

Кинетика и катализ, 2022, T. 63, № 6, стр. 798-807
Высокоактивные моноатомные сплавные PdAg-катализаторы на церийсодержащих носителях для селективного гидрирования алкинов в алкены
А. В. Рассолов a, Г. О. Брагина a, Г. Н. Баева a, И. С. Машковский a, Н. С. Смирнова a, Е. Ю. Герасимов b, А. В. Бухтияров b, Я. В. Зубавичус b, А. Ю. Стахеев a, *
a ФГБУН Институт органической химии им. Н.Д. Зелинского РАН
119991 Москва, Ленинский просп., 47, Россия
b ФГБУН ФИЦ Институт катализа им. Г.К. Борескова СО РАН
630090 Новосибирск, просп. Акад. Лаврентьева, 5, Россия
* E-mail: st@ioc.ac.ru
Поступила в редакцию 28.06.2022
После доработки 01.07.2022
Принята к публикации 02.07.2022
- EDN: GZMTKX
- DOI: 10.31857/S0453881122060119
Аннотация
При исследовании серии моноатомных сплавных катализаторов Pd1Ag3/Al2O3, Pd1Ag3/CeO2–Al2O3 и Pd1Ag3/CeO2–ZrO2 в селективном гидрировании дифенилацетилена (ДФА) показан существенный (пятикратный) рост активности для образца Pd1Ag3/CeO2–ZrO2 в сравнении с Pd1Ag3/Al2O3. Особо отмечается, что увеличение активности не сопровождается снижением селективности образования целевого продукта. Такое каталитическое поведение можно объяснить двумя факторами: 1) более чем двукратным увеличением дисперсности катализатора PdAg3/CeO2–ZrO2 и 2) изменением электронного состояния наночастиц, установленным по результатам ИК-спектроскопии адсорбированного СО. Сохранение высокой селективности синтезированных катализаторов указывает на стабильность структуры моноатомных центров Pd1 в катализаторах, приготовленных нанесением на Ce-содержащие носители, что также подтверждают данные ИК-спектроскопии адсорбированного СО. Полученные результаты свидетельствуют о перспективности использования Ce-содержащих носителей для синтеза катализаторов селективного гидрирования замещенных алкинов.
ВВЕДЕНИЕ
Реакция селективного гидрирования алкинов – одна из ключевых и наиболее актуальных в современном катализе [1–4]. Показательным примером ее промышленного использования являются многотоннажные процессы селективного гидрирования ацетиленовых углеводородов в пиролизном этилене или стироле перед их направлением на полимеризацию [1, 5, 6] Применение селективного гидрирования алкинов в тонком органическом синтезе позволяет с высокой регио- и стереоселективностью получать цис- и транс-олефины – исходное сырье для пищевой, фармацевтической (например, синтез витаминов А, Е и K) промышленности, а также для производства растворителей, моющих средств и т.д. [7].
В качестве активного компонента в катализаторах селективного гидрирования, как правило, используют палладий, что обеспечивает высокую активность, однако селективность процесса при этом зачастую недостаточна [8, 9]. Одно из перспективных направлений разработки высокоселективных катализаторов, активно изучаемое в последние несколько лет, связано с концепцией моноатомных сплавных катализаторов. В зарубежной литературе они получили название “Single-Atom-Alloy catalysts” (сокращенно SAA catalysts или SAAC) [10–12]. В этих системах активными каталитическими центрами являются атомы Pd1, изолированные друг от друга атомами металла-модификатора (M = Ag, Au, Zn, In), активность которого в гидрировании пренебрежимо мала. Применение моноатомных сплавных катализаторов обеспечивает чрезвычайно высокую селективность по целевому продукту, однако их активность, как правило, несущественна ввиду малого количества таких центров на поверхности наночастиц Pd–M, поэтому актуальной задачей является ее повышение.
На активность катализатора значительное влияние может оказывать носитель, который также вносит свой вклад в соотношение “активность/селективность” [13, 14]. Важный параметр при выборе носителя – степень взаимодействия наночастиц металла с поверхностью носителя. Известно, что все носители можно разделить на инертные (слабо взаимодействующие с нанесенными металлами) и активные (сильно взаимодействующие с нанесенной активной фазой). В последнем случае возможно как изменение электронного состояния поверхностных металлических частиц, так и увеличение их дисперсности [15, 16].
В последние годы большой интерес исследователей привлекает оксид церия, использование которого в качестве носителя позволяет повысить дисперсность нанесенного металла, а также варьировать электронную плотность на металлических наночастицах [5, 13, 17]. В ряде случаев важную роль могут играть центры на периферии наночастиц на границе раздела металл–носитель, в состав которых входят как атомы металла самой наночастицы, так и поверхностные атомы носителя. К настоящему времени для ряда металлнанесенных катализаторов, полученных с использованием оксида церия, было обнаружено существенное увеличение каталитической активности, хотя причины этого до сих пор остаются невыясненными. Так, в работах [18, 19] повышенная активность Pd/CeO2 была установлена в гидрировании фенола. Авторы предположили, что возрастанию каталитической активности при превращении фенола в циклогексанон может способствовать увеличение дисперсности Pd в катализаторе, нанесенном на CeO2.
Следует отметить, что CeO2 часто используется в комбинации с другими оксидами. Значительно улучшить структурные и текстурные характеристики оксида церия можно при использовании в качестве модификатора диоксида циркония. Из литературы известно, что добавление ZrO2 даже в небольших количествах позволяет повысить термостабильность CeO2 [20, 21] в результате образования твердого раствора CeO2–ZrO2.
В цикле работ Рединой и сотр. наблюдалось увеличение активности монометаллического Pt катализатора в гидрировании различных органических кислород- и азотсодержащих субстратов при нанесении Pt на носитель CeO2–ZrO2 [22–25]. Авторы предположили, что высокая каталитическая активность мезопористого катализатора Pt/CeO2–ZrO2 обусловлена спилловером активного водорода, за счет которого происходит частичное восстановление носителя CeO2–ZrO2 с образованием центров адсорбции и активации исходного субстрата. Кроме того, в реакции гидрирования коричного альдегида была отмечена зависимость выхода непредельного спирта от площади поверхности носителей синтезированных Pt-катализаторов.
Аналогичные результаты в гидрировании коричного альдегида были ранее получены на Pt/CeO2–ZrO2 [26, 27] и Pd/CeO2–ZrO2 [28]. Во всех случаях образцы на основе CeO2–ZrO2 превосходили катализаторы сравнения по активности и селективности.
В приведенных выше примерах эффект увеличения активности был установлен для катализаторов, содержащих монометаллические наночастицы. К сожалению, возможность повышения активности биметаллических катализаторов, нанесенных на CeO2–ZrO2, остается практически неисследованной. В этой связи можно упомянуть лишь работу [29], в которой гидрирование коричного альдегида изучали на модифицированных катализаторах Ni–M/CeO2–ZrO2 (M = Pt, Pd).
Следует отметить, что в открытой печати полностью отсутствуют данные об активности моноатомных сплавных катализаторов на Ce-содержащих носителях. В связи с вышесказанным целью настоящей работы являлось исследование влияния носителей CeO2–ZrO2 и CeO2–Al2O3 на активность моноатомных сплавных Pd1Ag3-катализаторов в реакции гидрирования дифенилацетилена (ДФА). При этом особое внимание было уделено изучению влияния носителя на селективность катализатора. Ранее аналогичный PdAg3-катализатор, полученный нанесением активного компонента на Al2O3, показал чрезвычайно высокую селективность в гидрировании ДФА [30, 31] и 1-фенил-1-пропина [31, 32], однако его активность была существенно ниже монометаллического аналога.
Для определения влияния носителя на соотношение активность/селективность моноатомных катализаторов была получена серия образцов Pd1Ag3/CeO2–ZrO2, Pd1Ag3/CeO2–Al2O3 и Pd1Ag3/Al2O3, структурные характеристики которых были изучены методами ИК-спектроскопии адсорбированного СО и просвечивающей электронной микроскопии высокого разрешения. Элементный состав синтезированных катализаторов оценивали по данным энергодисперсионной спектроскопии. Каталитические характеристики были исследованы в реакции жидкофазного гидрирования ДФА.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Приготовление катализаторов
Все образцы получены методом совместной пропитки носителей растворами Pd(NO3)2 и AgNO3. В качестве носителей использованы γ-Al2O3 (“Sasol”, Германия, SБЭТ = 56 м2/г), 20% CeO2–80% Al2O3 (Puralox SCFa-160 Ce20, “Sasol”, Германия) и 80% CeO2–20% ZrO2 (C20Z, ООО “Экоальянс”, Свердловская область, Новоуральск). Перед нанесением активного компонента все носители предварительно прокаливали в токе воздуха при 550°С в течение 4 ч. После пропитки образцы сушили при комнатной температуре, затем прокаливали в токе воздуха (300 мл/мин) при 550°С и восстанавливали в токе 5% H2/Ar при 550°С. По окончании восстановления образцы охлаждали до 200°C в токе 5% H2/Ar, а затем – до комнатной температуры в токе N2-ОСЧ. Содержание металлов в готовых катализаторах составило 2 веc. % Pd и 6 вес. % Ag. В тексте статьи катализаторы обозначены следующим образом: Pd1Ag3/Al2O3 – PdAg3/A; Pd1Ag3/CeO2–Al2O3 – PdAg3/CA; Pd1Ag3/CeO2–ZrO2 – PdAg3/CZ.
Характеризация катализаторов
Просвечивающая электронная микроскопия (ПЭМ). Исследование структуры и микроструктуры образцов осуществляли методом просвечивающей электронной микроскопии высокого разрешения (ПЭМВР) на электронном микроскопе ThemisZ (“Thermo Fisher Scientific”, США) c ускоряющим напряжением 200 кВ и предельным разрешением 0.07 нм. Запись изображений осуществляли с помощью ПЗС матрицы Ceta 16 (“Thermo Fisher Scientific”, США). Прибор оснащен энергодисперсионным спектрометром рентгеновского характеристического излучения (EDX) SuperX (“Thermo Fisher Scientific”, США) с полупроводниковым Si-детектором с разрешением по энергии 128 эВ.
Для проведения исследований на электронном микроскопе частицы образцов наносили из спиртового раствора на дырчатые углеродные подложки, закрепленные на медных сетках, с использованием ультразвукового диспергатора УЗД-1УЧ2 (Россия) что позволяло добиться равномерного распределения частиц по поверхности подложки.
ИК-спектроскопия адсорбированного СО. Для регистрации ИК-спектров диффузного отражения адсорбированного CO использовали ИК-спектрометр Tensor 27 (“Bruker”, Германия) с in situ приставкой диффузного отражения Harrick Diffuse Reflectance Kit (“Harrick Scientific Products, Inc.”, Великобритания). Навеску образца (~0.02 г) помещали в ячейку со стеклами из CaF2 и нагревали в токе Ar до 550°С, после чего образец восстанавливали в токе 5% H2/Ar в течение 1 ч. После этого образец охлаждали сначала до 300°С в токе 5% H2/Ar, а затем в токе Ar до 50°С и регистрировали фоновый спектр. Съемку спектров адсорбированного СО осуществляли при 50°С в токе 0.5 об. % СО/He в течение 20 мин (250 сканов, разрешение – 4 см–1).
Жидкофазное гидрирование дифенилацетилена
Реакцию гидрирования проводили на каталитической установке автоклавного типа. Навеску катализатора загружали в стеклянный реактор с субстратом и растворителем. Реактор помещали в автоклав, оснащенный магнитной мешалкой, электронным датчиком давления, системой дозирования газов и отбора пробы. Процесс вели при температуре 25°С, начальном давлении водорода 5 бар и при постоянном перемешивании. В качестве субстрата применяли дифенилацетилен (98%, “Sigma-Aldrich”, Германия), в качестве растворителя – н-гексан (>99%, “Merck”, Германия). С целью корректного сопоставления результатов реакцию осуществляли в кинетическом режиме. Продукты реакции анализировали методом газовой хроматографии на хроматографе Кристалл 5000 (“Хроматэк”, Россия), оборудованном пламенно-ионизационным детектором, с использованием хроматографической колонки HP5-MS (5% фенилдиметилсилоксан) длиной 30 м, внутренним диметром 0.25 мм и толщиной пленки неподвижной фазы 0.25 мкм. По результатам газохроматографического анализа реакционной смеси была рассчитана селективность образования целевого дифенилэтилена (S=):
где n= и n– – мольные доли образующихся алкена и алкана соответственно.Эффективность работы синтезированных катализаторов оценивали по числу оборотов катализатора TOF на первой (TOF1) и на второй (TOF2) стадиях гидрирования исходя из величин скоростей реакции на этих стадиях. Ввиду сложности определения количества поверхностных атомов Pd в биметаллических PdAg-катализаторах методом электронной микроскопии, величины удельной активности находили в расчете на общее число атомов палладия в навеске катализатора.
Учитывая, что реакция гидрирования может протекать в две стадии (гидрирование исходного алкина в олефин и его последующее гидрирование в алкан), скорости реакции рассчитывали для первой и второй стадий (r1 и r2 соответственно). Величины r1 и r2 определяли по углу наклона касательных к кривым на графике зависимости количества поглощенного водорода от времени реакции: на первой стадии – в интервале 0.2–0.6 экв. поглощенного H2 для всех катализаторов, на второй стадии – в интервале 1.0–1.4 экв. для PdAg3/CZ и в интервале 1.0–1.2 экв. для PdAg3/CA и PdAg3/A. Скорость поглощения водорода находили в пересчете на 1 г катализатора (ммоль H2 мин–1${\text{г}}_{{{\text{кат}}}}^{{ - 1}}$). По соотношению скоростей гидрирования алкина и алкена (r1/r2) оценивали эффективность кинетического контроля протекания процесса.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Просвечивающая электронная микроскопия с картированием
На рис. 1 представлены результаты исследования катализаторов PdAg3/CZ, PdAg3/CA и PdAg3/A методом ПЭМВР, данные картирования по основным компонентам катализаторов, а также гистограммы распределения биметаллических PdAg-частиц по размерам. Для всех образцов наблюдается широкое распределение частиц по размерам (от 1 до 12 нм), при этом их средний размер составляет 3.5 нм для PdAg3/CZ, 5.4 нм для PdAg3/CA и 9.2 нм для PdAg3/A. Хорошо видно, что все частицы имеют сферическую форму. EDX-картирование различных участков образцов катализаторов позволило заключить, что PdAg-частицы равномерно распределены по поверхности носителя и соотношение Ag/Pd соответствует расчетному. По методике, предложенной в работе [33], была рассчитана дисперсность биметаллических PdAg-частиц, величина которой составила 0.32 для PdAg3/CZ, 0.21 для PdAg3/CA и 0.12 для PdAg3/A.
Рис. 1.
Данные ПЭМ, иллюстрирующие раcположение PdAg-частиц на поверхности носителя: для PdAg3/CZ (a) с картированием по Pd, Ag, Ce (б), Pd (в), Ag (г); для PdAg3/CA (д) с картированием по Pd, Ag, Ce, Al (е), Pd (ж), Ag (з); для PdAg3/A (и) с картированием по Pd, Ag, Al (к), Pd (л), Ag (м). На рис. 1а, 1д, и 1и приведены соответствующие гистограммы распределения частиц по размерам.

ИК-спектроскопия адсорбированного СО
Методом ИК-спектроскопии адсорбированного СО была изучена структура поверхности синтезированных катализаторов (рис. 2). В спектре PdAg3/CZ обнаружены две полосы поглощения: высокоинтенсивная с максимумом при 2055 см–1, характерная для молекул СО, адсорбированных на поверхности катализатора в линейной форме, и низкоинтенсивная (на уровне шума) при 1964 см–1, которую можно отнести к адсорбции СО в мостиковой форме (рис. 2, спектр 1). Похожая спектральная картина также наблюдается для катализаторов PdAg3/CA и PdAg3/A. В случае PdAg3/CA полосы поглощения, относящиеся к адсорбции СО в линейной и мостиковой формах, детектируются при 2045 и 1953 см–1 (рис. 2, спектр 2). Для PdAg3/A эти сигналы идентифицированы при 2047 и 1960 см–1 соответственно (рис. 2, спектр 3). Чрезвычайно низкая интенсивность полос поглощения в интервале 1953–1964 см–1 практически полностью исключает возможность формирования центров многоточечной адсорбции молекул СО на поверхности PdAg-наночастиц [34, 35] и свидетельствует об образовании PdAg-сплава, в котором активные центры представляют собой атомы Pd1, изолированные друг от друга атомами Ag. В результате формирования биметаллических PdAg-частиц может происходить увеличение электронной плотности на атомах палладия, что приводит к возрастанию донирования электронной плотности на π-разрыхляющую орбиталь молекулы адсорбированного СО и ослаблению связи С–О [34, 35].

Необходимо отметить, что симметричность полосы поглощения линейно адсорбированного CO и ее малая ширина также указывают на образование высокооднородных активных центров Pd1 в исследуемом катализаторе.
Селективное гидрирование дифенилацетилена
Основная цель настоящего исследования заключалась в изучении возможности увеличения активности моноатомного сплавного PdAg3-катализатора при сохранении высокой селективности. Кинетические кривые поглощения водорода в гидрировании ДФА на указанных катализаторах представлены на рис. 3. В табл. 1 систематизированы данные о скоростях гидрирования и числе оборотов на первой и второй стадиях реакции. Самая низкая каталитическая активность в гидрировании ДФА наблюдается для катализатора PdAg3/A, для которого значение скорости r1 составляет 5.41 ммоль Н2 мин–1${\text{г}}_{{{\text{кат}}}}^{{ - 1}}.$ Для PdAg3-катализатора, полученного нанесением активного компонента на Al2O3, модифицированный церием (PdAg3/CA), скорость гидрирования исходного ДФА несколько выше (7.32 ммоль Н2 мин–1${\text{г}}_{{{\text{кат}}}}^{{ - 1}}$). При использовании в качестве носителя CeO2–ZrO2 (катализатор PdAg3/CZ) скорость резко возрастает и достигает величины 28.76 ммоль Н2 мин–1${\text{г}}_{{{\text{кат}}}}^{{ - 1}}.$ Полученные данные находятся в хорошем согласии с ранее опубликованными для Pt/CeO2–ZrO2 и Pd/CeO2–ZrO2 катализаторов гидрирования органических кислород- и азот-содержащих субстратов [23, 25–28] и позволяют сделать вывод о возможности увеличении активности моноатомных PdAg-катализаторов путем нанесения биметаллических наночастиц на CeO2–ZrO2.
Рис. 3.
а – Кинетика поглощения водорода в реакции жидкофазного гидрирования ДФА на катализаторах PdAg3/CZ, PdAg3/CA и PdAg3/A; б – участок (*) в интервале 0–80 мин в увеличенном масштабе.

Таблица 1.
Влияние носителя на кинетические характеристики PdAg-катализаторов в жидкофазном гидрировании дифенилацетилена
| Катализатор | r1 | r2 | r1/r2 | TOF1 | TOF2 | TOF1/TOF2 |
|---|---|---|---|---|---|---|
| ммоль Н2 мин–1${\text{г}}_{{{\text{кат}}}}^{{ - 1}}$ | c–1 | |||||
| PdAg3/CZ | 28.76 | 2.07 | 13.9 | 2.55 | 0.18 | 13.9 |
| PdAg3/CA | 7.32 | 0.30 | 24.7 | 0.65 | 0.0263 | 24.7 |
| PdAg3/A | 5.41 | 0.14 | 38.3 | 0.48 | 0.0125 | 38.3 |
На изменение активности катализаторов, возможно, оказывает влияние дисперсность биметаллических наночастиц [36]. Так, наименьшая каталитическая активность в случае PdAg3/A может быть обусловлена формированием более крупных (до 9.2 нм) по сравнению с остальными катализаторами этой серии биметаллических частиц (ср. рис. 1а, 1д и 1и). При переходе от PdAg3/A к образцам, полученным нанесением на церийсодержащий носитель, средний размер частиц сокращается до 5.4 нм (PdAg3/CA) и 3.5 нм (PdAg3/CZ). Столь выраженное уменьшение размера биметаллических PdAg-частиц вероятно связано с увеличением энергии взаимодействия металл–носитель для PdAg3/CZ. Так, в ряде работ было показано, что наличие ионов Zr в кристаллической решетке оксида церия усиливает взаимодействие металл–носитель между подложкой и нанесенными на нее наночастицами, что может приводить к более высокой дисперсности металлических частиц [27, 37, 38]. Этот вывод находится в хорошем согласии с полученными данными ПЭМ.
Следует, однако, отметить, что расчет по формуле, предложенной в [33], показал, что величина дисперсности для PdAg3/CZ лишь в 2.6 раза превышает таковую для образца, полученного нанесением на Al2O3 (см. раздел “Просвечивающая электронная микроскопия с картированием”). Учитывая, что значения скоростей реакции для этих катализаторов отличаются друг от друга в 5 раз, бóльшую каталитическую активность катализатора PdAg3/CZ можно лишь частично объяснить увеличением дисперсности.
Отчасти повышение активности PdAg3/CZ может быть обусловлено изменением электронного состояния наночастиц в результате взаимодействия металл–носитель [39]. В ИК-спектрах адсорбированного СО (рис. 2) для образца PdAg3/CZ в сравнении с PdAg3/CA и PdAg3/A наблюдается сдвиг полосы СО, адсорбированного в линейной форме, в сторону бóльших волновых чисел – от 2045 до 2055 см–1. Это может быть связано с тем, что при введении циркония в структуру церия образуются кислотные центры Ce3+, взаимодействие с которыми способно приводить к снижению электронной плотности на наночастицах PdAg, которые в результате становятся более электрондефицитными. В свою очередь, увеличение степени электрондефицитности наночастиц PdAg может способствовать возрастанию активности катализатора в реакции гидрирования. Данное предположение согласуется с опубликованным ранее обзором [39], в котором рассмотрены вопросы влияния носителя на электронные свойства и каталитические характеристики металлнанесенных катализаторов.
После поглощения 1 экв. водорода на кинетических кривых всех образцов присутствует характерный излом (рис. 3), указывающий на замедление реакции при переходе от гидрирования тройной С≡С-связи к гидрированию олефинового интермедиата [40, 41]. Расчет значений скорости гидрирования, выполненный с использованием кинетических данных, показывает значительное уменьшение величины r2 в сравнении с r1 (табл. 1). На образце PdAg3/A скорость гидрирования падает с 5.41 до 0.14 ммоль Н2 мин–1${\text{г}}_{{{\text{кат}}}}^{{ - 1}},$ а на PdAg3/CA – с 7.32 до 0.30 ммоль Н2 мин–1${\text{г}}_{{{\text{кат}}}}^{{ - 1}}.$ В случае катализатора PdAg3/CZ скорость снижается с 28.76 до 2.07 ммоль Н2 мин–1${\text{г}}_{{{\text{кат}}}}^{{ - 1}}.$ Столь существеннное замедление реакции по завершении гидрирования связи С≡С способствует эффективному кинетическому контролю хода реакции.
Интересно сопоставить изменение состава продуктов от времени реакции для катализаторов PdAg3/CZ и PdAg3/A (рис. 4). В продуктах гидрирования детектируются цис- и транс-дифенилэтилен и дифенилэтан. Приведенные зависимости хорошо иллюстрируют вывод о существенном различии скорости гидрирования ДФА на разных катализаторах: так, на PdAg3/CZ-образце 100% конверсия ДФА достигается за 15 мин, в то время как на PdAg3/A для этого требуется 80 мин. В случае обоих катализаторов дифенилэтан присутствует в следовых количествах в самом начале реакции, и по достижении 100% конверсии ДФА содержание алкана в продуктах реакции не превышает 7–8%. Максимальный выход целевого олефина как для PdAg3/CZ, так и для PdAg3/A составляет 89%. Полученные экспериментальные данные свидетельствуют о том, что селективности катализаторов практически идентичны.
Рис. 4.
Состав реакционных продуктов при гидрировании дифенилацетилена на катализаторах PdAg3/CZ (а) и PdAg3/A (б). ДФА – дифенилацетилен, цис-/транс-ДФЭ – цис-/транс-дифенилэтилен, ДФЭт – дифенилэтан.

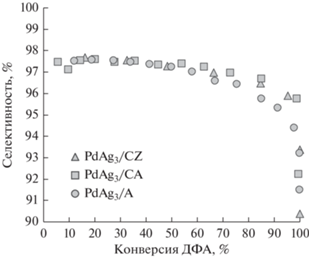
Для более детального анализа селективности было изучено влияние на нее конверсии исходного ДФА (рис. 5). Установлено, что для всех исследованных образцов вне зависимости от использованного носителя величина селективности равна ~96–98% практически во всем интервале конверсий. При этом по достижении 100% конверсии ДФА селективность катализаторов, нанесенных на Ce-содержащий носитель (~96%), несколько выше таковой для катализатора, нанесенного на Al2O3 (~93%).
Рис. 5.
Зависимость селективности по дифенилэтилену от конверсии дифенилацетилена на катализаторах PdAg3/CZ, PdAg3/CA и PdAg3/A.
Высокую селективность биметаллических PdAg-систем можно объяснить тем, что при их высокотемпературной активации водородом атомы Ag мигрируют к поверхности PdAg-сплава, в результате чего формируются центры Pd1, изолированные атомами Ag [42]. Это также приводит к преимущественному снижению энергии адсорбции олефина, что облегчает его десорбцию и предотвращает последующее нежелательное гидрирование в алкан.
Сохранение высокой селективности биметаллических катализаторов при использовании Ce-содержащих носителей, по-видимому, обусловлено сохранением структуры изолированных Pd1-центров при нанесении наночастиц. Об этом свидетельствуют данные ИК-спектроскопии адсорбированного СО, из которых видно, что доминирующей является линейная форма адсорбции, характерная для адсорбции СО на центрах Pd1, изолированных друг от друга атомами Ag, на которых СО при комнатной температуре не адсорбируется (рис. 2). Сигнал от полосы, характеризующей адсорбцию СО в мостиковой форме, незначителен или практически полностью отсутствует, что исключает возможность формирования центров многоточечной адсорбции молекул СО на поверхности PdAg-наночастиц. Данный вывод находится в хорошем согласии с результатами, опубликованными в [43]. Эффект увеличения селективности в результате формировании моноатомных Pd1-центров на поверхности биметаллических наночастиц был показан рядом авторов на примере селективного гидрирования ацетилена в этилен [44, 45], а также в наших предыдущих исследованиях [30, 31, 42].
ЗАКЛЮЧЕНИЕ
Полученные в настоящей работе результаты свидетельствуют, что применение Ce-содержащих носителей позволяет значительно увеличить активность моноатомных PdAg-катализаторов в селективном гидрировании замещенных алкинов в соответствующие алкены. Так, для PdAg3/CZ, приготовленного с использованием смешанного оксида CeO2–ZrO2, наблюдается пятикратный рост скорости реакции по сравнению с катализатором, приготовленным на основе традиционного Al2O3. Исключительно важным является тот факт, что рост активности не сопровождается снижением селективности образования целевого продукта.
Возрастание активности Ce-содержащих катализаторов отчасти можно объяснить более чем двукратным повышением дисперсности, что приводит к росту площади поверхности биметаллических наночастиц. Кроме того, согласно данным ИК-спектроскопии адсорбированного СО свою роль в увеличении активности может играть взаимодействие металл–носитель, приводящее к изменению электронного состояния PdAg-наночастиц. Так, сохранение высокой селективности достигается благодаря стабильности структуры моноатомных центров Pd1 на поверхности наночастиц PdAg, нанесенных на Ce-содержащие носители.
Список литературы
Глыздова Д.В., Смирнова Н.С., Шляпин Д.А., Цырульников П.Г. // Рос. хим. журн. (Журн. Рос. хим. об-ва им. Д.И. Менделеева). 2018. Т. 62. С. 89. (Glyzdova D.V., Smirnova N.S., Shlyapin D.A., Tsyrul’nikov P.G. Russ. J. Gen. Chem. 2020. V. 90. P. 1120.)
Sun Z., Wang S., Chen W. // J. Mater. Chem. A. 2021. V. 9. P. 5296.
Zhang L., Zhou M., Wang A., Zhang T. // Chem. Rev. 2020. V. 120. P. 683.
Takht Ravanchi M., Sahebdelfar S., Komeili S. // Rev. Chem. Eng. 2017. V. 34. P. 215.
Shittu T.D., Ayodele O.B. // Front. Chem. Sci. Eng. 2022. V. 16. P. 1031.
Zhao X., Chang Y., Chen W.-J., Wu Q., Pan X., Chen K., Weng B. // ACS Omega. 2022. V. 7. 1. P. 17.
Chen X., Shi C., Liang C. // Chin. J. Catal. 2021. V. 42. P. 2105.
Borodziski A., Bond G.C. // Catal. Rev. 2006. V. 48. P. 91.
Borodziski A., Bond G.C. // Catal. Rev. 2008. V. 50. P. 379.
Han Z.-K., Sarker D., Ouyang R., Mazheika A., Gao Y., Levchenko S.V. // Nat. Commun. 2021. V. 12. № 1833. https://doi.org/10.1038/s41467-021-22048-9
Han J., Lu J., Wang M., Wang Y., Wang F. // Chin. J. Chem. 2019. V. 37. P. 977.
Giannakakis G., Flytzani-Stephanopoulos M., Sykes E.C.H. // Acc. Chem. Res. 2019. V. 52. P. 237.
Razmgar K., Altarawneh M., Oluwoye I., Senanayake G. // Catal. Surv. Asia. 2021. V. 25. P. 27.
Пахомов Н.А. Научные основы приготовления катализаторов: введение в теорию и практику. Отв. ред. В.А. Садыков. Новосибирск: Издательство СО РАН, 2011. 262 с.
Abdel-Mageed A.M., Chen S., Fauth C., Häring T., Bansmann J. // ChemPhysChem. 2021. V. 22. P. 1302.
Schubert M.M., Hackenberg S., van Veen A.C, Muhler M., Plzakc V., Jürgen Behm R. // J. Catal. 2001. V. 197. P. 113.
Anand S., Pinheiro D., Sunaja Devi K.R. // Asian J. Org. Chem. 2021. V. 10. P. 3068.
Nelson N.C., Manzano J.S., Sadow A.D., Overbury S.H., Slowing I.I. // ACS Catal. 2015. V. 5. P. 2051.
Velu S., Kapoor M.P., Inagaki S., Suzuki K. // Appl. Catal. A: Gen. 2003. V. 245. P. 317.
Zhang X., Wang Q., Zhang J., Wang J., Guo M., Chen S., Li C., Hu C., Xie Y. // RSC Advances. 2015. V. 5. P. 89976.
Teng M., Luo L., Yang X. // Micropor. Mesopor. Mater. 2009. V. 119. P. 158.
Виканова К.В., Редина Е.А. // Журн. Физ. Химии. 2018. Т. 92. С. 1827. (Vikanova K.V., Redina E.A. Russ. J. Phys. Chem. A. 2018. V. 92. P. 2355.)
Виканова К.В., Редина Е.А., Капустин Г.И., Давшан Н.А., Кустов Л.М. // Журн. Физ. Химии. 2019. Т. 93. С. 208. (Vikanova K.V., Redina E.A., Kapustin G.I., Davshan N.A., Kustov L.M. Russ. J. Phys. Chem. A. 2019. V. 93. P. 231.)
Редина Е.А., Виканова К.В. // Журн. Физ. Химии. 2018. Т. 92. С. 1846. (Vikanova K.V., Redina E.A. Russ. J. Phys. Chem. A. 2018. V. 92. P. 2374.)
Redina E.A., Vikanova K.V., Kapustin G.I., Mishin I.V., Tkachenko O.P., Kustov L.M. // Eur. J. Org. Chem. 2019. P. 4159.
Bhogeswararao S., Srinivas D. // J. Catal. 2012. V. 285. P. 31.
Wei S., Zhao Y., Fan G., Yang L., Li F. // Chem. Eng. J. 2017. V. 322. P. 234.
Bhogeswararao S., Srinivas D. // Catal. Lett. 2010. V. 140. P. 55.
Bhogeswararao S., Pavan Kumar V., Chary K. V. R., Srinivas D. // Catal Lett 2013. V. 143. P. 1266.
Rassolov A.V., Mashkovsky I.S., Bragina G.O., Baeva G.N., Markov P.V., Smirnova N.S., Wärnå J., Stakheev A.Yu, Murzin D.Yu. // Mol. Catal. 2021. V. 506. № 111550. https://doi.org/10.1016/j.mcat.2021.111550
Рассолов А.В., Брагина Г.О., Баева Г.Н., Машковский И.С., Стахеев А.Ю. // Кинетика и катализ. 2020. Т. 61. С. 837. (Rassolov A.V., Bragina G.O., Baeva G.N., Mashkovsky I.S., Stakheev A.Yu. Kinet. Catal. 2020. V. 61. P. 869.)
Rassolov A.V., Mashkovsky I.S., Baeva G.N., Bragina G.O., Smirnova N.S., Markov P.V., Bukhtiyarov A.V., Wärnå J., Stakheev A.Yu, Murzin D.Yu. // Nanomater. 2021. V. 11. № 3286. https://doi.org/10.3390/nano11123286
Ichikawa S., Poppa H., Boudart M. // J. Catal. 1985. V. 91. P. 1.
Рассолов А.В., Брагина Г.О., Баева Г.Н., Смирнова Н.С., Казаков А.В., Машковский И.С., Бухтияров А.В., Зубавичус Я.В., Стахеев А.Ю. // Кинетика и катализ. 2020. Т. 61. С. 676. (Rassolov A.V., Bragina G.O., Baeva G.N., Smirnova N.S., Kazakov A.V., Mashkovsky I.S., Bukhtiyarov A.V., Zubavichus Ya.V., Stakheev A.Yu. Kinet. Catal. 2020. V. 61. P. 758.)
Рассолов А.В., Брагина Г.О., Баева Г.Н., Смирнова Н.С., Казакова А.В., Машковский И.С., Стахеев А.Ю. // Кинетика и катализ. 2019. Т. 60. С. 644. (Rassolov A.V., Bragina G.O., Baeva G.N., Smirnova N.S., Kazakov A.V., Mashkovsky I. S., Stakheev A. Yu. Kinet. Catal. 2019. V. 60. P. 642.)
Николаев С.А., Занавескин Л.Н., Смирнов В.В., Аверьянов В.А., Занавескин К.Л. // Успехи химии. 2009. Т. 78. С. 248. (Nikolaev S.A., Zanaveskin L.N., Smirnov V.V., Averyanov V.A., Zanaveskin K.L. Russ. Chem. Rev. 2009. V. 78. P. 231.)
Alifanti M., Baps B., Blangenois N., Naud J., Grange P., Delmon B. // Chem. Mater. 2003. V. 15. P. 395.
Liang Q., Wu X., Wu X., Weng D. // Catal. Lett. 2007. V. 119. P. 265.
Stakheev A.Yu., Kustov L.M. // Appl. Catal. A: Gen. 1999. V. 188. P. 3.
Choudary B.M., Lakshmi Kantam M., Mahender Reddy N., Koteswara Rao K., Haritha Y., Bhaskar V., Figueras F., Tuel A. // Appl. Catal. A: Gen. 1999. V. 181. P. 139.
Marín-Astorga N., Alvez-Manoli G., Reyes P. // J. Mol. Catal. A. Chem. 2005. V. 226. P. 81.
Pei G.X., Liu X.Y., Wang A., Lee A.F., Isaacs M.A., Li L., Pan X., Yang X., Wang X., Tai Z., Wilson K., Zhang T. // ACS Catal. 2015. V. 5. P. 3717.
Tang J., Deng L., Deng H., Xiao S., Zhang X., Hu W. // J. Phys. Chem. C. 2014. V. 118. P. 27850.
Jin Y., Datye A.K., Rightor E., Gulotty R., Waterman W., Smith M., Holbrook M., Maj J. // J. Catal. 2001. V. 203. P. 292.
Sheth P.A., Neurock M., Smith C.M. // J. Phys. Chem. B. 2005. V. 109. P. 12449.
Дополнительные материалы отсутствуют.
Инструменты
Кинетика и катализ