

Кинетика и катализ, 2022, T. 63, № 6, стр. 808-815
Влияние циркуляции хвостовых газов на активность и селективность Сo/SiO2-катализатора синтеза Фишера‒Тропша
В. Н. Соромотин a, Р. Е. Яковенко a, Т. В. Краснякова a, b, Р. Д. Светогоров c, С. А. Митченко a, b, *
a ФГБОУ ВО Южно-Российский государственный политехнический университет (НПИ) им. М.И. Платова
346128 Новочеркасск, ул. Просвещения, 132, Россия
b Институт физико-органической химии и углехимии им. Л.М. Литвиненко
283114 Донецк, ул. Р. Люксембург, 70, ДНР
c ФГБУ Национальный исследовательский центр “Курчатовский институт”
123182 Москва, пл. Акад. Курчатова, 1, Россия
* E-mail: samit_rpt@mail.ru
Поступила в редакцию 10.07.2022
После доработки 20.07.2022
Принята к публикации 21.07.2022
- EDN: DSYKAH
- DOI: 10.31857/S0453881122060144
Аннотация
Изучено влияние режима циркуляции газа на селективность и скорость дезактивации катализатора Со-Al2O3/SiO2 синтеза Фишера‒Тропша при высоком давлении (6 МПа). Применение циркуляции с кратностью до 2.2 способствует образованию углеводородов С19+ за счет реадсорбции газообразных олефинов и вовлечения их в процесс роста цепи. Повышенная селективность по углеводородным воскам С19+ обусловливает ускоренную дезактивацию катализатора. Увеличение кратности циркуляции до 8 приводит к росту селективности по олефинам С5+ за счет эвакуации их паров из реакционной зоны и снижению выхода парафинов С19+. Последнее связано с уменьшением вклада реадсорбции высших олефинов С5+ в удлинение углеродной цепи. При высокой кратности циркуляции зафиксировано восстановление СоО до металла, что частично компенсирует дезактивацию катализатора в результате блокирования восками С19+ активных центров роста цепи.
ВВЕДЕНИЕ
Технология синтеза Фишера‒Тропша (СФТ) позволяет производить широкий спектр ценных органических веществ (парафины, олефины, в небольших количествах оксигенаты [1]) из синтез-газа (смеси СО и Н2). В силу разных причин СФТ в промышленных условиях удобно осуществлять в трубчатых реакторах с неподвижным слоем катализатора [2]. Во избежание ускоренной дезактивации катализатора из-за высокого парциального давления паров воды приходится ограничивать конверсию СО [3], что снижает производительность процесса. Для повышения общей степени превращения синтез-газа в промышленном СФТ используют циркуляцию, при которой хвостовые газы повторно возвращают на вход реактора [4]. Такой режим способствует снижению парциального давления паров воды за счет меньшей конверсии синтез-газа за один проход и последующей конденсации водяного пара в охлаждаемой ловушке. Важным параметром режима циркуляции газа является ее кратность Kц – отношение суммы объемов свежего и рециркулируемого хвостового газов к объему подаваемого в единицу времени свежего синтез-газа. Однако в литературе имеется очень мало сведений о влиянии кратности циркуляции на активность катализатора и селективность процесса. Выяснение влияния циркуляции на активность и селективность катализатора СФТ Со-Al2O3/SiO2 в изотермических (210°С) условиях при повышенном давлении (6.0 МПа) явилось целью настоящего исследования.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
В работе использовали приготовленный по методике [5, 6] кобальт-силикагелевый катализатор Co-Al2O3/SiO2 (содержание кобальта ‒ 20 мас. %), промотированный оксидом алюминия (1 мас. %), который охарактеризован в ряде работ [7‒9]. Каталитические испытания синтеза углеводородов осуществляли проточно-циркуляционным методом в трубчатом реакторе (внутренний диаметр ‒ 16 мм) с неподвижным слоем катализатора (объем загрузки ‒ 10 см3, фракция 1‒2 мм с разбавлением кварцем 30 см3) при температуре 210°С, давлении 6.0 МПа, объемной скорости газа ОСГ = = 1000 ч–1, мольном соотношении компонентов подаваемого свежего синтез-газа Н2/СО = 1.85 при варьировании кратности циркуляции. Для каждого испытания при разных кратностях циркуляции загружали свежую порцию катализатора. Перед проведением экспериментов катализаторы восстанавливали в течение 1 ч в токе Н2 при атмосферном давлении, температуре 400°С и ОСГ = = 1000 ч–1. Мониторинг основных показателей процесса СФТ осуществляли спустя 50 ч после достижения температуры синтеза в течение 100 ч в непрерывном режиме. Скорость дезактивации катализатора Rcd (%/ч) при разных кратностях циркуляции рассчитывали из наклона нормализованной активности (конверсия СО в текущий момент времени, приведенная к начальной) от времени в потоке [10‒12].
Состав синтез-газа и газообразных продуктов синтеза анализировали методом газо-адсорбционной хроматографии на хроматографе Кристалл 5000 (“Хроматэк”, Россия) с детектором по теплопроводности [13]. Продукты синтеза фракционировали, выделяя три фракции в зависимости от температуры кипения: до 180°С ‒ бензиновая фракция (С5‒С10); 180‒330°С ‒ дизельная фракция (С11‒С18); кубовый остаток – парафиновые воски С19+. Состав фракции конденсированных углеводородов С5‒С18 определяли методом капиллярной газожидкостной хромато-масс-спектрометрии на газовом хроматографе GC 7890А (“Agilent”, США) с масс-детектором MSD 5975С и капиллярной колонкой HP-5MS. Количественный хроматографический анализ выполняли с применением программного обеспечения GCforFT [14] методом внутренней нормализации с использованием калибровочных коэффициентов для углеводородов.
Парциальное давление водяных паров в реакторе рассчитывали с помощью программного обеспечения “Hysys” с применением термодинамического пакета Пенг-Робинсон. Состав подаваемой в реактор газовой смеси определяли исходя из имеющихся экспериментальных данных о составе хвостовых газов для каждой кратности циркуляции.
Часть приготовленного катализатора Co-Al2O3/SiO2 в оксидной форме использовали для получения зауглероженного по реакции диспропорционирования оксида углерода образца сравнения. Окисленную форму катализатора восстанавливали in situ в трубчатом реакторе с неподвижным слоем как описано выше. После удаления током аргона (ОСГ = 1000 ч–1) в течение 1 ч остатков водорода поток газа переключали на окись углерода (ОСГ = 500 ч–1), повышали давление до 2.0 МПа, постепенно поднимали температуру (5°С/мин) до 220°С и выдерживали в течение 16 ч. Как показано в [15, 16], при этих условиях формируется карбид кобальта.
С поверхности отработавших в СФТ катализаторов предварительно экстрагировали в аппарате Сокслета оставшиеся углеводороды. Экстракцию осуществляли гептаном в атмосфере аргона в течение 6 ч, после чего отмытые образцы сушили еще 6 ч в токе аргона при температуре 130°C.
Рентгенофазовый анализ (РФА) катализаторов проводили на оборудовании станции “РСА” синхротрона “КИСИ-Курчатов” [17]; измерения выполняли при комнатной температуре с использованием монохроматического излучения с длинами волн 0.074 или 0.079 нм. Фазовый состав определяли с помощью базы данных PDF-2 [18] в программном комплексе Crystallographica.
Размер d кристаллитов металлического кобальта оценивали из данных рентгенофазового анализа (РФА) по уравнению Шеррера d = = Kλ/β cos θ, где K = 0.89, λ – длина волны рентгеновского излучения, β – ширина рефлекса на полувысоте, θ – положение рентгеновского рефлекса.
Исследования методом термопрограммированного окисления (ТПО) осуществляли с применением комплекса STA 449F5 Jupiter (“NETZSCH”, Германия), совмещенного с масс-спектрометром QMS 403 Aeolos. Условия эксперимента: реакционный газ ‒ 20 об. % O2–He, скорость подачи – 50 см3/мин; защитный газ ‒ Не, скорость подачи ‒ 20 см3/мин; навеска образца – 50 мг; скорость нагрева – 10°С/мин в температурном интервале 50–130°С, термостатирование при 130°С в течение 30 мин, дальнейший нагрев со скоростью 10°С в минуту в температурном интервале 130–800°С.
ПОЛУЧЕННЫЕ РЕЗУЛЬТАТЫ
Каталитические испытания
Усредненные по времени в потоке показатели процесса при использовании режима циркуляции газа и варьировании ее кратности приведены в табл. 1.
Таблица 1.
Усредненные по времени в потоке показатели процесса в зависимости от кратности циркуляции*
| Kц | Соотношение Н2/СО | ${{P}_{{{{{\text{H}}}_{{\text{2}}}}{\text{O}}}}}$ на выходе из реактора, МПа | Конверсия СО, % | Селективность, % | ||||
|---|---|---|---|---|---|---|---|---|
| свежий газ | на входе | CH4 | C2–C4 | C5+ | CO2 | |||
| 1 (проточный режим) | 1.85 | 1.85 | 1.74 | 56.5 | 11.8 | 21.1 | 66.8 | 0.3 |
| 1.5 | 1.85 | 1.75 | 1.51 | 51.6 | 18.1 | 21.6 | 59.9 | 0.3 |
| 2.2 | 1.85 | 1.75 | 0.43 | 53.1 | 12.2 | 16.6 | 70.8 | 0.4 |
| 8 | 1.85 | 1.60 | 0.11 | 53.1 | 19.2 | 16.3 | 64.0 | 0.5 |
Применение режима циркуляции газа приводит к возрастанию селективности образования метана за счет небольшого снижения выхода конденсированных продуктов С5+. При увеличении кратности циркуляции соотношение Н2/СО на входе в реактор уменьшается вследствие разбавления свежего синтез-газа хвостовыми газами. Парциальное давление паров воды на выходе из реактора также снижается (табл. 1) из-за отделения образующейся воды в охлаждаемом пробосборнике и циркуляции хвостовых газов.
Скорость дезактивации катализатора, полученная из зависимости его нормализованной активности от времени в потоке (рис. 1), при разных кратностях циркуляции приведена в табл. 3. Включение режима циркуляции (Kц = 1.5) и незначительное повышение ее кратности (Kц = 2.2) приводит к ускорению дезактивации по сравнению с проточным режимом (Kц = 1), однако дальнейшее увеличение Kц до 8 существенно замедляет скорость дезактивации.
Рис. 1.
Зависимость нормализованной активности от времени в потоке при разных кратностях циркуляции.

Таблица 2.
Состав конденсированных продуктов в зависимости от кратности циркуляции Kц*
| Kц | Продукты | Состав продуктов, мас. % | изо/н | О/П | ||
|---|---|---|---|---|---|---|
| C5–C10 | C11–С18 | С19+ | ||||
| 1 | н-Парафины | 20.5 | 24.2 | 34.4 | 0.01 | 0.18 |
| изо-Парафины | 0.4 | 0.6 | 0.1 | |||
| Олефины | 9.0 | 5.0 | 0.5 | |||
| Оксигенаты | 3.0 | 2.2 | 0.1 | |||
| Сумма | 32.9 | 32.0 | 35.1 | |||
| 1.5 | н-Парафины | 16.2 | 30.4 | 37.4 | 0.01 | 0.15 |
| изо-Парафины | 0.2 | 0.5 | – | |||
| Олефины | 6.6 | 6.1 | 0.3 | |||
| Оксигенаты | 2.0 | 0.4 | – | |||
| Сумма | 25.0 | 37.3 | 37.7 | |||
| 2.2 | н-Парафины | 13.0 | 24.2 | 43.5 | 0.01 | 0.18 |
| изо-Парафины | 0.2 | 0.6 | 0.5 | |||
| Олефины | 6.4 | 7.7 | 1.0 | |||
| Оксигенаты | 1.9 | 1.0 | – | |||
| Сумма | 21.5 | 33.5 | 45.0 | |||
| 8 | н-Парафины | 13.5 | 28.4 | 33.3 | 0.01 | 0.26 |
| изо-Парафины | 0.4 | 0.6 | – | |||
| Олефины | 7.7 | 11.7 | 0.5 | |||
| Оксигенаты | 2.2 | 1.7 | – | |||
| Сумма | 23.8 | 42.4 | 33.8 | |||
Групповой и фракционный составы конденсированных продуктов СФТ в зависимости от кратности циркуляции показаны в табл. 2. Применение режима циркуляции сдвигает селективность в сторону образования парафинов с большей длиной цепи: повышение Kц сопровождается ростом выхода углеводородов дизельной фракции (С11–С18) и восков С19+ за счет бензиновой фракции (C5–C10), что согласуется с результатами нашей предыдущей работы [7]. Селективность образования восков вначале растет, достигая максимума при Kц = 2.2, но дальнейшее увеличение кратности циркуляции до Kц = 8 снижает выход парафиновых восков.
С повышением кратности циркуляции заметно возрастает селективность образования олефинов, что проявляется в росте соотношения олефинов к парафинам О/П в составе конденсированных продуктов, а также увеличивается средняя длина синтезированных олефинов (табл. 2).
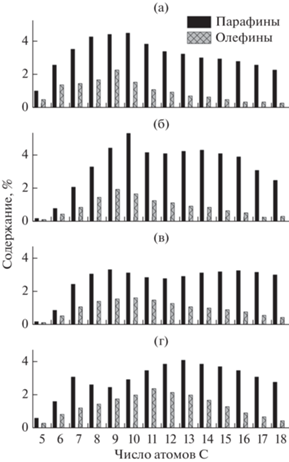
Распределение продуктов синтеза по длине углеродной цепи приведено на рис. 2. В отсутствие циркуляции (Kц = 1, проточный режим) имеет место унимодальное распределение парафинов Сn с максимумом при n = 10 (рис. 2а). При включении режима циркуляции (Kц = 1.5) распределение насыщенных углеводородов Сn становится бимодальным с максимумами при n = 10 и 14 (рис. 2б). Увеличение кратности циркуляции до Kц = 2.2 смещает первый максимум в область более коротких цепей – n = 9 (рис. 2в), а при дальнейшем повышении до Kц = 8 наблюдается существенный сдвиг обоих максимумов до n = 7 и 13 (рис. 2г). Распределение конденсированных олефинов с ростом кратности циркуляции остается унимодальным, но максимум смещается в сторону более длинных цепей, n = 9, 10 и 11 при Kц = 1, 2.2 и 8 соответственно.
Характеристика катализаторов
Исходный восстановленный, специально зауглероженный и отработавшие по 100 ч при разной кратности циркуляции (Kц = 2.2 и 8) катализаторы были исследованы методами РФА и ТПО с масс-спектральным анализом двуокиси углерода – продукта окисления карбида кобальта, а также остаточных продуктов СФТ и углеродных отложений в виде сажи и графита.
Дифрактограммы указанных образцов приведены на рис. 3. В составе зауглероженного катализатора (кривая 4) кобальт находится только в виде карбида Co2C и оксида СоО, отвечающих металлическому кобальту рефлексов не наблюдается. Дифрактограммы восстановленного образца (кривая 1) и отработавших по 100 ч при Kц равной 2.2 (кривая 2) и 8 (кривая 3) катализаторов содержат дифракционные пики металлического кобальта ГЦК-модификации и фазы СоО; рефлексов карбида кобальта в отработавших катализаторов не зафиксировано.
Рис. 3.
Дифрактограммы катализаторов: исходного восстановленного (1), λ = 0.079 нм; отработавших по 100 ч при кратностях циркуляции 2.2 (2) и 8 (3), λ = = 0.074 нм; зауглероженного (4), λ = 0.074 нм.

С увеличением кратности циркуляции от 2.2 до 8 (кривые 2 и 3 соответственно) наблюдается уширение и снижение интенсивности рефлексов фазы СоО, а линии, отвечающие Со0, заметно сужаются при росте их интенсивности. Оцененные по уравнению Шеррера размеры областей когерентного рассеяния (ОКР) металлического кобальта для исходного восстановленного и отработавших катализаторов приведены в табл. 3. ОКР Со0 для исходного восстановленного и отработавшего при Kц = 2.2 катализаторов одинаковы, тогда как в испытанном при Kц = 8 образце кристаллиты металлического кобальта несколько укрупняются.
Таблица 3.
Скорость дезактивации катализатора Rcd, содержание восков С19+ в конденсированных продуктах и средний размер кристаллитов Co0 катализаторов в зависимости от кратности циркуляции Kц*
| Катализатор | Средний размер** кристаллитов Со0, нм | Содержание C19+, % | Rcd, %/ч | |
|---|---|---|---|---|
| Исходный восстановленный | 8 ± 2 | – | – | |
| Отработавший | Kц = 1 | н/o | 35.1 | 0.16 ± 0.01 |
| Kц = 1.5 | н/o | 37.7 | 0.19 ± 0.01 | |
| Kц = 2.2 | 8 ± 2 | 45.0 | 0.23 ± 0.03 | |
| Kц = 8 | 11 ± 3 | 33.8 | 0.044 ± 0.003 | |
Таким образом, из данных порошковой рентгеновской дифракции следует:
1) в отработавших 100 ч в потоке катализаторах образования фазы карбида кобальта не наблюдается;
2) за 100 ч работы при небольшой кратности циркуляции (2.2) фазовый состав катализатора не меняется, размеры ОКР Со0 также остаются неизменными; с увеличением кратности циркуляции до 8 вклад фазы металлического кобальта и размеры кристаллитов Со0 возрастают за счет восстановления оксида кобальта(II).
Отсутствие карбида кобальта после 100 ч работы независимо подтверждается сравнением профилей ТПО (рис. 4) катализаторов, отработавших при разных кратностях циркуляции, и предварительно зауглероженного образца.
Рис. 4.
Профили ТПО катализаторов, отработавших в проточном режиме без циркуляции (1) и при Kц 1.5 (2), 2.2 (3) и 8 (4), а также предварительно зауглероженного образца (5).

Для зауглероженного образца зафиксированы два связанных с окислением карбида кобальта пика выделения СО2 (молекулярный ион, m/z = = 44) при 200 и 290°С, а также сигнал в области температур 300–500°С, который обычно относят к окислению аморфного и графитизированного углерода [11, 19]. На кривых ТПО всех отработавших катализаторов отсутствуют характерные для окисления Со2С пики выделения СО2. Наблюдающийся в интервале температур 265–275°С пик отвечает окислению углеводородов, оставшихся в порах катализатора после процедуры экстракции в аппарате Сокслета [19], а широкий сигнал в интервале 350–450°С обусловлен сгоранием углеродных отложений (сажа, графит), накопленных за время СФТ [11, 19].
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Линейные олефины, содержащие четыре или более атомов углерода, являются ценными строительными блоками в нефтехимическом синтезе [20, 21]. Это обстоятельство мотивирует поиск способов прямого производства олефинов из синтез-газа посредством СФТ путем так называемого процесса FTO (Fischer–Tropsch to olefins) [22, 23]. Увеличение селективности образования олефинов обычно достигается двумя методами – подбором подходящих катализаторов (промоутеров) [24, 25] и/или технологических условий [22, 26]. Находкой в настоящей работе является обнаружение неожиданного эффекта заметного возрастания селективности по олефинам при применении в СФТ на недорогом кобальт-силикагелевом катализаторе известного технологического приема – циркуляции хвостовых газов.
Высокую селективность образования олефинов в СФТ на кобальтовых катализаторах обычно связывают с формированием на поверхности катализатора карбида кобальта [24, 25, 27]. Однако исследования отработавших катализаторов высокочувствительными методами порошковой рентгеновской дифракции с использованием синхротронного излучения и ТПО с масс-спектральным анализом выделяющегося углекислого газа позволяют исключить эту возможность. Причины изменения селективности процесса в нашем случае, очевидно, следует искать в особенностях применения циркуляции хвостовых газов. Циркуляционный режим возвращает на вход реактора хвостовой газ, содержащий, в том числе, газообразные олефины, которые могут [28] реадсорбироваться на катализаторе и повторно включаться в процесс роста цепи. Действительно, мы нашли, что объемное содержание олефинов С2–С4 в хвостовых газах монотонно убывает с увеличением кратности циркуляции и достигает предельных значений, начиная с Kц ≈ 3 (рис. 5). Факт наличия таких предельных значений, очевидно, является следствием установления стационарного состояния по газообразным олефинам: скорость их образования становится равной скорости реадсорбции.

Включение газообразных олефинов в процесс роста цепи в результате их повторной адсорбции объясняет появление бимодальности в распределении парафинов по длине цепи и повышение селективности по воскам С19+ при небольших значениях кратности циркуляции (табл. 2). Вместе с тем дальнейшее увеличение Kц до 8 приводит к снижению селективности образования восков. Возможная причина этого состоит в том, что начиная с Kц ≈ 3 на вход реактора подается неизменное количество газообразных олефинов, отвечающее стационарному состоянию (рис. 5), поэтому они не вносят дополнительного вклада в процесс роста цепи. Однако возросший газовый поток уносит из реакционной зоны пары конденсированных олефинов С5+, что уменьшает время контакта последних и, соответственно, вероятность их реадсорбции с удлинением углеродной цепи или гидрированием.
Ранее было показано [8], что быстрая (за первые 100–150 ч в потоке) дезактивация катализатора СФТ Со-Al2O3/SiO2 при давлении 6.0 МПа и Kц = 2.2 обусловлена блокированием центров роста цепи синтезированными углеводородными восками: обнаружена корреляция скорости дезактивации катализатора с содержанием С19+ в составе конденсированных продуктов. В настоящей работе такая тенденция сохраняется для экспериментов в проточном режиме (Kц = 1) и при небольших значениях Kц (1.5 и 2.2): в указанных условиях увеличение Kц приводит к симбатному росту содержания восков в конденсированных продуктах СФТ и, как следствие, к ускорению дезактивации катализатора (табл. 3).
Дальнейшее повышение кратности циркуляции до 8 снижает селективность образования С19+ до значения, близкого к полученному в проточном режиме (Kц = 1), но при этом скорость дезактивации оказывается в 4 раза меньше величины Rcd для проточного режима. Такое замедление дезактивации при Kц = 8 (рис. 3) может быть обусловлено ростом поверхности активного металла за счет восстановления СоО реакционной смесью в ходе СФТ. Возможность восстановления оксида кобальта синтез-газом была продемонстрирована в работах [29, 30]. Восстановителями в системе могут служить СО и Н2, причем под действием моноксида углерода образуется Со0 в ГПУ-модификации, а под действием водорода – в ГЦК [29, 30]. Поскольку в нашем случае наблюдается исключительно ГЦК Со0, восстановителем в системе является водород. Причиной наблюдаемого при Kц = 8 восстановления оксида кобальта может быть существенное падение с ростом кратности циркуляции парциального давления паров воды (табл. 1), способной окислять металлический кобальт до его оксида [3].
Наблюдаемое с увеличением Kц возрастание селективности по метану также может быть следствием снижения парциального давления паров воды (табл. 1). Известно [31, 32], что метанообразование в СФТ на кобальт-силикагелевых катализаторах заметно подавляется при соотношении парциальных давлений паров воды и водорода ${{{{P}_{{{{{\text{H}}}_{{\text{2}}}}{\text{O}}}}}} \mathord{\left/ {\vphantom {{{{P}_{{{{{\text{H}}}_{{\text{2}}}}{\text{O}}}}}} {{{P}_{{{{{\text{H}}}_{{\text{2}}}}}}}}}} \right. \kern-0em} {{{P}_{{{{{\text{H}}}_{{\text{2}}}}}}}}}$ > 0.4. В нашем случае указанное условие выполняется только для проточного режима, Kц = 1.
ЗАКЛЮЧЕНИЕ
Использование режима циркуляции хвостовых газов при небольших значениях кратности (Kц < 3) способствует увеличению содержания восков С19+ в конденсированных продуктах синтеза за счет реадсорбции газообразных олефинов и включения их в процесс роста цепи. В свою очередь рост селективности по воскам приводит к ускорению дезактивации катализатора.
Повышение кратности циркуляции до 8 способствует эвакуации паров высших олефинов из реакционной зоны. Это проявляется в наблюдаемом возрастании селективности образования олефинов и в снижении выхода восков С19+ за счет уменьшения вероятности вторичных превращений олефинов С5+. В ходе СФТ при этих условиях отмечается восстановление СоО до активного в реакции металлического кобальта, что обусловливает замедление дезактивации катализатора.
Список литературы
Martinelli M., Gnanamani M.K., LeViness S., Jacobs G., Shafer W.D. // Appl. Catal. A: Gen. 2020. V. 608. № 117 740. https://doi.org/10.1016/j.apcata.2020.117740
Teimouri Z., Abatzoglou N., Dalai A.K. // Catalysts. 2021. V. 11. № 3. P. 330.
Wolf M., Gibson E.K., Olivier E.J., Neethling J.H., Catlow C.A., Fischer N., Claeys M. // Am. Chem. Soc. Catal. 2019. V. 9. № 6. P. 4902.
Wang Y.N., Xu Y.Y., Li Y.W., Zhao Y., Zhang B.J. // Chem. Eng. Sci. 2003. V. 58. P. 867.
Savost’yanov A.P., Yakovenko R.E., Sulima S.I., Bakun V.G., Narochnyi G.B., Chernyshev V.M., Mitchenko S.A. // Catal. Today. 2017. V. 279. P. 107.
Яковенко Р.Е., Зубков И.Н., Нарочный Г.Б., Папета О.П., Денисов О.Д., Савостьянов А.П. // Кинетика и катализ. 2020. Т. 61. № 2. С. 278. (Yakovenko R.E., Zubkov I.N., Narochniy G.B., Papeta O.P., Denisov O.D., Savost’yanov A.P. // Kinet. Catal. 2020. V. 61. № 2. P. 310.)
Savost’yanov A.P., Yakovenko R.E., Narochniy G.B., Sulima S.I., Bakun V.G., Soromotin V.N., Mitchenko S.A. // Catal. Commun. 2017. V. 99. P. 25.
Соромотин В.Н., Яковенко Р.Е., Медведев А.В., Митченко С.А. // Кинетика и катализ. 2021. Т. 62. № 6. С. 881. (Soromotin V.N., Yakovenko R.E., Medvedev A.V., Mitchenko S.A. // Kinet. Catal. 2021. V. 62. № 6. P. 845.)
Savost'yanov A.P., Eliseev O.L., Yakovenko R.E., Narochniy G.B., Maslakov K.I., Zubkov I.N., Soromotin V.N., Kozakov A.T., Nicolskii A.V., Mitchenko S.A. // Catal. Lett. 2020. V. 150. № 7. P. 1932.
Rahmati M., Safdari M., Fletcher T., Argyle M., Bartholomew C.H. // Chem. Rev. 2020. V. 120. № 10. P. 4455.
Moodley D., loosdrecht J., Saib A., Overett M., Datye A., Niemantsverdriet J. // Appl. Catal. A: Gen. 2009. V. 354. № 1–2. C. 102.
Choudhury H., Cheng X., Afzal S., Prakash A.V., Tatarchuk B.J., Elbashir N.O. // Catal. Today. 2020. V. 343. P. 112.
Yakovenko R.E., Savost’yanov A.P., Narochniy G.B., Soromotin V.N., Zubkov I.N., Papeta O.P., Svetogorov R.D., Mitchenko S.A. // Catal. Sci. Technol. 2020. V. 10. № 22. P. 7613.
Соромотин В.Н., Соколов А.Н., Яковенко Р.Е., Савостьянов А.П. // Изв. вузов. Северо-Кавказский регион. Технические науки. 2019. Т. 3. С. 38.
Ravenhorst I.K., Hoffman A.S., Vogt C., Boubnov A., Patra N., Oord R., Akatay C., Meirer F., Bare S.R., Weckhuysen B. // ACS Catal. 2021. V. 11. P. 2956.
Claeys M., Dry M.E., van Steen E., du Plessis E., van Berge P., Saib A., Moodley D. // J. Catal. 2014. V. 318. P. 193.
Svetogorov R.D., Dorovatovskii V.A., Lazarenko V.A. // Cryst. Res. Technol. 2020. V. 55. № 5. № 1900184. https://doi.org/10.1002/crat.201900184
PDF-2. The powder diffraction file TM. International Center for Diffraction Data (ICDD). 2012. URL: www.icdd.com
Keyvanloo K., Fisher M.J., Hecker W.C., Lancee R.J., Jacobs G., Bartholomew C.H. // J. Catal. 2015. V. 327. P. 33.
Голубь Ф.С., Болотов В.А., Пармон В.Н. // Катализ в промышленности. 2020. Т. 20. № 6. С. 433. (Golub F.S., Blolotov V.A., Parmon V.N. // Catal. Indust . 2021. V. 13. № 3. P. 203.)
Prasanseang W., Choojun K., Poo-arporn Y., Huang A., Lin Y., Sooknoi T. // Appl. Catal. A: Gen. 2022. V. 635. № 118555.
Yang J., Rodriguez C.L., Qi Y., Ma H., Holmen A., Chen D. // Appl. Catal. A: Gen. 2020. V. 598. № 117 564.
Jeske K., Kizilkaya C., Lopez-Luque I., Pfander N., Bartsch M., Concepcion P., Prieto G. // ACS Catal. 2021. V. 11. P. 4784.
Li Z., Yu D., Yang L. C., J., Xiao K., Yao N., Li X. // Am. Chem. Soc. Catal. 2021. V. 11. P. 2746.
Wang X., Lin T., Li J., Yu F., Lv D., Qi X., Wang H., Zhong L., Sun Y. // RCS Adv. 2019. V. 9. P. 4131.
Zhang Y., Yao Y., Shen J., Chang J., Gorimbo J., Liu X., Hildebrandt D. // Fuel. 2021. V. 302. № 121146.
Liu Y., He S., Yang R., Sun F., Yang Y., Mei B., Kang J., Wu D., Jiang Z. // Energy Mater. 2020. V. 55. № 21. P. 9037.
Petersen A.P., Claeys M., Kooyman P.J., van Steen E. // Catalysts. 2019. V. 9. № 10. 794. https://doi.org/10.3390/catal9100794
Pan Z., Parvari M., Bukur D.B. // Top. Catal. 2014. V. 57. № 6–9. P. 470.
Paterson J., Peacock M., Ferguson E., Purves R., Ojeda M. // ChemCatChem. 2017. V. 9. № 18. P. 3463.
Okoye-Chine C.G., Moyo M., Liu X.,№№ Hildebrandt D. // Fuel Process. Technol. 2019. V. 192. P. 105.
Dalai A.K., Davis B.H. // Appl. Catal. A: Gen.2008. V. 348. № 1. P. 1.
Дополнительные материалы отсутствуют.
Инструменты
Кинетика и катализ