

Кинетика и катализ, 2022, T. 63, № 6, стр. 727-735
Соотношение между коэффициентами Бренстеда стадий гетерогенных каталитических реакций
А. П. Герасев *
ФГБУН ФИЦ Институт катализа им. Г.К. Борескова СО РАН
630090 Новосибирск, просп. Акад. Лаврентьева, 5, Россия
* E-mail: a.gerasev@ngs.ru
Поступила в редакцию 08.04.2022
После доработки 11.07.2022
Принята к публикации 20.07.2022
- EDN: XRFWAQ
- DOI: 10.31857/S0453881122060041
Аннотация
Рассмотрена термодинамика каталитического цикла в условиях воздействия реакционной среды на катализатор. Изменение энергии активации реакции в зависимости от изменения теплоты стадии реакции описывали корреляцией Бренстеда–Эванса–Поляни. Для двухстадийной схемы каталитической реакции (нелинейный механизм Или–Ридила) показана взаимосвязь коэффициентов Бренстеда стадий каталитической реакции. Для трехстадийной схемы каталитической реакции (нелинейный механизм Ленгмюра–Хиншельвуда) получено уравнение баланса, содержащее коэффициенты Бренстеда и приращения теплот стадий каталитической реакции, на основе которого были проанализированы некоторые частные случаи.
ВВЕДЕНИЕ
Наиболее существенной и специфической особенностью гетерогенного катализа является то, что не только катализатор воздействует на реагент, вызывая его химическое превращение, но и реагенты также воздействуют на катализатор и могут изменять его кинетические свойства [1–6]. Учет воздействия реакционной среды на катализатор весьма существенен для правильной трактовки многих явлений катализа. В то же время, как отмечал Г.К. Боресков [6], “бросается в глаза противоречие между распространенностью явлений неоднородности поверхности реальных катализаторов и очень малой долей кинетических уравнений, учитывающих неоднородность”. Неоднородность поверхности катализатора часто проявляется в изменении теплот адсорбции и энергий активации процессов адсорбции и химических превращений адсорбированных веществ. Во многих областях химической кинетики обнаружена взаимосвязь между кинетической стандартной свободной энергией активации и термодинамической стандартной свободной энергией реакции. Если пренебречь энтропийными эффектами, мы имеем дело только с активационным барьером элементарной стадии химической реакции и теплотой реакции. Если существует линейная зависимость между кинетикой и термодинамическими свойствами катализатора, изменение энергии активации может быть выражено уравнением [1, 3, 4, 7–13]:
Здесь ν – стехиометрический коэффициент расходования кислорода, Кв и Ко – объединяют величины, не зависящие от степени восстановленности катализатора, рв и ${{p}_{{{{{\text{O}}}_{2}}}}}$ – парциальные давления восстанавливающего реактанта и кислорода, α и β – коэффициенты Бренстеда стадий восстановления и реокисления катализатора. При этом важно отметить, что поверхностные свойства железосурьмяного катализатора исследовали импульсным микрокаталитическим методом с применением проточного калориметра [18, 19], а скорость каталитических реакций измеряли при постоянном известном составе катализатора, используя импульсную технику [20]. Для реакций окисления СО и окислительного дегидрирования бутена-1 на железосурьмяном катализаторе были определены энергия связи кислорода и параметры кинетических уравнений [6, 17]. Более того, в [21] было получено удовлетворительное совпадение экспериментальных и расчетных данных по динамике окисления СО в реакторе с виброожиженным слоем катализатора при вариациях состава реакционной смеси после выхода на стационарный режим. Важно отметить, что во всех приведенных выше публикациях вопрос о взаимосвязи коэффициентов Бренстеда стадий каталитической реакции (α и β) даже не обсуждался. Заметим также, что каталитические системы могут демонстрировать критические явления (множественность стационарных состояний, медленные релаксации, сложное динамическое поведение, автоколебания скорости реакции), для описания которых используются сложные нелинейные механизмы реакций [22–24]. При этом в основном используются математические модели с постоянными значениями энергии активации стадий реакции.
Основная цель настоящей работы – анализ термодинамики каталитического цикла в условиях воздействия реакционной среды на катализатор и выявление взаимосвязи между коэффициентами Бренстеда стадий каталитической реакции для простейших нелинейных механизмов.
ДВУХСТАДИЙНАЯ СХЕМА КАТАЛИТИЧЕСКОЙ РЕАКЦИИ
Каталитический цикл является важным принципом каталитического действия [1, 4]. Особое значение имеет тот факт, что в каталитическом цикле происходит промежуточное химическое взаимодействие отдельных веществ с реагентами. Из того факта, что свободная энергия катализатора не изменяется во время каталитического действия (каталитического цикла), следует, что сдвинуть равновесие химической реакции под действием катализаторов невозможно [4, 13]. Однако свободная энергия некоторых катализаторов может изменяться при вариации состава реакционной среды [3, 4, 8]. Например, в реакциях окисления на оксидах металлов энергия связи хемосорбированных атомов кислорода может возрастать при восстановлении катализатора [4, 8], что приводит к повышению энергии активации реакции.
Строго говоря, все химические реакции обратимы, но при этом следует обращать внимание на значения равновесных концентраций реагентов, которые могут быть исчезающе малыми. С практической точки зрения, различают реакции обратимые, протекающие одновременно в двух направлениях, и реакции необратимые, протекающие в одном направлении до тех пор, пока по крайней мере один из реагентов не будет полностью исчерпан. Рассмотрим для простоты необратимую экзотермическую реакцию типа 2A + O2 → → 2AO + q, которая протекает на поверхности катализатора по двухстадийной схеме (механизм Или–Ридеала или Марса–Ван-Кревелена) с тепловыми эффектами стадий, зависящими от степени покрытия поверхности катализатора адсорбированным (хемосорбированным) реагентом (интермедиатом)
Здесь ${{\theta }}$ – доля свободных участков поверхности катализатора, * – активный центр на поверхности катализатора, O* – адсорбированный атом вещества О2 (под веществом О2 понимаем двухатомные молекулы, включая кислород), q1(θ) и q2(θ) – тепловые эффекты стадий (I) и (II). Поскольку тепловой эффект брутто реакции является постоянной величиной $q = - {{{{\Delta }}}_{{\text{r}}}}H_{T}^{^\circ }$ = const (${{{{\Delta }}}_{{\text{r}}}}H_{T}^{^\circ }~$ – энтальпия реакции), в стационарном состоянии выполняется баланс(1)
$\begin{gathered} q = {{q}_{1}}\left( {{\theta }} \right) + 2{{q}_{2}}\left( {{\theta }} \right) = \\ = {{q}_{{1,0}}} + \Delta {{q}_{1}}\left( {{\theta }} \right) + 2\left( {{{q}_{{2,0}}} + \Delta {{q}_{2}}\left( {{\theta }} \right)} \right), \\ \end{gathered} $Важно отметить, что при анализе термодинамики каталитического цикла исходят из энергетического баланса, в отличие от кинетических (математических) моделей, где допустимо использовать различные порядки по реагентам (см., например, [25]).
Пусть теплота первой стадии химической реакции возрастает с увеличением доли свободной поверхности катализатора $~{{\theta }}{\text{.}}$ Эта теплота, по определению, равна сумме энтальпий образования исходных веществ (реакционная группа i) с вычетом суммы энтальпий образования продуктов реакции (реакционная группа j) (о понятии реакционная группа см., например, [26])
(3)
$\begin{gathered} {{q}_{1}}\left( {{\theta }} \right) = {{q}_{{1,0}}} + \Delta {{q}_{1}}\left( {{\theta }} \right) = - {{{{\Delta }}}_{{\text{r}}}}H_{{ij}}^{^\circ }\left( {{\theta }} \right) = \\ = - 2{{\Delta }_{{\text{f}}}}H_{{{\text{O}}*}}^{^\circ }\left( {{\theta }} \right) + 2{{\Delta }_{{\text{f}}}}H_{*}^{^\circ }\left( {{\theta }} \right) + {{\Delta }_{{\text{f}}}}H_{{{{{\text{O}}}_{2}}}}^{^\circ } = \\ = - 2\left( {{{\Delta }_{{\text{f}}}}H_{{{\text{O}}*,0}}^{^\circ } - {{\Delta }}\left( {{{\Delta }_{{\text{f}}}}H_{{{\text{O}}*}}^{^\circ }\left( {{\theta }} \right)} \right)} \right. - \\ - \,\,\left. {{{\Delta }_{{\text{f}}}}H_{{*,0}}^{^\circ } - {{\Delta }}\left( {{{\Delta }_{{\text{f}}}}H_{*}^{^\circ }\left( {{\theta }} \right)} \right)} \right) + {{\Delta }_{{\text{f}}}}H_{{{{{\text{O}}}_{2}}}}^{^\circ }. \\ \end{gathered} $(4)
$\Delta {{q}_{1}}\left( {{\theta }} \right) = 2\left( {{{\Delta }}\left( {{{\Delta }_{{\text{f}}}}H_{{{\text{O}}*}}^{^\circ }\left( {{\theta }} \right)} \right) + {{\Delta }}\left( {{{\Delta }_{{\text{f}}}}H_{*}^{^\circ }\left( {{\theta }} \right)} \right)} \right).$Как видно из уравнения (4), увеличение теплоты первой стадии химической реакции возможно только за счет повышения стандартной энтальпии образования свободных активных центров на поверхности катализатора и уменьшения стандартной энтальпии образования адсорбированных атомов О* (рис. 1).
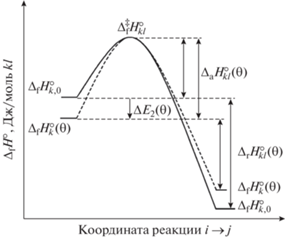
Рис. 1.
Схематичное представление протекания элементарной реакции ij с активационным барьером в традиционных энергетических координатах (стандартные значения параметров).

Эмпирическое уравнение Аррениуса устанавливает зависимость константы скорости химической реакции от температуры. Это уравнение содержит два параметра – предэкспоненциальный коэффициент, учитывающий вероятность и число столкновений, и энергию активации химической реакции, которые определяются по экспериментальным данным. Сходство функциональной формы уравнений Аррениуса и Эйринга [13, 26, 27] обеспечивает мост между кинетикой и термодинамикой. Для первой стадии химической реакции (адсорбции/хемосорбции) связь между энергией активации ${{E}_{1}}\left( {{\theta }} \right)$ и энтальпией активации ${{\Delta }_{{\text{a}}}}H_{{ij}}^{^\circ }\left( {{\theta }} \right)$ выразим уравнением
(5)
$\begin{gathered} {{E}_{1}}\left( {{\theta }} \right) = {{E}_{{1,0}}} + {{\Delta }}{{E}_{1}}\left( {{\theta }} \right) = {{\Delta }_{{\text{a}}}}H_{{ij}}^{^\circ }\left( {{\theta }} \right) + RT = \\ = {{\Delta }^{\ddag }}H_{{ij}}^{^\circ } - {{\Delta }_{{\text{f}}}}H_{{{{{\text{O}}}_{2}}}}^{^\circ } - 2{{\Delta }_{{\text{f}}}}H_{*}^{^\circ }\left( {{\theta }} \right) + RT = \\ = {{\Delta }^{\ddag }}H_{{ij}}^{^\circ } - {{\Delta }_{{\text{f}}}}H_{{{{{\text{O}}}_{2}}}}^{^\circ } - 2{{\Delta }_{{\text{f}}}}H_{{*,0}}^{^\circ } - \\ - \,\,2{{\Delta }}\left( {{{\Delta }_{{\text{f}}}}H_{*}^{^\circ }\left( {{\theta }} \right)} \right) + RT, \\ \end{gathered} $(6)
${{\Delta }}{{E}_{1}}\left( {{\theta }} \right) = - 2{{\Delta }}\left( {{{\Delta }_{{\text{f}}}}H_{*}^{^\circ }\left( {{\theta }} \right)} \right).$Далее представим изменение энергии активации первой стадии реакции в виде соотношения БЭП [1, 3, 4, 6–11]
(7)
${{\Delta }}{{E}_{1}}\left( {{\theta }} \right) = - {{{{\alpha }}}_{1}}{{\Delta }}{{q}_{1}}\left( {{\theta }} \right),$(8)
${{{{\alpha }}}_{1}}{{\Delta }}{{q}_{1}}\left( {{\theta }} \right) = 2{{\Delta }}\left( {{{\Delta }_{{\text{f}}}}H_{*}^{^\circ }\left( {{\theta }} \right)} \right).$Вычитая уравнение (8) из (4), приходим к выражению
(9)
$\left( {1 - {{{{\alpha }}}_{1}}} \right)\Delta {{q}_{1}}\left( {{\theta }} \right) = 2{{\Delta }}\left( {{{\Delta }_{{\text{f}}}}H_{{{\text{O}}*}}^{^\circ }\left( {{\theta }} \right)} \right).$Таким образом, уравнения (8) и (9) связывают изменение теплоты первой стадии реакции и коэффициент Бренстеда ${{{{\alpha }}}_{1}}$ с изменениями стандартных энтальпий образования активного центра поверхности и адсорбированного атома О*.
Далее рассмотрим вторую стадию реакции, энергия активации E2 и энтальпия активации ${{\Delta }_{{\text{a}}}}H_{{kl}}^{^\circ }$ которой связаны уравнением
(10)
$\begin{gathered} {{E}_{2}}\left( {{\theta }} \right) = {{E}_{{2,0}}} + {{\Delta }}{{E}_{2}}\left( {{\theta }} \right) = {{\Delta }_{{\text{a}}}}H_{{kl}}^{^\circ }\left( {{\theta }} \right) + RT = \\ = {{\Delta }^{\ddag }}H_{{kl}}^{^\circ } - {{\Delta }_{{\text{f}}}}H_{{\text{A}}}^{^\circ } - {{\Delta }_{{\text{f}}}}H_{{{\text{O}}*}}^{^\circ }\left( \theta \right) + RT = \\ = {{\Delta }^{\ddag }}H_{{kl}}^{^\circ } - {{\Delta }_{{\text{f}}}}H_{{\text{A}}}^{^\circ } - {{\Delta }_{{\text{f}}}}H_{{{\text{О}}*,0}}^{^\circ } + \\ + \,\,{{\Delta }}\left( {{{\Delta }_{{\text{f}}}}H_{{{\text{O}}*}}^{^\circ }\left( {{\theta }} \right)} \right) + RT, \\ \end{gathered} $Предположим, что стандартная энтальпия образования активированного комплекса второй стадии реакции не изменяется ${{\Delta }^{\ddag }}H_{{kl}}^{^\circ }$ = const (рис. 2). Тогда из уравнения (10) следует
(11)
${{\Delta }}{{E}_{2}}\left( {{\theta }} \right) = {{\Delta }}\left( {{{\Delta }_{{\text{f}}}}H_{{{\text{O}}*}}^{^\circ }\left( {{\theta }} \right)} \right).$Рис. 2.
Схематичное представление протекания элементарной реакции kl с активационным барьером в традиционных энергетических координатах (стандартные значения параметров).
Далее представим изменение энергии активации второй стадии реакции в виде соотношения БЭП [1, 3, 4, 6–11]
(12)
${{\Delta }}{{E}_{2}}\left( {{\theta }} \right) = {{{{\alpha }}}_{2}}{{\Delta }}{{q}_{2}}\left( {{\theta }} \right),$(13)
${{{{\alpha }}}_{2}}{{\Delta }}{{q}_{2}}\left( {{\theta }} \right) = {{\Delta }}\left( {{{\Delta }_{{\text{f}}}}H_{{{\text{O}}*}}^{^\circ }\left( {{\theta }} \right)} \right).$Из уравнений (2) и (13) следует, что
(14)
${{{{\alpha }}}_{2}}{{\Delta }}{{q}_{1}}\left( {{\theta }} \right) = - 2{{\Delta }}\left( {{{\Delta }_{{\text{f}}}}H_{{{\text{O}}*}}^{^\circ }\left( {{\theta }} \right)} \right).$Обратим внимание на знак коэффициента Бренстеда в уравнении (15) (${{{{\alpha }}}_{{2~}}} < 0$). Выше мы отмечали, что коэффициент Бренстеда α является эмпирической величиной между нулем и единицей. В нашем случае знак минус коэффициента Бренстеда обусловлен тем, что теплота второй стадии реакции с ростом ${{\theta }}$ уменьшается (${{\Delta }}{{q}_{2}}\left( {{\theta }} \right)~$ < 0), в то время как ${{\Delta }}{{E}_{2}}\left( {{\theta }} \right) > 0.$ Единственное предположение, которое мы использовали для получения уравнения (15), – это постоянство стандартных энтальпий образования активированных комплексов ${{\Delta }^{\ddag }}H_{{ij}}^{^\circ }$ и ${{\Delta }^{\ddag }}H_{{kl}}^{^\circ }.$ Обоснованность этого предположения может быть оценена только в каждом конкретном случае. В то же время следует отметить противоположное влияние приращений стандартных энтальпии образования активных центров и адсорбированных атомов на поверхности катализатора на возможные изменения стандартных энтальпий образования активированных комплексов. Исходя из этого, мы делаем вывод, что изменения стандартных энтальпий образования активированных комплексов не могут быть большими. В таком случае равенство (15) будет приближенным. Следует также отметить, что на рис. 1 и 2 оси ординат и абсцисс имеют разные размерности (на рис. 1 – Дж/(моль ij), на рис. 2 – Дж/(моль kl)), поэтому невозможно представить эти зависимости на одном рисунке.
Если реакция или отдельная стадия реакции (адсорбция/десорбция) является обратимой и тепловой эффект этой стадии зависит от степени покрытия поверхности катализатора адсорбированными веществами, то возникает вопрос о взаимосвязи коэффициентов Бренстеда стадии реакции в прямом и обратном направлении. Следует отметить, что этот вопрос рассматривался М.И. Тёмкиным в работе [7]. Для случая логарифмической изотермы адсорбции вещества в области средних степеней покрытия поверхности было показано, что сумма коэффициентов Бренстеда стадии реакции в прямом и обратном направлении равна единице. При этом М.И. Тёмкин исходил из связи констант скоростей реакции в прямом и обратном направлении с константой равновесия стадии. Он также указал [7], что коэффициенты Бренстеда были ранее введены А.Н. Фрумкиным в статье [28] по электрохимической кинетике для обратимой реакции и его результат является переносом результатов А.Н. Фрумкина на каталитические реакции. Заметим, что метод М.И. Тёмкина основывается на закономерностях протекания обратимой реакции, поэтому он не пригоден при рассмотрении последовательных стадий реакции. Продемонстрируем работоспособность рассмотренного выше метода для обратимой стадии каталитической реакции. Для этого рассмотрим обратимую экзотермическую реакцию типа 2A + + B2$ \rightleftarrows $ 2AB + q, которая протекает на поверхности катализатора по нелинейному двухстадийному механизму
(IV)
${\text{A}} + {\text{B}}* \rightleftarrows {\text{AB}} + * + \,\,~{{q}_{4}}\left( {{\theta }} \right).$Энергии активации прямой (E+) и обратной (E–) реакций для любой из этих стадии связаны соотношением [4, 29]
Здесь M – молекулярность реакции, равная числу молекул реагирующего вещества, вступающих в реакцию при превращении одного активированного комплекса. В качестве примера рассмотрим стадию (III), для которой запишем соотношение (16) в следующем виде(17)
$\begin{gathered} {{E}_{{ - 3.0}}} + {{\Delta }}{{E}_{{ - 3}}}\left( {{\theta }} \right) - {{E}_{{ + 3.0}}} - {{\Delta }}{{E}_{{ + 3}}}\left( {{\theta }} \right) = \\ = {{q}_{{3.0}}} + {{\Delta }}{{q}_{3}}\left( {{\theta }} \right). \\ \end{gathered} $Здесь ${{q}_{{3.0}}} = {{q}_{3}}\left( {{{\theta }} = 0} \right),$ $\Delta {{q}_{3}}\left( {{\theta }} \right)$ – приращение теплоты стадий (III). Далее используем уравнение БЭП для описания уменьшения энергии активации стадии (III) в прямом направлении и увеличения энергии активации в обратном направлении [1, 3, 4]
(18)
${{\Delta }}{{E}_{{ + 3}}}\left( {{\theta }} \right) = - {{{{\alpha }}}_{3}}{{\Delta }}{{q}_{3}}\left( {{\theta }} \right),\,\,\,\,{{\Delta }}{{E}_{{ - 3}}}\left( \theta \right) = {{{{\beta }}}_{3}}{{\Delta }}{{q}_{3}}\left( {{\theta }} \right),$Согласно уравнению (19) значение коэффициентами Бренстеда и заключены между нулем и единицей. Из приведенных выше рассуждений ясно, что соотношение типа (19) справедливо для любой обратимой стадии реакции, при этом никакие ограничения на вид функции, кроме непрерывности, не накладывали.
ТРЕХСТАДИЙНАЯ СХЕМА КАТАЛИТИЧЕСКОЙ РЕАКЦИИ
Механизм Ленгмюра–Хиншелвуда является еще одним распространенным механизмом реакций, катализируемых поверхностью. Рассмотрим для простоты необратимую экзотермическую реакцию типа A2 + 2B → 2AB + q, которая протекает на поверхности катализатора по трехстадийному механизму
(V)
$~{{{\text{A}}}_{2}} + 2* \to 2{\text{A}}{\kern 1pt} * + \,\,{{q}_{5}}\left( {{{{{\theta }}}_{{{\text{A*}}}}},{{\theta }_{{{\text{B*}}}}}} \right){\kern 1pt} ,$(VI)
$~{\text{B}} + * \to {\text{B}}{\kern 1pt} * + \,\,{{q}_{6}}\left( {{{{{\theta }}}_{{{\text{A*}}}}},{{{{\theta }}}_{{{\text{B*}}}}}} \right),$(VII)
${\text{A}}{\kern 1pt} * + {\text{ B}}{\kern 1pt} * \to 2{\kern 1pt} * + {\text{ AB}} + {{q}_{7}}\left( {{{{{\theta }}}_{{{\text{A*}}}}},{{{{\theta }}}_{{{\text{B*}}}}}} \right).$Здесь A* и B* – адсорбированные частицы, ${{{{\theta }}}_{{{\text{A*}}}}}$ и ${{{{\theta }}}_{{{\text{B*}}}}}$ – доли поверхности катализатора, занятые адсорбированными частицами A* и B*, ${{\theta }}$ – доля свободной поверхности катализатора, $~{{q}_{5}}\left( {{{{{\theta }}}_{{{\text{A*}}}}},{{{{\theta }}}_{{{\text{B*}}}}}} \right),$ ${{q}_{6}}\left( {{{{{\theta }}}_{{{\text{A*}}}}},{{{{\theta }}}_{{{\text{B*}}}}}} \right)$ и ${{q}_{7}}\left( {{{{{\theta }}}_{{{\text{A*}}}}},{{{{\theta }}}_{{{\text{B*}}}}}} \right)$ – тепловые эффекты стадий (V), (VI) и (VII), которые будем считать непрерывными функциями концентраций адсорбированных веществ. Очевидно, что (${{{{\theta }}}_{{{\text{A*}}}}} + {{{{\theta }}}_{{{\text{B*}}}}} + {{\theta }} = 1$). Поскольку тепловой эффект брутто реакции является постоянной величиной ($q$ = const), в стационарном состоянии должен выполняться баланс
(20)
$\begin{gathered} q = {{q}_{5}}\left( {{{{{\theta }}}_{{{\text{A*}}}}},{{{{\theta }}}_{{{\text{B*}}}}}} \right) + 2{{q}_{6}}\left( {{{{{\theta }}}_{{{\text{A*}}}}},{{{{\theta }}}_{{{\text{B*}}}}}} \right) + 2{{q}_{7}}\left( {{{{{\theta }}}_{{{\text{A*}}}}},{{{{\theta }}}_{{{\text{B*}}}}}} \right) = \\ = {{q}_{{5,0}}} + \Delta {{q}_{5}}\left( {{{{{\theta }}}_{{{\text{A*}}}}},{{{{\theta }}}_{{{\text{B*}}}}}} \right) + 2\left( {{{q}_{{6,0}}} + \Delta {{q}_{6}}\left( {{{{{\theta }}}_{{{\text{A*}}}}},{{{{\theta }}}_{{{\text{B*}}}}}} \right)} \right) + \\ + \,\,2\left( {{{q}_{{7,0}}} + \Delta {{q}_{7}}\left( {{{{{\theta }}}_{{{\text{A*}}}}},{{{{\theta }}}_{{{\text{B*}}}}}} \right)} \right), \\ \end{gathered} $(21)
$\begin{gathered} \Delta {{q}_{5}}\left( {{{{{\theta }}}_{{{\text{A*}}}}},{{{{\theta }}}_{{{\text{B*}}}}}} \right) + 2\Delta {{q}_{6}}\left( {{{{{\theta }}}_{{{\text{A*}}}}},{{{{\theta }}}_{{{\text{B*}}}}}} \right) + \\ + \,\,2\Delta {{q}_{7}}\left( {{{{{\theta }}}_{{{\text{A*}}}}},{{{{\theta }}}_{{{\text{B*}}}}}} \right) = 0. \\ \end{gathered} $Пусть теплота стадии (V) реакции уменьшается с увеличением доли адсорбированных частиц и ${{{{\theta }}}_{{{\text{A*}}}}}$ и ${{{{\theta }}}_{{{\text{B*}}}}}.$ Эта теплота, по определению, равна
(22)
$\begin{gathered} {{q}_{5}}\left( {{{{{\theta }}}_{{{\text{A*}}}}},{{{{\theta }}}_{{{\text{B*}}}}}} \right) = {{q}_{{5,0}}} + \Delta {{q}_{5}}\left( {{{{{\theta }}}_{{{\text{A*}}}}},{{{{\theta }}}_{{{\text{B*}}}}}} \right) = \\ = - {{{{\Delta }}}_{r}}H_{{ij}}^{^\circ }\left( {{{{{\theta }}}_{{{\text{A*}}}}},{{{{\theta }}}_{{{\text{B*}}}}}} \right) = - 2{{\Delta }_{{\text{f}}}}H_{{{\text{A*}}}}^{^\circ }\left( {{{{{\theta }}}_{{{\text{A*}}}}},{{{{\theta }}}_{{{\text{B*}}}}}} \right) + \\ + \,\,2{{\Delta }_{{\text{f}}}}H_{*}^{^\circ }\left( {{{{{\theta }}}_{{{\text{A*}}}}},{{{{\theta }}}_{{{\text{B*}}}}}} \right) + {{\Delta }_{{\text{f}}}}H_{{{{{\text{A}}}_{2}}}}^{^\circ } = \\ = - \left( {2{{\Delta }_{{\text{f}}}}H_{{{\text{A*}},0}}^{^\circ } + {{\Delta }}\left( {{{\Delta }_{{\text{f}}}}H_{{{\text{A*}}}}^{^\circ }\left( {{{{{\theta }}}_{{{\text{A*}}}}},{{{{\theta }}}_{{{\text{B*}}}}}} \right)} \right)} \right. - \\ - \,\,\left. {{{\Delta }_{{\text{f}}}}H_{{*,0}}^{^\circ } - {{\Delta }}\left( {{{\Delta }_{{\text{f}}}}H_{*}^{^\circ }\left( {{{{{\theta }}}_{{{\text{A*}}}}},{{{{\theta }}}_{{{\text{B*}}}}}} \right)} \right)} \right). \\ \end{gathered} $(23)
$\begin{gathered} \Delta {{q}_{5}}\left( {{{{{\theta }}}_{{{\text{A*}}}}},{{{{\theta }}}_{{{\text{B*}}}}}} \right) = \\ = - 2{{\Delta }}\left( {{{\Delta }_{{\text{f}}}}H_{{{\text{A*}}}}^{^\circ }\left( {{{{{\theta }}}_{{{\text{A*}}}}},{{{{\theta }}}_{{{\text{B*}}}}}} \right)} \right) + 2{{\Delta }}\left( {{{\Delta }_{{\text{f}}}}H_{{\text{*}}}^{^\circ }\left( {{{{{\theta }}}_{{{\text{A*}}}}},{{{{\theta }}}_{{{\text{B*}}}}}} \right)} \right). \\ \end{gathered} $Для стадии (V) химической реакции связь между энергией активации ${{E}_{5}}\left( {{{{{\theta }}}_{{{\text{A*}}}}},{{{{\theta }}}_{{{\text{B*}}}}}} \right)$ и энтальпией активации ${{\Delta }_{{\text{a}}}}H_{{ij}}^{^\circ }\left( {{{{{\theta }}}_{{{\text{A*}}}}},{{{{\theta }}}_{{{\text{B*}}}}}} \right)$ выразим уравнением
(24)
$\begin{gathered} {{E}_{5}}\left( {{{{{\theta }}}_{{{\text{A*}}}}},{{{{\theta }}}_{{{\text{B*}}}}}} \right) = {{E}_{{5,0}}} + {{\Delta }}{{E}_{5}}\left( {{{{{\theta }}}_{{{\text{A*}}}}},{{{{\theta }}}_{{{\text{B*}}}}}} \right) = \\ = {{\Delta }_{{\text{a}}}}H_{{ij}}^{^\circ }\left( {{{{{\theta }}}_{{{\text{A*}}}}},{{{{\theta }}}_{{{\text{B*}}}}}} \right) + RT = \\ = {{\Delta }^{\ddag }}H_{{ij}}^{^\circ } - {{\Delta }_{{\text{f}}}}H_{i}^{^\circ }\left( {{{{{\theta }}}_{{{\text{A*}}}}},{{{{\theta }}}_{{{\text{B*}}}}}} \right) + RT = \\ = {{\Delta }^{\ddag }}H_{{ij}}^{^\circ } - {{\Delta }_{{\text{f}}}}H_{{{{{\text{A}}}_{2}}}}^{^\circ } - 2{{\Delta }_{{\text{f}}}}H_{{*,0}}^{^\circ } - \\ - \,\,2{{\Delta }}\left( {{{\Delta }_{{\text{f}}}}H_{*}^{^\circ }\left( {{{{{\theta }}}_{{{\text{A*}}}}},{{{{\theta }}}_{{{\text{B*}}}}}} \right)} \right) + RT, \\ \end{gathered} $(25)
${{\Delta }}{{E}_{5}}\left( {{{{{\theta }}}_{{{\text{A*}}}}},{{{{\theta }}}_{{{\text{B*}}}}}} \right) = - 2{{\Delta }}\left( {{{\Delta }_{{\text{f}}}}H_{*}^{^\circ }\left( {{{{{\theta }}}_{{{\text{A*}}}}},{{{{\theta }}}_{{{\text{B*}}}}}} \right)} \right).$Представим приращение энергии активации стадии (V) каталитической реакции уравнением БЭП
(26)
${{\Delta }}{{E}_{5}}\left( {{{{{\theta }}}_{{{\text{A*}}}}},{{{{\theta }}}_{{{\text{B*}}}}}} \right) = {{{{\alpha }}}_{5}}{{\Delta }}{{q}_{5}}\left( {{{{{\theta }}}_{{{\text{A*}}}}},{{{{\theta }}}_{{{\text{B*}}}}}} \right).$Поскольку теплота стадии (V) реакции уменьшается ${{\Delta }}{{q}_{5}}\left( {{{{{\theta }}}_{{{\text{A*}}}}},{{{{\theta }}}_{{{\text{B*}}}}}} \right) < 0,$ а энергия активации увеличивается ${{\Delta }}{{E}_{5}}\left( {{{{{\theta }}}_{{{\text{A*}}}}},{{{{\theta }}}_{{{\text{B*}}}}}} \right) > 0,$ коэффициент Бренстеда должен быть отрицательным ${{{{\alpha }}}_{5}} < 0.$ Из уравнений (25) и (26) находим
(27)
${{{{\alpha }}}_{5}}{{\Delta }}{{q}_{5}}\left( {{{{{\theta }}}_{{{\text{A*}}}}},{{{{\theta }}}_{{{\text{B*}}}}}} \right) = - 2{{\Delta }}\left( {{{\Delta }_{{\text{f}}}}H_{*}^{^\circ }\left( {{{{{\theta }}}_{{{\text{A*}}}}},{{{{\theta }}}_{{{\text{B*}}}}}} \right)} \right).$Складывая уравнения (27) и (23), получаем
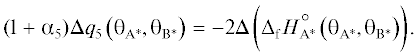
Для стадии (VI) связь между энергией активации ${{E}_{6}}\left( {{{{{\theta }}}_{{{\text{A*}}}}},{{{{\theta }}}_{{{\text{B*}}}}}} \right)$ и энтальпией активации ${{\Delta }_{{\text{a}}}}H_{{kl}}^{^\circ }\left( {{{{{\theta }}}_{{{\text{A*}}}}},{{{{\theta }}}_{{{\text{B*}}}}}} \right)$ выразим уравнением
(29)
$\begin{gathered} {{E}_{6}}\left( {{{{{\theta }}}_{{{\text{A*}}}}},{{{{\theta }}}_{{{\text{B*}}}}}} \right) = {{E}_{{6,0}}} + {{\Delta }}{{E}_{6}}\left( {{{{{\theta }}}_{{{\text{A*}}}}},{{{{\theta }}}_{{{\text{B*}}}}}} \right) = \\ = {{\Delta }_{{\text{a}}}}H_{{kl}}^{^\circ }\left( {{{{{\theta }}}_{{{\text{A*}}}}},{{{{\theta }}}_{{{\text{B*}}}}}} \right) + RT = \\ = {{\Delta }^{\ddag }}H_{{kl}}^{^\circ } - {{\Delta }_{{\text{f}}}}H_{k}^{^\circ }\left( {{{{{\theta }}}_{{{\text{A*}}}}},{{{{\theta }}}_{{{\text{B*}}}}}} \right) + RT = \\ = {{\Delta }^{\ddag }}H_{{kl}}^{^\circ } - {{\Delta }_{{\text{f}}}}H_{{\text{B}}}^{^\circ } - {{\Delta }_{{\text{f}}}}H_{{{\text{*}},0}}^{^\circ } - \\ - \,\,{{\Delta }}\left( {{{\Delta }_{{\text{f}}}}H_{{\text{*}}}^{^\circ }\left( {{{{{\theta }}}_{{{\text{A*}}}}},{{{{\theta }}}_{{{\text{B*}}}}}} \right)} \right) + RT, \\ \end{gathered} $(30)
${{\Delta }}{{E}_{6}}\left( {{{{{\theta }}}_{{{\text{A*}}}}},{{{{\theta }}}_{{{\text{B*}}}}}} \right) = - {{\Delta }}\left( {{{\Delta }_{{\text{f}}}}H_{{\text{*}}}^{^\circ }\left( {{{{{\theta }}}_{{{\text{A*}}}}},{{{{\theta }}}_{{{\text{B*}}}}}} \right)} \right).$Пусть теплота стадии (VI) уменьшается с увеличением доли адсорбированных частиц ${{{{\theta }}}_{{{\text{A*}}}}}$ и ${{{{\theta }}}_{{{\text{B*}}}}}.$ Эта теплота, по определению, равна разности стандартных энтальпий продуктов реакции и исходных реагентов
(31)
$\begin{gathered} {{q}_{6}}\left( {{{\theta }_{{A*}}},{{\theta }_{{B*}}}} \right) = {{q}_{{6,0}}} + \Delta {{q}_{6}}\left( {{{\theta }_{{{\text{A*}}}}},{{\theta }_{{{\text{B*}}}}}} \right) = \\ = - {{{{\Delta }}}_{{\text{r}}}}H_{{kl}}^{^\circ }\left( {{{{{\theta }}}_{{{\text{A*}}}}},{{{{\theta }}}_{{{\text{B*}}}}}} \right) = \\ = - {{\Delta }_{{\text{f}}}}H_{{{\text{B*}}}}^{^\circ }\left( {{{{{\theta }}}_{{{\text{A*}}}}},{{{{\theta }}}_{{{\text{B*}}}}}} \right) + {{\Delta }_{{\text{f}}}}H_{{\text{*}}}^{^\circ }\left( {{{{{\theta }}}_{{{\text{A*}}}}},{{{{\theta }}}_{{{\text{B*}}}}}} \right) + {{\Delta }_{{\text{f}}}}H_{{\text{B}}}^{^\circ } = \\ = {{\Delta }_{{\text{f}}}}H_{{\text{B}}}^{^\circ } - \left( {{{\Delta }_{{\text{f}}}}H_{{{\text{B*}},0}}^{^\circ } + {{\Delta }}\left( {{{\Delta }_{{\text{f}}}}H_{{{\text{B*}}}}^{^\circ }\left( {{{{{\theta }}}_{{{\text{A*}}}}},{{{{\theta }}}_{{{\text{B*}}}}}} \right)} \right)} \right. - \\ - \,\,\left. {{{\Delta }_{{\text{f}}}}H_{{{\text{*}},0}}^{^\circ } - {{\Delta }}\left( {{{\Delta }_{{\text{f}}}}H_{{\text{*}}}^{^\circ }\left( {{{{{\theta }}}_{{{\text{A*}}}}},{{{{\theta }}}_{{{\text{B*}}}}}} \right)} \right)} \right). \\ \end{gathered} $(32)
$\begin{gathered} \Delta {{q}_{6}}\left( {{{{{\theta }}}_{{{\text{A*}}}}},{{{{\theta }}}_{{{\text{B*}}}}}} \right) = \\ = - {{\Delta }}\left( {{{\Delta }_{{\text{f}}}}H_{{{\text{B*}}}}^{^\circ }\left( {{{{{\theta }}}_{{{\text{A*}}}}},{{{{\theta }}}_{{{\text{B*}}}}}} \right)} \right) + {{\Delta }}\left( {{{\Delta }_{{\text{f}}}}H_{{\text{*}}}^{^\circ }\left( {{{{{\theta }}}_{{{\text{A*}}}}},{{{{\theta }}}_{{{\text{B*}}}}}} \right)} \right). \\ \end{gathered} $Представим приращение энергии активации стадии (VI) уравнением БЭП в следующем виде
(33)
${{\Delta }}{{E}_{6}}\left( {{{{{\theta }}}_{{{\text{A*}}}}},{{{{\theta }}}_{{{\text{B*}}}}}} \right) = {{{{\alpha }}}_{6}}{{\Delta }}{{q}_{6}}\left( {{{\theta }_{{{\text{A*}}}}},{{\theta }_{{{\text{B*}}}}}} \right).$Отметим, что в уравнении (33) ${{\Delta }}{{E}_{6}}\left( {{{{{\theta }}}_{{{\text{A*}}}}},{{{{\theta }}}_{{{\text{B*}}}}}} \right) > 0,$ ${{\Delta }}{{q}_{6}}\left( {{{{{\theta }}}_{{{\text{A*}}}}},{{{{\theta }}}_{{{\text{B*}}}}}} \right) < 0,$ следовательно, коэффициент Бренстеда ${{{{\alpha }}}_{6}} < 0.$
Из уравнения (30) и (33) следует
(34)
${{{{\alpha }}}_{6}}{{\Delta }}{{q}_{6}}\left( {{{{{\theta }}}_{{{\text{A*}}}}},{{{{\theta }}}_{{{\text{B*}}}}}} \right) = - {{\Delta }}\left( {{{\Delta }_{{\text{f}}}}H_{{\text{*}}}^{^\circ }\left( {{{{{\theta }}}_{{{\text{A*}}}}},{{{{\theta }}}_{{{\text{B*}}}}}} \right)} \right).$Складывая уравнения (34) и (32), получаем

Для стадии (VII) связь между энергией активации ${{E}_{7}}\left( {{{{{\theta }}}_{{{\text{A*}}}}},{{{{\theta }}}_{{{\text{B*}}}}}} \right)$ и энтальпией активации ${{\Delta }_{{\text{a}}}}H_{{nm}}^{^\circ }\left( {{{{{\theta }}}_{{{\text{A*}}}}},{{{{\theta }}}_{{{\text{B*}}}}}} \right)$ выразим уравнением
(36)
$\begin{gathered} {{E}_{7}}\left( {{{{{\theta }}}_{{{\text{A*}}}}},{{{{\theta }}}_{{{\text{B*}}}}}} \right) = {{E}_{{7,0}}} + {{\Delta }}{{E}_{7}}\left( {{{{{\theta }}}_{{{\text{A*}}}}},{{{{\theta }}}_{{{\text{B*}}}}}} \right) = \\ = {{\Delta }_{{\text{a}}}}H_{{nm}}^{^\circ }\left( {{{{{\theta }}}_{{{\text{A*}}}}},{{{{\theta }}}_{{{\text{B*}}}}}} \right) + RT = \\ = {{\Delta }^{\ddag }}H_{{nm}}^{^\circ } - {{\Delta }_{{\text{f}}}}H_{n}^{^\circ }\left( {{{{{\theta }}}_{{{\text{A*}}}}},{{{{\theta }}}_{{{\text{B*}}}}}} \right) + RT = \\ = {{\Delta }^{\ddag }}H_{{nm}}^{^\circ } - {{\Delta }_{{\text{f}}}}H_{{{\text{A*}},0}}^{^\circ } - \Delta \left( {{{\Delta }_{{\text{f}}}}H_{{{\text{A*}}}}^{^\circ }\left( {{{{{\theta }}}_{{{\text{A*}}}}},{{{{\theta }}}_{{{\text{B*}}}}}} \right)} \right) - \\ - \,\,{{\Delta }_{{\text{f}}}}H_{{{\text{B*}},0}}^{^\circ } - \Delta \left( {{{\Delta }_{{\text{f}}}}H_{{{\text{B*}}}}^{^\circ }\left( {{{{{\theta }}}_{{{\text{A*}}}}},{{{{\theta }}}_{{{\text{B*}}}}}} \right)} \right) + RT, \\ \end{gathered} $(37)
$\begin{gathered} {{\Delta }}{{E}_{7}}\left( {{{{{\theta }}}_{{{\text{A*}}}}},{{{{\theta }}}_{{{\text{B*}}}}}} \right) = \\ = - {{\Delta }}\left( {{{\Delta }_{{\text{f}}}}H_{{{\text{A*}}}}^{^\circ }\left( {{{{{\theta }}}_{{{\text{A*}}}}},{{{{\theta }}}_{{{\text{B*}}}}}} \right)} \right) - {{\Delta }}\left( {{{\Delta }_{{\text{f}}}}H_{{{\text{B*}}}}^{^\circ }\left( {{{{{\theta }}}_{{{\text{A*}}}}},{{{{\theta }}}_{{{\text{B*}}}}}} \right)} \right). \\ \end{gathered} $Пусть теплота стадии (VII) увеличивается с уменьшением доли адсорбированных частиц ${{{{\theta }}}_{{{\text{A*}}}}}$ и ${{{{\theta }}}_{{{\text{B*}}}}}.$ Это теплота, по определению, равна разности стандартных энтальпий образования продуктов реакции и исходных реагентов
(38)
$\begin{gathered} {{q}_{7}}\left( {{{{{\theta }}}_{{{\text{A*}}}}},{{{{\theta }}}_{{{\text{B*}}}}}} \right) = {{q}_{{7,0}}} + \Delta {{q}_{7}}\left( {{{{{\theta }}}_{{{\text{A*}}}}},{{{{\theta }}}_{{{\text{B*}}}}}} \right) = \\ = - {{{{\Delta }}}_{{\text{r}}}}H_{{nm}}^{^\circ }\left( {{{{{\theta }}}_{{{\text{A*}}}}},{{{{\theta }}}_{{{\text{B*}}}}}} \right) = \\ = - 2{{\Delta }_{{\text{f}}}}H_{{\text{*}}}^{^\circ }\left( {{{{{\theta }}}_{{{\text{A*}}}}},{{{{\theta }}}_{{{\text{B*}}}}}} \right) - {{\Delta }_{{\text{f}}}}H_{{{\text{AB}}}}^{^\circ } + \\ + \,\,{{\Delta }_{{\text{f}}}}H_{{{\text{A*}}}}^{^\circ }\left( {{{{{\theta }}}_{{{\text{A*}}}}},{{{{\theta }}}_{{{\text{B*}}}}}} \right) + {{\Delta }_{{\text{f}}}}H_{{{\text{B*}}}}^{^\circ }\left( {{{{{\theta }}}_{{{\text{A*}}}}},{{{{\theta }}}_{{{\text{B*}}}}}} \right) = \\ = - 2\left( {{{\Delta }_{{\text{f}}}}H_{{{\text{*}},0}}^{^\circ } - {{\Delta }}\left( {{{\Delta }_{{\text{f}}}}H_{{\text{*}}}^{^\circ }\left( {{{{{\theta }}}_{{{\text{A*}}}}},{{{{\theta }}}_{{{\text{B*}}}}}} \right)} \right)} \right) - \\ - \,\,{{\Delta }_{{\text{f}}}}H_{{{\text{AB}}}}^{^\circ } + {{\Delta }_{{\text{f}}}}H_{{{\text{A*}},0}}^{^\circ } + {{\Delta }}\left( {{{\Delta }_{{\text{f}}}}H_{{{\text{A*}}}}^{^\circ }\left( {{{{{\theta }}}_{{{\text{A*}}}}},{{{{\theta }}}_{{{\text{B*}}}}}} \right)} \right) + \\ + \,\,{{\Delta }_{{\text{f}}}}H_{{{\text{B*}},0}}^{^\circ } + {{\Delta }}\left( {{{\Delta }_{{\text{f}}}}H_{{{\text{B*}}}}^{^\circ }\left( {{{{{\theta }}}_{{{\text{A*}}}}},{{{{\theta }}}_{{{\text{B*}}}}}} \right)} \right). \\ \end{gathered} $Из уравнения (38) находим
(39)
$\begin{gathered} \Delta {{q}_{7}}\left( {{{{{\theta }}}_{{{\text{A*}}}}},{{{{\theta }}}_{{{\text{B*}}}}}} \right) = - 2{{\Delta }}\left( {{{\Delta }_{{\text{f}}}}H_{{\text{*}}}^{^\circ }\left( {{{{{\theta }}}_{{{\text{A*}}}}},{{{{\theta }}}_{{{\text{B*}}}}}} \right)} \right) + \\ {\text{ + }}\,\,{{\Delta }}\left( {{{\Delta }_{{\text{f}}}}H_{{{\text{A*}}}}^{^\circ }\left( {{{{{\theta }}}_{{{\text{A*}}}}},{{{{\theta }}}_{{{\text{B*}}}}}} \right)} \right) + {{\Delta }}\left( {{{\Delta }_{{\text{f}}}}H_{{{\text{B*}}}}^{^\circ }\left( {{{{{\theta }}}_{{{\text{A*}}}}},{{{{\theta }}}_{{{\text{B*}}}}}} \right)} \right). \\ \end{gathered} $Представим уменьшение энергии активации стадии (VII) уравнением БЭП в следующем виде
(40)
${{\Delta }}{{E}_{7}}\left( {{{{{\theta }}}_{{{\text{A*}}}}},{{{{\theta }}}_{{{\text{B*}}}}}} \right) = - {{{{\alpha }}}_{7}}{{\Delta }}{{q}_{7}}\left( {{{{{\theta }}}_{{{\text{A*}}}}},{{{{\theta }}}_{{{\text{B*}}}}}} \right).$Отметим, что в уравнении (40) ${{\Delta }}{{E}_{7}}\left( {{{{{\theta }}}_{{{\text{A*}}}}},{{{{\theta }}}_{{{\text{B*}}}}}} \right) < 0,$ ${{\Delta }}{{q}_{7}}\left( {{{{{\theta }}}_{{{\text{A*}}}}},{{{{\theta }}}_{{{\text{B*}}}}}} \right) > 0,$ следовательно, коэффициент Бренстеда ${{{{\alpha }}}_{7}} > 0.$
Из уравнений (37) и (40) находим
(41)
$\begin{gathered} {{{{\alpha }}}_{7}}{{\Delta }}{{q}_{7}}\left( {{{{{\theta }}}_{{{\text{A*}}}}},{{{{\theta }}}_{{{\text{B*}}}}}} \right) = \\ = {{\Delta }}\left( {{{\Delta }_{{\text{f}}}}H_{{{\text{A*}}}}^{^\circ }\left( {{{{{\theta }}}_{{{\text{A*}}}}},{{{{\theta }}}_{{{\text{B*}}}}}} \right)} \right) + {{\Delta }}\left( {{{\Delta }_{{\text{f}}}}H_{{{\text{B*}}}}^{^\circ }\left( {{{{{\theta }}}_{{{\text{A*}}}}},{{{{\theta }}}_{{{\text{B*}}}}}} \right)} \right). \\ \end{gathered} $Вычитая уравнение (41) из (39), получаем
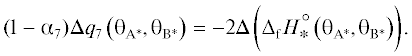
Складывая уравнения (28), (39) и (41), приходим к уравнению

Как видно из уравнения (43), в нем содержатся только коэффициенты Бренстеда и приращения теплот стадий каталитической реакции. При этом приращения теплот стадий каталитической реакции являются функциями только от концентраций адсорбированных веществ А* и В* без каких-либо ограничений на них, кроме непрерывности. Рассматривая уравнение (43) совместно с уравнением баланса (21), можно исключить одну из функций, но этого недостаточно, чтобы установить связь только между коэффициентами Бренстеда. Очевидно, что для этого потребуется дополнительная информация о теплотах стадий реакции. Далее рассмотрим несколько примеров.
Случай 1. Предположим, что ${{\Delta }}{{q}_{6}}\left( {{{{{\theta }}}_{{{\text{A*}}}}},{{{{\theta }}}_{{{\text{B*}}}}}} \right) = 0,$ тогда из уравнений (21) и (43) мы находим связь между коэффициентами Бренстеда первой и третьей стадий реакции
Интересно отметить, что уравнение (44) эквивалентно уравнению (15) с учетом знака коэффициентов ${{{{\alpha }}}_{5}}$ и ${{{{\alpha }}}_{7}}.$
Случай 2. Если предположить, что ${{\Delta }}{{q}_{5}}\left( {{{{{\theta }}}_{{{\text{A*}}}}},{{{{\theta }}}_{{{\text{B*}}}}}} \right) = 0,$ то из уравнений (21) и (43) находим связь между коэффициентами Бренстеда (VI) и (VII) стадий реакции
Случай 3. Если предположить, что ${{\Delta }}{{q}_{5}}\left( {{{{{\theta }}}_{{{\text{A*}}}}},{{{{\theta }}}_{{{\text{B*}}}}}} \right) = $ = $2{{\Delta }}{{q}_{6}}\left( {{{{{\theta }}}_{{{\text{A*}}}}},{{{{\theta }}}_{{{\text{B*}}}}}} \right),$то из уравнений (21) и (43) находим связь между коэффициентами Бренстеда всех трех стадий каталитической реакции
ЗАКЛЮЧЕНИЕ
Каталитический цикл является одним из основополагающих принципов катализа. Теоретически катализатор остается неизменным в стационарном состоянии, но это может быть не так в нестационарном состоянии, когда каталитический цикл не завершен. Идея о влиянии реакционной среды на катализатор получила убедительное подтверждение в многочисленных экспериментальных исследованиях. Катализатор и реакционная среда рассматриваются как единая система, в которой катализатор может изменять свою структуру, химический состав и каталитические свойства по мере приближения реакции к стационарному состоянию. Разнообразие явлений, происходящих с катализатором в реагирующей системе, чрезвычайно велико. В представленных результатах рассматривались термодинамические аспекты воздействия реакционной среды на катализатор в рамках простейших схем каталитических реакций, которые допускали лишь изменения энергий активации стадий реакции. Для описания этих изменений использовалось соотношение Бренстеда–Эванса–Поляни, устанавливающее взаимосвязь между изменением энергии активации реакции и теплотой реакции. При этом какие-либо ограничения на функцию, описывающую зависимость приращения теплоты реакции от степени покрытия поверхности катализатора адсорбированными веществами, не накладывались. Предположение о постоянстве стандартных энтальпий образования активированных комплексов каталитической реакции, протекающей по двухстадийной схеме (механизм Или–Ридеала), позволило установить взаимосвязь между коэффициентами Бренстеда этих стадий. Для трехстадийной схемы (механизм Ленгмюра–Хиншелвуда) в отсутствие информации о теплотах стадий каталитической реакции установить взаимосвязь между коэффициентами Бренстеда этих стадий в общем виде не удается. В этом случае возможно лишь получить уравнение баланса, содержащее коэффициенты Бренстеда и приращения теплот стадий каталитической реакции, на основе которого были проанализированы некоторые частные случаи.
Список литературы
Boudar M. / In: Handbook of Heterogeneous Catalysis. Eds. G. Ertl, H. Knözinger, J. Weitkamp, Wiley-VCH, Weinheim, 1977. P. 1.
Taylor H.S. // Proc. Roy. Soc. A (London). 1925. V. 108. P. 105.
Боресков Г.К. // Кинетика и катализ. 1980. Т. 21. С. 5.
Боресков Г.К. Гетерогенный катализ. М.: Наука, 1986. 304 с.
Somorjai G.A. // Ann. Rev. Phys. Chem. 1994. V. 45. P. 721.
Marin G.B., Galvita V.V., Yablonsky G.S. // J. Catal. 2021. V. 404. P. 745.
Темкин М.И. // Журн. физ. химии. 1957. Т. 31. С. 3.
Боресков Г.К., Веньяминов С.А., Сазонова Н.Н., Панкратьев Ю.Д., Питаева А.Н. // Кинетика и катализ. 1975. Т. 16. С. 1442.
Panov G.I., Parfenov M.V., Parmon V.N. // Catal. Rev. Sci. Eng. 2015. V. 57. P. 436.
Logadottir A., Rod T.H., Nørskov J.K., Hammer B., Dahl S., Jacobsen C.J.H. // J. Catal. 2001. V. 197. P. 229.
Nørskov J.K., Bligaard T., Logadottir A., Bahn S., Hansen L.B., Bollinger M., Bengaard H., Hammer B., Sljivancanin Z., Mavrikakis M., Dahl Y., Xu S., Jacobsen C.J.H. // J. Catal. 2002. V. 209. P. 275.
Cheng J., Hu P., Ellis P., French S., Kelly G., Lok C.M. // J. Phys. Chem. C (Letter). 2008. V. 112. P. 1308.
Dumesic J.A., Huber G.W., Boudar M. / In: Handbook of Heterogeneous Catalysis, 2nd Ed. Eds. G. Ertl, H. Knözinger, J. Weitkamp, WIL-VCH Verlag GmbH & Co. KGaA, Weinheim. 2008. P. 1445.
Brönsted J.N., Pedersen Kai. // Zeitschrift für Physikalische Chemie. 1924. Bd. 108 (Helf 3, 4). P. 185.
Pallassana V., Neurock M. // J. Catal. 2000. V. 191. P. 301.
Liu Z.P., Hu P.J. // J. Chem. Phys. 2001. V. 114. P. 8244.
Веньяминов С.А., Боресков Г.К. // Докл. АН СССР. 1986. Т. 289. № 2. С. 389.
Веньяминов С.А., Сазонова Н.Н., Баранник Г.Б., Панкратьев Ю.Д. // Кинетика и катализ. 1988. Т. 29. С. 854.
Боресков Г.К., Веньяминов С.А., Сазонова Н.Н., Панкратьев Ю.Д. // Докл. АН СССР. 1971. Т. 196. № 3. С. 621.
Щукин В.П., Веньяминов С.А., Боресков Г.К. // Кинетика и катализ. 1971. Т. 12. С. 621.
Боресков Г.К., Веньяминов С.А., Сазонова И.И., Герасев А.П., Матрос Ю.Ш. // Материалы II Всесоюзной конференции “Нестационарные процессы в катализе”. Новосибирск. 1983. Ч. 1. С. 181.
Колебания и бегущие волны в химических системах. Ред. Филд Р., Бургер. М. М.: Мир, 1988. 720 с.
Яблонский Г.С., Быков В.И., Елохин В.И. Кинетика модельных реакций гетерогенного катализа. Новосибирск: Наука, 1984. 223 с.
Yablonskii G.S., Bykov V.I., Gorban A.N., Elokhin V.I. Kinetic Models of Catalytic Reactions, in series, Comprehensive Chemical Kinetics. V. 32. Amsterdam-Oxford-New York-Tokyo: Elsevier, 1991, 392 pp.
Синев М.Ю. // Кинетика и катализ. 2019. Т. 60. С. 450.
Пармон В.Н. Термодинамика неравновесных процессов для химиков. С приложением к химической кинетике, катализу, материаловедению и биологии. Долгопрудный: Издательский дом “Интеллект”. 2015. 472 с.
Ertl G. / In: Handbook of Heterogeneous Catalysis, 2nd Ed. Eds. G. Ertl, H. Knözinger, J. Weitkamp, WIL-VCH Verlag GmbH & Co. KGaA, Weinheim. 2008. P. 1462.
Frumkin A. HYPERLINK “http://www.elch.chem. msu.ru/cgi-bin/getpdf.cgi?pdf=zpc1932.pdf" \t "pdfwindow” // Z. Phys. Chem. (A). 1932. V. 160. P. 116.
Боресков Г.К. // Журн. физ. химии. 1945. Т. 19. С. 92.
Дополнительные материалы отсутствуют.
Инструменты
Кинетика и катализ