерода, так и с окислением этилена до оксидов углерода. Для этого образца также характерно образование достаточно большого количества метана, которое снижается с температурой. Для оксидной системы Ni–Mg–O наблюдается наименьшая селективность по этилену. Это объясняется относительно высокой склонностью никеля к закоксовыванию. Отметим, однако, что среди никелевых катализаторов, нанесенных на различные носители, самая низкая способность к образованию углерода отмечена в случае MgO [38]. Максимальная селективность по этилену для образца Ni–Mg–O зафиксирована при 650°C. Селективность образования метана возрастает с температурой, что может быть связано с рядом причин. Во-первых, поскольку реакция каталитического разложения метана (I) является обратимой, углеродные отложения могут взаимодействовать с водородом, появляющимся в результате дегидрирования этана. Другой вероятной причиной увеличения селективности по метану может быть реакция гидрогенолиза этана на металлическом никеле (II).
(I)
${\text{C}} + 2{{{\text{H}}}_{{\text{2}}}} \rightleftarrows {\text{C}}{{{\text{H}}}_{{\text{4}}}}{\text{,}}$(II)
${{{\text{C}}}_{{\text{2}}}}{{{\text{H}}}_{6}} + {{{\text{H}}}_{2}} \to 2{\text{C}}{{{\text{H}}}_{4}}.$При сопоставлении значений селективности образования этилена для исследованных образцов можно отметить, что совместная система Ni–Mg–O/V–Mg–O превосходит по данному параметру образец Ni–Mg–O и практически приближается к катализатору V–Mg–O. Селективность по метану для Ni–Mg–O/V–Mg–O заметно ниже по сравнению c V–Mg–O при температурах 600–650°C, однако становится выше в интервале 675–700°C.
Можно выдвинуть несколько предположений, объясняющих наблюдаемое повышение выхода этилена в случае системы Ni–Mg–O/V–Mg–O. Во-первых, оксид никеля способен поглощать водород, отдавая решеточный кислород и восстанавливаясь до металлического состояния. Особенности поведения системы в циклах восстановления–окисления описаны в следующем разделе. Во-вторых, каталитическое дегидрирование этана может также проходить на поверхности металлического никеля. С другой стороны, никель способен катализировать реакцию гидрогенолиза этана, что должно приводить к снижению селективности по этилену и увеличению количества образующегося метана. Не следует также исключать из рассмотрения вклад никеля в процесс коксообразования. Протекание этих побочных процессов на системе Ni–Mg–O планируется рассмотреть на дальнейших этапах работы.
Изучение окислительно-восстановительного поведения системы Ni–Mg–O
Метод температурно-программированного восстановления водородом широко используется для характеризации катализаторов и наноструктурированных материалов. В некоторых случаях литературные данные позволяют четко соотнести температурные интервалы поглощения в ТПВ-H2 и восстанавливаемые в этом интервале формы активного компонента. Поведение образцов в ТПВ-H2 зависит от размера частиц активного компонента, фазового состава и типа взаимодействия с носителем.
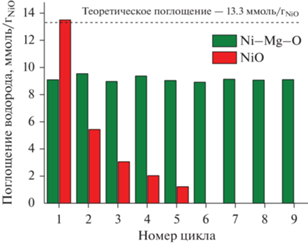
На рис. 5 показана динамика изменения величины поглощения водорода (ммоль/гNiO) в нескольких последовательных циклах ТПВ-H2 для системы Ni–Mg–O в сравнении с массивным оксидом никеля. Как видно из диаграммы, в случае массивного NiO происходит существенное снижение количества поглощаемого водорода от цикла к циклу. Это обусловлено спеканием частиц никеля, из-за чего он не может быть полностью окислен кислородом воздуха на стадии регенерации. В случае оксидной системы Ni–Mg–O матрица оксида магния препятствует спеканию дисперсных форм никеля, в результате чего наблюдается воспроизводимое поведение образца. Поглощение водорода лежит в интервале 8.9–9.6 ммоль/гNiO, что составляет ~70% от расчетного содержания кислорода в NiO (13.3 ммоль/гNiO). Поскольку конечная температура в экспериментах ТПВ-H2 составляет 700°C, часть никеля остается в виде твердого раствора NixMg1–xO, для восстановления которого требуются более высокие температуры.
Монооксид углерода является более мягким восстановителем, что позволяет получить дополнительную информацию о свойствах исследуемых образцов. На рис. 6а представлены результаты циклических экспериментов по температурно-программированному восстановлению монооксидом углерода (ТПВ-CO) для образца массивного оксида никеля. В первом цикле наблюдается один пик в интервале температур 300–450°C, связанный с восстановлением оксида никеля в металлический никель по реакции (III). Профиль выделения CO2 близок к профилю поглощения CO, однако значения концентрации CO2 оказываются ниже, чем ожидаемые при полном превращении CO по указанной реакции. Это свидетельствует о том, что часть монооксида углерода расходуется на образование кокса по реакции Будуара (IV), которая может протекать на металлических частицах никеля [39, 40]. Соответствующие расчетные данные представлены в табл. 1. Начиная с третьего цикла на кривой становятся видны максимумы поглощения, соответствующие, предположительно, восстановлению частиц оксида никеля разного размера. Суммарное поглощение за пять циклов снижается примерно в два раза, что связано со спеканием частиц, в результате которого часть оксида никеля становится недоступна для восстановителя.
Рис. 6.
Кривые поглощения CO (сплошные линии) и выделения CO2 (пунктирные линии) в циклических экспериментах по ТПВ-CO для массивного NiO (а) и оксидной системы Ni–Mg–O (б). Номера кривых соответствуют номерам циклов.

Таблица 1.
Результаты циклических экспериментов по ТПВ-CO на образцах массивного NiO и Ni–Mg–O
| Цикл | Массивный NiO | Ni–Mg–O | |||
|---|---|---|---|---|---|
| поглощение CO, ммоль/г | выделение CO2, ммоль/г | поглощение CO, ммоль/г | выделение CO2, ммоль/г | выделение CO2 за счет разложения карбонатов (оценка), ммоль/г | |
| 1 | 20.4 | 17.9 | 7.45 | 5.54 | 1.12 |
| 2 | 21.8 | 18.3 | 11.6 | 7.19 | 0.69 |
| 3 | 16.1 | 13.4 | 12.4 | 7.82 | 0.92 |
| 4 | 12.9 | 11.2 | 13.1 | 8.04 | 0.79 |
| 5 | 11.7 | 9.89 | 13.1 | 8.09 | 0.84 |
| 6 | – | – | 13.2 | 8.14 | 0.84 |
| 7 | – | – | 13.4 | 8.14 | 0.74 |
Образец Ni–Mg–O демонстрирует несколько иное поведение в циклах ТПВ-CO (рис. 6б). Поглощение монооксида углерода происходит в широком температурном интервале 360–700°C, а максимум наблюдается при 460°C. Выделение CO2 при этом находится на уровне 74% от ожидаемого. В последующих циклах восстановления–окисления наблюдается смещение максимума поглощения в низкотемпературную область. Количество поглощаемого CO увеличивается с каждым циклом. Так, в седьмом цикле поглощается в 1.8 раз больше CO, чем в первом. Можно сделать вывод о том, что взаимодействие оксида никеля с носителем ослабляется в результате восстановления и последующей регенерации. Это упрощает восстановление NiO в последующих циклах. Стоит также отметить, что в каждом последующем цикле увеличивается доля CO, затрачиваемого на реакцию диспропорционирования (IV). Предположительно в ходе циклов происходит разрушение фазы твердого раствора NixMg1–xO и образование дисперсных частиц NiO, что приводит к повышению активности образца в реакции (IV) и способствует росту углерода на этих частицах [40]. Представленные данные также свидетельствуют о том, что температура стадии окисления 500°C является достаточной для отжига кокса и регенерации катализатора, а дисперсность частиц NiO не претерпевает изменений в результате отжига.
ЗАКЛЮЧЕНИЕ
В настоящей работе изучен подход к реализации процесса дегидрирования этана, в котором совмещаются преимущества окислительного и неокислительного дегидрирования. Он заключается в использовании катализатора окислительного дегидрирования V–Mg–O совместно с оксидной системой Ni–Mg–O, выполняющей функцию акцептора водорода, в результате чего стадии дегидрирования этана и окисления выделяемого водорода разделяются. Для приготовления катализатора и оксидной системы был применен золь–гель-метод, что позволило обеспечить равномерность распределения активного компонента в матрице оксида магния, достичь высоких значений удельной поверхности и объема пор. Методом ЭСДО показано, что часть никеля в образце Ni–Mg–O находится в виде твердого раствора NixMg1–xO.
Показано, что совместное использование V–Mg–O и Ni–Mg–O ведет к существенному увеличению выхода этилена в диапазоне температур 600–650°C. Несмотря на то, что металлический никель также проявляет активность в исследуемой реакции, существенный вклад в наблюдаемый рост выхода этилена вносит поглощение водорода оксидом никеля. При более высоких температурах (675–700°C) происходит снижение конверсии до уровня, соответствующего катализатору V–Mg–O, что свидетельствует о закоксовывании частиц никеля и их исключении из участия в процессе.
Способность системы Ni–Mg–O воспроизводимо поглощать водород была подтверждена в циклических экспериментах ТПВ-H2. Наличие в реакционном объеме углерода (углеводороды, кокс) и кислорода (в составе оксида никеля) может приводить к образованию оксидов углерода. В связи с этим образцы были исследованы в ТПВ-CO. Показано, что система Ni–Mg–O способна поглощать CO с протеканием реакции восстановления NiO и реакции Будуара (диспропорционирования CO) с образованием углерода. При этом часть CO2 на стадии окислительной регенерации обратимо связывается в карбонаты магния.
Список литературы
Riley C.R., de La Riva A., Ibarra I.L., Datye A.K., Chou S.S. // Appl. Catal. A: Gen. 2021. V. 624. P. 118309.
Liu S., Zhang B., Liu G. // React. Chem. Eng. 2021. V. 6. № 1. P. 9.
Vincent R.S., Lindstedt R.P., Malik N.A., Reid I.A.B., Messenger B.E. // J. Catal. 2008. V. 260, № 1. P. 37.
Rao T.V.M., Zahidi E.M., Sayari A. // J. Mol. Catal. A: Chem. 2009. V. 301. № 1–2. P. 159.
Galvita V., Siddiqi G., Sun P., Bell A.T. // J. Catal. 2010. V. 271. № 2. P. 209.
Weckhuysen B.M., Schoonheydt R.A. // Catal. Today. 1999. V. 51. № 2. P. 223.
Karnaukhov T.M., Veselov G.B., Cherepanova S.V., Vedyagin A.A. // Materials. 2022. V. 15. № 6. P. 2021.
Veselov G.B., Karnaukhov T.M., Stoyanovskii V.O., Vedyagin A.A. // Nanomater. 2022. V. 12. № 6. P. 952.
Cavani F., Ballarini N., Cericola A. // Catal. Today. 2007. V. 127. № 1–4. P. 113.
Najari S., Saeidi S., Concepcion P., Dionysiou D.D., Bhargava S.K., Lee A.F., Wilson K. // Chem. Soc. Rev. 2021. V. 50. № 7. P. 4564.
Melzer D., Mestl G., Wanninger K., Zhu Y., Browning N.D., Sanchez-Sanchez M., Lercher J.A. // Nature Commun. 2019. V. 10. № 1. P. 4012.
Yusuf S., Haribal V., Jackson D., Neal L., Li F. // Appl. Catal. B: Env. 2019. V. 257. P. 117885.
Vedyagin A.A., Mishakov I. V., Ilyina E. V. // J. Sol–Gel Sci. Technol. 2020. V. 97. P. 117.
Tian Y., Dudek R.B., Westmoreland P.R., Li F. // Chem. Eng. J. 2020. V. 398. P. 125583.
Neal L.M., Haribal V.P., Li F. // Science. 2019. V. 19. P. 894.
Neal L.M., Yusuf S., Sofranko J.A., Li F. // Energy Technol. 2016. V. 4. № 10. P. 1200.
Fairuzov D., Gerzeliev I., Maximov A., Naranov E. // Catalysts. 2021. V. 11. № 7. P. 833.
Yusuf S., Neal L., Haribal V., Baldwin M., Lamb H.H., Li F. // Appl. Catal. B: Env. 2018. V. 232. P. 77.
Yusuf S., Haribal V., Jackson D., Neal L., Li F. // Appl. Catal. B: Env. 2019. V. 257. P. 117885.
Tian Y., Dudek R.B., Westmoreland P.R., Li F. // Chem. Eng. J. 2020. V. 398. P. 125583.
Ding W., Zhao K., Jiang S., Zhao Z., Cao Y., He F. // Appl. Catal. A: Gen. 2021. V. 609. P. 117910.
Wang T., Gao Y., Liu Y., Song M., Liu J., Guo Q. // Fuel. 2021. V. 303. P. 121286.
Luongo G., Donat F., Krödel M., Cormos C.-C., Müller C.R. // Renew. Sustain. Energy Rev. 2021. V. 149. P. 111403.
Tian X., Zheng C., Zhao H. // Appl. Catal. B: Env. 2022. V. 303. P. 120894.
Arastoopour H., Abbasian J. / Handbook of Chemical Looping Technology. 2019. P. 435.
Shelepova E.V., Vedyagin A.A., Mishakov I.V., Noskov A.S. // Int. J. Hydrogen Energy. 2015. V. 40. № 8. P. 3592.
Shelepova E.V., Vedyagin A.A., Mishakov I.V., Noskov A.S. // Chem. Eng. J. 2011. V. 176–177. P. 151.
Champagnie A.M., Tsotsis T.T., Minet R.G., Webster A.I. // Chem. Eng. Sci. 1990. V. 45. № 8. P. 2423.
Wang C., Yang B., Gu Q., Han Y., Tian M., Su Y., Pan X., Kang Y., Huang C., Liu H., Liu X., Li L., Wang X. // Nature Commun. 2021. V. 12. № 1. P. 5447.
Ilyina E.V., Mishakov I.V., Vedyagin A.A., Bedilo A.F. // J. Sol–Gel Sci. Technol. 2013. V. 68. № 3. P. 423.
Mishakov I.V., Ilyina E.V., Bedilo A.F., Vedyagin A.A. // React. Kinet. Catal. Lett. 2009. V. 97. № 2. P. 355.
Sokolov S., Stoyanova M., Rodemerck U., Linke D., Kondratenko E.V. // J. Catal. 2012. V. 293. P. 67.
Gu Y., Liu H., Yang M., Ma Z., Zhao L., Xing W., Wu P., Liu X., Mintova S., Bai P., Yan Z. // Appl. Catal. B: Env. 2020. V. 274. P. 119089.
Xie Y., Luo R., Sun G., Chen S., Zhao Z.-J., Mu R., Gong J. // Chem. Sci. 2020. V. 11. № 15. P. 3845.
Karnaukhov T., Vedyagin A., Mishakov I., Bedilo A., Volodin A. // Mater. Sci. Forum. 2018. V. 917. P. 157.
Karnaukhov T.M., Vedyagin A.A., Cherepanova S. V., Rogov V.A., Mishakov I.V. // J. Sol–Gel Sci. Technol. 2019. V. 92. № 1. P. 208.
Niedermeier C.A., Råsander M., Rhode S., Kachkanov V., Zou B., Alford N., Moram M.A. // Sci. Rep. 2016. V. 6. P. 31230.
Park C., Keane M.A. // J. Catal. 2004. V. 221. № 2. P. 386.
Trimm D.L. // Catal. Rev. 1977. V. 16. № 1. P. 155.
Chen P., Zhang H.B., Lin G.D., Hong Q., Tsai K.R. // Carbon. 1997. V. 35. № 10–11. P. 1495.
Li J., Zhang H., Gao Z., Fu J., Ao W., Dai J. // Energy & Fuels. 2017. V. 31. № 4. P. 3475.
Zhang L., Zheng Y., Guo Y., Bai S., Song M., Huang P., Wei X., Sun J., Li C., Zhang J., Zhao C. // ACS Appl. Energy Mater. 2021. V. 4. № 9. P. 9513.
Дополнительные материалы отсутствуют.
Инструменты
Кинетика и катализ