

Кинетика и катализ, 2023, T. 64, № 1, стр. 86-96
О новом режиме каталитического окисления сульфита в присутствии Mn(II) в избытке ионов металла
А. Н. Ермаков *
Институт энергетических проблем химической физики РАН им. В.Л. Тальрозе ФИЦ ХФ им. Н.Н. Семенова РАН
119334 Москва, Ленинский просп., 38, корп. 2, Россия
* E-mail: polclouds@yandex.ru
Поступила в редакцию 23.08.2022
После доработки 04.10.2022
Принята к публикации 04.10.2022
- EDN: KGEKTE
- DOI: 10.31857/S045388112301001X
Аннотация
В работе рассматриваются данные о кинетике катализа ионами марганца(II) окисления сульфита в избытке ионов металла. Наряду с результатами опытов в растворах привлекалась информация и о динамике реакции в аэрозольных частицах. Впервые выявлен быстрый вырождено-разветвленный (ВР) режим реакции. Его динамику определяет скорость разветвления цепи с участием полупродукта ${\text{HSO}}_{5}^{ - }$ и ионов Mn(II). В работе приводятся оценки константы скорости этой реакции, и рассматривается критерий перехода реакции в ВР-режим. Показано, что наблюдаемое ускорение образования сульфатов в ВР-режиме в опытах с аэрозолем является результатом сопряжения каталитической реакции и захвата SO2 из газа. Расчеты в этих рамках динамики реакции находят согласие с данными опытов в smog chambers, а также с результатами мониторинга атмосферного аэрозоля.
ВВЕДЕНИЕ
Окисление сульфита (ОС) – исторически первая цепная жидкофазная реакция [1]. Расшифровкой ее механизма, в том числе в присутствии ионов переходных металлов (ПМ), долгое время занимались многие исследователи, см. обзоры [2, 3] и ссылки в них. Важным результатом изучения механизма катализа данного процесса ионами марганца стал вывод авторов настоящей работы о том, что эти ионы сами по себе каталитически не активны [4, 5]. Наблюдавшийся “катализ” ионами Mn окисления сульфита в [6–20] и др. связан с усилением действия неконтролируемых, ничтожных примесей ионов железа, [Fe]0 = (2–50) × 10–8 моль/л [4, 14, 18], содержащихся в воде или других реагентах любой степени очистки.
Каталитический процесс ОС широко применяется в сероочистке дымовых газов [21] и др. Ведущее место он занимает и в самоочищении атмосферы от естественных и антропогенных выбросов SO2 (так называемые кислотные дожди и другие виды загрязнений [22]). С ним связывают и участившиеся с 2013 г. случаи катастрофического обволакивания аэрозольной дымкой в зимнее время Пекина и других городов Юго-Востока Китая (концентрация аэрозоля доходит до ∼103 мкг/м3) [23–26]. Внимание привлекает аномально высокие скорости образования сульфатов в частицах: в расчете на газ WS(VI)_атм составляет десятки мкг м–3 ч–1 [23, 24]. И это вопреки обусловленным дымкой низким концентрациям в воздухе О3, ОН, Н2О2 и низкому уровню инсоляции, а также несмотря на экстремально малое содержание влаги в частицах (100–300 мкг/м3). В работе [26] в этой связи указывалось на нефотохимическое происхождение сульфатов в частицах дымки. О причастности ионов ПМ к образованию в них сульфатов говорят данные прямого контроля изотопного состава атомарного кислорода (Δ17O) в сульфатах аэрозоля не морского происхождения (non-seasalt, NSS) [27]. В работе [23] сообщалось также о совпадении по времени пиковых концентраций сульфатов (∼30 мкг/м3) и ионов марганца (∼70 нг/м3) в аэрозоле (Baoding, апрель, 2015). Вместе с тем экстраполяция данных о динамике каталитической реакции к атмосферным условиям не воспроизводит результаты натурных наблюдений [23, 26]. Найденные скорости жидкофазной реакции (wS(VI), моль л–1 с–1) при характерном содержании капельной влаги в аэрозоле и в пересчете на газ оказываются по крайней мере на порядок более низкими. На этом фоне загадочными и необъяснимыми остаются результаты недавних опытов по моделированию динамики каталитической реакции в аэрозольных частицах в темновых условиях так называемых smog chambers [23, 24, 28], которые указывают на многократно бóльшие значения wS(VI) в присутствии ионов марганца (“enhanced sulfate formation”). Их расхождение с данными экспериментов в растворах (далее в bulk условиях) было связано в [29] с формированием особых зон в частицах (“surface effects and potentially aerosol pH gradients”). Цель настоящей работы заключалась в том, чтобы, опираясь на опыт изучения авторами [4, 5, 30] катализа ионами ПМ окисления сульфита, раскрыть действительные причины ускорения этого процесса в аэрозольных частицах.
ПРЕДВАРИТЕЛЬНЫЕ СВЕДЕНИЯ
Ранее кинетику каталитического ОС в избытке ионов металла (α ≥ 1, рН ∼ 3), включая данные опытов с мелкими каплями на подложках [8, 10, 13], удалось непротиворечиво описать в рамках цепного механизма с участием в качестве переносчиков цепи ${\text{SO}}_{{3 - 5}}^{{ - \,}}$ и Mn(III) [4, 18]. При рассматриваемых условиях ионы 3-х валентного марганца присутствуют в растворе в виде MnOH2+ [18]. Здесь и далее α = [Mn(II)]/[S(IV)], а [S(IV)] ≈ [SO2(aq)] + [${\text{HSO}}_{3}^{ - }$], где SO2(aq) – H2SO3. Каталитическое действие ионов Mn обусловлено быстрыми реакциями (X), (XI) с участием переносчиков цепи ${\text{SO}}_{{3 - 5}}^{ - }$ [18] и Mn(III) [31] (табл. 1), а также смещением распределения ионов железа по зарядовым формам (ζMn = [Fe(III)]/[Fe(II)]) в пользу Fe(III) – участника инициирования (I) цепей [4]. Основной валентной формой ионов примесного железа в кислых растворах оказываются ионы Fe(III), т.е. ζMn ≈ 1 и [Fe(III)] ≈ [Fe]0 [4, 19]. В стационарных условиях для скорости реакции было найдено: wS(VI)= k3/2_набл[S(IV)]3/2 [30]. Здесь k3/2_набл = 2kXI(kIχ*[Fe]0/kXIII)1/2 – наблюдаемая константа скорости полуторного порядка, л1/2 моль–1/2 с–1, где χ* = χ/[S(IV)] и χ = = [Fe(OH)SO3H+]/[Fe(III)] – доля сульфитных комплексов ионов 3-х валентного железа в их общем содержании в растворе в виде Fe3+, FeOH2+, ${\text{FeHSO}}_{3}^{{2 + }}$, Fe(OH)SO3H+, ${\text{Fe(OH}})_{2}^{ + }$, ${\text{FeSO}}_{3}^{ + }$ и Fe(ОН)SO3, [2, 30]. Происхождение полуторного по сульфиту и нулевого по ионам марганца порядков реакции в этих рамках связывалось в [30] с лимитирующей ролью реакций с участием сульфита при инициировании (I) и продолжении цепи (XI). Рассчитанные k3/2_набл [30] остаются близкими к постоянству: ∼12 л1/2 моль–1/2 с–1 в диапазоне концентраций сульфита 10–6 ≤ ≤ [S(IV)] ≤ 10–3 моль/л ионов марганца 10–5 ≤ ≤ [Mn(II)] ≤ 10–2 моль/л.
Таблица 1.
Механизм катализа ионами марганца окисления сульфита, ВР-режим реакции
| № | Реакция | Константа скорости, л моль–1 с–1 |
|---|---|---|
| I | Fe(OH)SO3H+ → Fe2+ + ${\text{SO}}_{3}^{{ - \,\centerdot }}$+ H2O | *0.2 |
| II | ${\text{SO}}_{3}^{ - }$ + O2 → ${\text{SO}}_{5}^{{ - \,\centerdot }}$ | 2.5 × 109 |
| IIIа | ${\text{SO}}_{5}^{{ - \,\centerdot }}$ + HSO3 → ${\text{HSO}}_{5}^{ - }$ + ${\text{SO}}_{3}^{{ - \,\centerdot }}$ | 3.4 × 103 |
| IIIb | ${\text{SO}}_{5}^{{ - \,\centerdot }}$ + H${\text{SO}}_{3}^{ - }$ → ${\text{SO}}_{4}^{{2 - }}$ + ${\text{SO}}_{4}^{{ - \,\centerdot }}$ + H+ | 2 × 102 |
| IV | ${\text{SO}}_{4}^{{ - \,\centerdot }}$ + H${\text{SO}}_{3}^{ - }$ → ${\text{SO}}_{4}^{{2 - }}$ + ${\text{SO}}_{3}^{{ - \,\centerdot }}$+ H+ | 7.5 × 108 |
| Va | ${\text{SO}}_{5}^{{ - \,\centerdot }}$ + ${\text{SO}}_{5}^{{ - \,\centerdot }}$ → ${\text{SO}}_{4}^{{ - \,\centerdot }}$+ ${\text{SO}}_{4}^{ - }$ + O2 | 8.7 × 107 |
| Vb | ${\text{SO}}_{5}^{{ - \,\centerdot }}$ + ${\text{SO}}_{5}^{{ - \,\centerdot }}$ → ${{{\text{S}}}_{{\text{2}}}}{\text{O}}_{8}^{{2 - }}$ + O2 | 1.3 × 107 |
| VI | ${\text{HSO}}_{5}^{ - }$ + ${\text{HSO}}_{3}^{ - }$ + H + → 2${\text{SO}}_{4}^{{2 - }}$ + 3H+ | **107 |
| VII | Fe2+ + ${\text{SO}}_{5}^{ - }$$\xrightarrow{{{{{\text{H}}}^{ + }}}}$ Fe3+ + ${\text{HSO}}_{5}^{ - }$ | 3.2 × 106 |
| VIII | Fe2+ + ${\text{SO}}_{4}^{{ - \,\centerdot }}$→ Fe3+ + ${\text{SO}}_{4}^{{2 - }}$ | 3.0 × 108 |
| IX | Fe2+ + ${\text{HSO}}_{5}^{ - }$ → Fe3+ + ${\text{SO}}_{4}^{{ - \,\centerdot }}$+ OН– | 3.5 × 104 |
| X | Mn2+ + ${\text{SO}}_{5}^{{ - \,\centerdot }}$$\xrightarrow{{{{{\text{H}}}^{ + }}}}$ ***Mn(III) + ${\text{HSO}}_{5}^{ - }$ | 108 |
| XIa | Mn(III) + ****SO2(aq) → Mn2+ + ${\text{SO}}_{3}^{{ - \,\centerdot }}$ + H2O + H+ | ∼3 × 105 |
| XIb | Mn(III) + H${\text{SO}}_{3}^{ - }$ → Mn2+ + ${\text{SO}}_{3}^{{ - \,\centerdot }}$ + H2O | ∼106 |
| XII | Mn(III) + ${\text{SO}}_{5}^{ - }$$\xrightarrow{{{{{\text{H}}}^{ + }}}}$ Mn(IV) + ${\text{HSO}}_{5}^{ - }$ | ∼108 |
| XIII | Mn(III) + Mn(III) → Mn2+ + Mn(IV) | ∼105 |
| XIV | Mn2+ + ${\text{HSO}}_{5}^{ - }$ → Mn(III) + ${\text{SO}}_{4}^{{ - \,\centerdot }}$ | ∼15 |
| XV | Mn2+ + ${\text{SO}}_{4}^{{ - \,\centerdot }}$→ Mn(III) + ${\text{SO}}_{4}^{{2 - }}$ | 2 × 107 |
Тем удивительнее оказываются результаты опытов, в которых, несмотря на избыток ионов металла, наблюдаются отклонения от этой картины. Так, в [18] при рН 4 и α ≥ 5 сообщалось об ускорении реакции с увеличением [Mn(II)]. Отмечались также признаки роста порядка реакции по ионам металла от 0-го к 1-ому при одновременном снижении порядка реакции и по сульфиту (wS(VI) ≈ [Mn(II)][S(IV)]). Подобное поведение реакции отмечается и в более кислых растворах, но при бóльшем избытке ионов металла над сульфитом (α ≈ 106). О росте wS(VI) с повышением [Mn(II)] и изменении порядков по ионам металла от 0-го до 1-го и сульфита (до 1-го) в опытах в аэрозоле сообщалось в [28] и при рН ≤ 1.5. Это притом, что в работе в [18] при рН 2.4 в bulk условиях нулевой по ионам марганца порядок реакции наблюдался и при α ≈ 30! Постоянство скорости реакции с ростом [Mn(II)] при рН 2.4 авторы [18] относили на полное связывание сульфита в комплексы ${\text{MnHSO}}_{3}^{ + }$ (β ≈ 3 × 104 л/моль) при [S(IV)]0 ≈ β–1. Здесь β – константа равновесия образования комплекса ${\text{MnHSO}}_{3}^{ + }$. Наблюдавшийся в исследовании [18] в избытке ионов металла рост скорости ОС с увеличением [Mn(II)] при рН 4 приписывался этими же авторами в [32] реакциям с участием смешанного комплекса (ОН)MnIIHSO3–ОMnIII. Подчеркнем, что прямых свидетельств существования таких комплексов в сульфитных растворах нет. Их участие в ОС ставит под сомнение данные [8] и [18] о соотношении скоростей реакций ОС в указанных работах (wS(VI)_8/wS(VI)_18 ≈ 180! [30]). И это несмотря на одинаковый (1-ый согласно [8] и [18]) по сульфиту порядок и лишь ∼30-ти кратное превышение концентрации сульфита в [8] в сравнении с [18]. Здесь цифры в подстрочнике отражают номера ссылок на источники.
О НОВОМ РЕЖИМЕ РЕАКЦИИ В ИЗБЫТКЕ ИОНОВ МЕТАЛЛА
Отмеченные отклонения в поведении реакции в избытке ионов металла в bulk условиях и в опытах с аэрозолем [18, 28] не противоречат, однако, выявленному нами в [30] режиму реакции при α ≥ 1, см. выше. Приводившимся выше результатам [18, 28], как и данным опытов [23, 24], удается найти естественное объяснение, рассматривая при вариациях рН и [Mn(II)] переход реакции в быстрый и неизученный ранее новый вырождено-разветвленный (ВР) режим. Ключевым элементом его механизма является разветвление цепи:
(XIVa), (XIVb)
${\text{HSO}}_{5}^{ - }{\text{ + M}}{{{\text{n}}}^{{2 + }}} \to {\text{MnO}}{{{\text{H}}}^{{2 + }}}({\text{Mn(III)}}) + {\text{OH/SO}}_{4}^{{ - \,}} + {\text{HSO}}_{4}^{ - }.$Взамен расходуемого одного активного центра ${\text{HSO}}_{5}^{ - }$ в этой реакции в растворе возникает два новых переносчика цепи: Mn(III) и ${\text{OH/SO}}_{4}^{{ - \,}}$ [33], что сопровождает рост их концентраций, а вместе с тем и увеличение wS(VI). При низких рН (2.4) и невысоких [Mn(II)], как в работе [18], их рост ограничивает расход ${\text{HSO}}_{5}^{ - }$ в основном в реакции (VI). Каталитический процесс описывается при этом полуторным по сульфиту и нулевым по ионам марганца порядками реакции [8, 18, 30], см. выше. При более высоких рН [32] или [Mn(II)], как в [28], все бóльшая часть ${\text{HSO}}_{5}^{ - }$ исчезает в параллельном разветвлении цепи с участием ионов марганца (XIV), ведущем в конечном итоге к переходу реакции в ВР-режим. Именно в перераспределении по этим каналам расходования ${\text{HSO}}_{5}^{ - }$, вызванном ростом рН/концентрации ионов марганца, и видятся причины отклонений в поведении реакции в избытке ионов металла по данным [18, 28], происхождение которых в действительности служит отражением перехода реакции в ВР-режим.
О КИНЕТИКЕ РЕАКЦИИ ${\text{HSO}}_{5}^{ - }$ + Mn2+
Для идентификации перехода реакции в ВР-режим необходимы данные о константе скорости реакции разветвления цепей (XIV). В их отсутствие приводившееся значение α ≈ 5 (рН 4), при котором в [18] отмечались признаки прироста wS(VI) с увеличением [Mn(II)], можно в первом приближении связать с началом перераспределения расходования ${\text{HSO}}_{5}^{ - }$ по реакциям (VI) и (XIV). Для константы скорости разветвления цепи будем иметь отсюда:
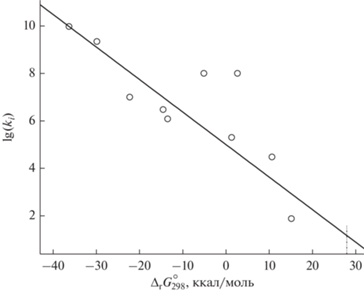
Независимую оценку kXIV получим, основываясь на известной линейной корреляции между константами скорости (lg ki) и изменением энергии Гиббса (Δr(i)$G_{{298}}^{^\circ }$, ккал/моль) в ряду однотипных реакций, см. [33].С этой целью рассматривали реакции с участием ионов металлов (Fe2+, Cu+, Mn2+) и соединений (H2O2, ${{{\text{S}}}_{{\text{2}}}}{\text{O}}_{8}^{{2 - }}$, ${\text{H}}{{{\text{O}}}_{2}}{\text{/O}}_{2}^{ - }$, ${\text{SO}}_{5}^{ - }$, ${\text{HSO}}_{5}^{ - }$) с перекисной группой. При этом Δr(i)$G_{{298}}^{^\circ }$ (–45–27.8 ккал/моль) рассчитывали по данным о Δf(i)$G_{{298\_i}}^{^\circ }$ и величинам стандартных редокс потенциалов $E_{{298(i)}}^{^\circ }$ (В) реагентов [34]. Различающиеся на ∼9 порядков константы скорости реакций (1.11010–12 л моль–1 с–1) заимствовали из [35]. Из сопоставления lg(ki) и Δr(i)$G_{{298}}^{^\circ }$ следует, что, несмотря на определенный разброс, прослеживается неплохая в целом линейная их корреляция, рис 1. Для константы разветвления, не противоречащей приводимой выше ее оценке по данным опытов [18], будем иметь отсюда: kXIV ≈ 15 л моль–1 с–1. Заметим, что в [33] и др. отмечалось, что в (XIV) возможны два канала реакции разветвления. Первый (XIVa) сопровождает выделение (Δr$G_{{298}}^{^\circ }$ < 0), а второй (XIVb) – поглощение энергии Гиббса (Δr$G_{{298}}^{^\circ }$ > 0), их значения составляют ∼–15.6 и ∼27.8 ккал/моль соответственно. Доминирующим оказывается, по-видимому, канал образования ${\text{SO}}_{4}^{ - }$ и Mn(III). На это указали опыты с добавками бензола к растворам S(IV)/Mn(II)/${\text{HSO}}_{5}^{ - }$ в присутствии трет-бутанола, не вступающего в реакцию с радикалами ${\text{SO}}_{4}^{ - }$, но взаимодействующего с радикалами ОН. Полученные результаты подтвердили, что формирование в продуктах фенола происходит в основном по реакции ${\text{SO}}_{4}^{ - }$ + + С6Н6 → С6Н5ОН [36].
Рис. 1.
Корреляция констант скорости ki (л моль–1 с–1) и изменений энергии Гиббса Δr(i)$G_{{298}}^{^\circ }$ (ккал/моль) по ходу однотипных реакций с участием ионов переходных металлов и соединений, содержащих перекисную группу, по данным [34, 35]. Пунктир отвечает изменению энергии Гиббса (∼27.8 ккал/моль) в реакции разветвления цепи с участием полупродукта ${\text{HSO}}_{5}^{ - }$ и ионов Mn2+ (XIVb).
О КРИТЕРИИ ПЕРЕХОДА РЕАКЦИИ В ВР-РЕЖИМ
Для разграничения медленного и быстрого режимов каталитического процесса в избытке ионов металла будем рассматривать конкуренцию между разветвлением цепи (XIV) и автокаталитической реакцией с участием ${\text{HSO}}_{5}^{ - }$ (VI). Приравнивая их скорости, находим:
Здесь K – константа равновесия ионизации диоксида серы, моль/л (табл. 2). На рис. 2 в координатах “α–рН” показана зависимость αкр = f(pH) при µ ≈ 0 и Т = 298 К, где µ – ионная сила раствора, моль/л. Разные значки соответствуют концентрационным условиям ряда bulk опытов (светлые кружки), опытов с аэрозолем (темные кружки) и опытов по контролю частиц дымки в реальной атмосфере [37] (крестики). При этом считали, что kVI = kVI × [${\text{H}}_{{{\text{aq}}}}^{ + }$] [38], где [${\text{H}}_{{{\text{aq}}}}^{ + }$] – концентрация свободных протонов в растворе, моль/л (табл. 1). Отчетливо видно различие условий для медленного (α < αкр) и быстрого (α > αкр) режимов реакции в избытке ионов металла. Видно, что ВР-режим реакции характеризуют, во-первых, бóльшие по величине [Mn(II)]/[S(IV)], чем в медленном цепном режиме. Во-вторых, соотношения [Mn(II)]/[S(IV)] оказываются в ВР-режиме тем выше, чем ниже рН, т.е., чем быстрее протекает реакция (VI), ведущая к снижению содержания ${\text{HSO}}_{5}^{ - }$ и подавлению разветвления цепи (XIV), подтверждая, таким образом, наличие конкуренции реакций (VI) и (XIV) в переходе в ВР-режим.
Таблица 2.
Равновесия растворения и ионизации компонентов каталитического окисления сульфита
| Параметр | Равновесие | Константа равновесия; [ссылка] |
|---|---|---|
| ${{H}_{{{\text{S}}{{{\text{O}}}_{{\text{2}}}}}}}$(T) | SO2 ⇆ SO2(aq) | 1.23 exp(3145.3(1/T – 1/298), моль л–1 атм–1; [21] |
| K(T) | SO2(aq) ⇆ H${\text{SO}}_{3}^{ - }$ + H+ | 1.310–2 exp(1960(1/T – 1/298), моль/л; [24] |
| ${{H}_{{{\text{S}}{{{\text{O}}}_{{\text{2}}}}}}}$(μ) | SO2 ⇆ SO2(aq) | lg($H_{{{\text{S}}{{{\text{O}}}_{{\text{2}}}}}}^{\mu }$/$H_{{{\text{S}}{{{\text{O}}}_{{\text{2}}}}}}^{{\mu = 0}}$) = (22.3/T – 0.0997)μ; [24, 39] |
| K(μ) | SO2(aq) ⇆ H${\text{SO}}_{3}^{ - }$ + H+ | lg(Kμ/Kμ=0 ) = 0.5μ1/2 – 0.31μ; [24, 39] |
Рис. 2.
Кинетическая диаграмма каталитического окисления сульфита в присутствии ионов марганца. Кривой αкр = f(pH) показан критерий перехода каталитической реакции в избытке ионов металла в ВР-режим (μ ≈ 0, Т = 298 К). Светлые кружки – опыты в растворах [8, 18]; темные кружки – опыты в аэрозольных частицах [10, 13]; крестики – концентрационные условия контроля частиц дымки в атмосфере [37]. Кривая ϕкр = f(pH) разграничивает области с [${\text{HSO}}_{5}^{ - }$]/[S(IV)] > 1 (выше кривой) и [${\text{HSO}}_{5}^{ - }$]/[S(IV)] < 1 (ниже кривой) при подпитке сульфитом.

При постоянстве рН этот переход реакции по мере увеличения [Mn(II)]/[S(IV)] сопровождает, как отмечалось, не только рост wS(VI), но и изменение порядка реакции по ионам металла. Так, по данным [18] при рН 2.4 и α ≈ 25 ($ \ll $αкр) для скорости реакции в bulk условиях имеем нулевой по ионам марганца порядок и wS(VI)_18 ≈ 10–6 моль л–1 с–1. В тоже время работе в [24], где были описаны опыты с аэрозолем при не слишком отличном рН (2.8), но при α ≈ 105 (>αкр), сообщалось о почти на 3 порядка бóльшей скорости реакции (wS(VI)_24 ≈ ≈ 6 × 10–4 моль л–1 с–1) и первом(!) по ионам металла порядке реакции. При заданной кислотности влаги в частицах такой рост [Mn(II)]/[S(IV)] в [24] потребовался для смещения конкуренции (VI), (XIV) к (XIV). Это вновь подтверждает не только ключевую роль разветвления цепи с участием ионов марганца для перехода реакции в ВР-режим, но также указывает и на не слишком высокое значение kXIV.
Из рис. 2 с очевидностью следует также, что вопреки “surface effects and potentially aerosol pH gradients” [29] режим ВР-реакции в аэрозольных частицахпри α < αкр невозможен, см. темные кружки, располагающиеся ниже кривой αкр = = f(pH). Этот вывод вытекает и из результатов сравнения в сопоставимых условиях данных о кинетике ОС в bulk условиях [8, 18] и в неподвижных мелких каплях [10, 13] при рН ≤ 3 и α < 102, которые указывают на близкие wS(VI) и, следовательно, тождественность механизма образования S(VI). Подытоживая вышесказанное, можно утверждать, что наблюдаемое в опытах с аэрозолем усиление каталитической активности ионов марганца не связано с формированием реакционных зон в частицах. Это повышение их активности при рассматриваемых рН и α, т.е. специфика реакции в аэрозоле по данным [29], отражает единственно переход реакции в ВР-режим, рис. 2.
О КИНЕТИКЕ РЕАКЦИИ В ВР-РЕЖИМЕ
Ранее при рассмотрении кинетики каталитического ОС в избытке ионов металла (α < αкр) авторами [30] было показано, что стационарный режим реакции в таких условиях достигается при равенстве скоростей разветвления цепи с участием ионов железа (IX) и гибели переносчиков цепи в квадратичной реакции рекомбинации (XIII) [30]. По аналогии в ВР-режиме прирост концентрации переносчиков цепи в разветвлении цепи (XIV) уравновешивает их гибель в (XIII). При этом накопление сульфатов осуществляется в основном по реакции (XV), скорость которой лимитирует реакция (XIV). Для скорости наработки сульфатов в аэрозоле в этих рамках находим:
(1)
$\begin{gathered} {{w}_{{S({\text{VI}})}}} = k_{{{\text{XIа}}}}^{2}{{k}_{{{\text{XIV}}}}}{{\left( {1 + \delta K\left( {{\mu }} \right){\text{/}}\left[ {{\text{H}}_{{{\text{aq}}}}^{ + }} \right]} \right)}^{2}}\left[ {{\text{Mn}}\left( {{\text{II}}} \right)} \right] \times \\ \times \,\,{{H}_{{{\text{S}}{{{\text{O}}}_{{\text{2}}}}}}}{{P}_{{{\text{S}}{{{\text{O}}}_{{\text{2}}}}}}}{\text{/}}{{k}_{{{\text{VI}}}}}{{k}_{{{\text{XIII}}}}}K\left( \mu \right). \\ \end{gathered} $Здесь ${{H}_{{{\text{S}}{{{\text{O}}}_{{\text{2}}}}}}}$ – коэффициент Генри, описывающий физическую растворимость SO2 (1.23 моль л–1 атм–1 при Т = 298 К [33]), табл. 2, а ${{P}_{{{\text{S}}{{{\text{O}}}_{{\text{2}}}}}}}$ – парциальное давление SO2, атм. Было принято, что реакция (XI) проходит как с участием SO2(aq) (XIa), так и ${\text{HSO}}_{3}^{ - }$ (XIb) [18] и δ = kXIb/kXIa, тогда как автокаталитическая реакция (VI) считалась протекающей только с участием ${\text{HSO}}_{3}^{ - }$ [38], табл. 1. Предполагалось также, что по ходу реакции [Mn(III)]/[Mn(II)] $ \ll $ 1.
Видно, что кинетику реакции в избытке ионов металла в ВР-режиме характеризует первые порядки по ионам металла и сульфиту: wS(VI) ≈ ≈ [Mn(II)]${{H}_{{{\text{S}}{{{\text{O}}}_{{\text{2}}}}}}}{{P}_{{{\text{S}}{{{\text{O}}}_{{\text{2}}}}}}}$. Внимание привлекает отсутствие в выражении для wS(VI) концентрации ионов железа – участника инициирования реакции (I), табл. 1. Напомним, что при α < αкр их концентрация в явном виде фигурирует в выражении для k3/2_набл, см. выше. Сказанное означает, что ионы железа в ВР-режиме проявляют себя лишь на стадии зарождения первичных переносчиков цепи (${\text{SO}}_{3}^{ - }$) (I). Для темпов прироста концентраций переносчиков цепи в разветвлении цепей (XIV) будем иметь: f = kXIV × [Mn(II)], с–1. При [Mn(II)] = = 0.2 моль/л (α ≈ 106), например, как в [28], их концентрации нарастают по ходу реакции почти втрое за ∼0.3 с! Столь бурный их рост маскирует, очевидно, участие ионов Fe в медленной генерации переносчиков цепи в (I). Об этом свидетельствует и отмечавшийся в опытах [28] слабый отклик wS(VI) на вариации [Fe(III)] (≤1.7 моль/л) при низких рН (≤1.5). В работе [28] это послужило поводом для выражения скепсиса в отношении значимости синергизма пары ионов Mn/Fe при образовании S(VI) в частицах атмосферной дымки. Напомним, однако, что в отсутствие ионов железа катализ ОС ионами марганца(II) был бы невозможен [4, 5]. Именно вместе они образуют синергическую пару, в которой ионы Mn(II) многократно ускоряют каталитическую реакцию при переходе в ВР-режим.
Из уравнения (1) для наблюдаемой константы скорости реакции в ВР-режиме находим:
(2)
${{k}_{{{\text{набл}}}}} = k_{{{\text{XIa}}}}^{2}{{k}_{{{\text{XIV}}}}}{{(1 + \delta K\left( {{\mu }} \right){\text{/}}[{\text{H}}_{{{\text{aq}}}}^{ + }])}^{2}}{\text{/}}{{k}_{{{\text{VI}}}}}K\left( {{\mu }} \right).$Становится очевидной сильная зависимость wS(VI) от рН для каталитической реакции в ВР-режиме. При этом необходимо считаться и с влиянием ионной силы на комбинацию констант скорости реакций ($k_{{{\text{XIa}}}}^{2}$ kXIV/kVIkXIII), а также на величины δ, K(µ), [${\text{H}}_{{{\text{aq}}}}^{ + }$] и содержание сульфита в растворе (${{H}_{{{\text{S}}{{{\text{O}}}_{{\text{2}}}}}}}$ = f(µ) [39]). Незнание вида этой зависимости послужило в [28] поводом для необоснованных сомнений авторов в корректности наблюдаемых в [24] на несколько порядков более высоких wS(VI) при рН 2.8 в сравнении с измеренными их значениями в [28] при рН ≤ 1.5, см. выше.
При кислотности растворов δ × K298(µ)/[${\text{H}}_{{{\text{aq}}}}^{ + }$] $ \ll $ 1 будем иметь:
При рН ≤ 1.5, например, как в опытах [28], приходим к:
Здесь 105 л моль–1 с–1 – константа скорости реакции (XIII) [30], а 5 × 10–3 моль/л – константа равновесия ионизации SO2 при Т = 298 К и μ ≈ ≈ 5 моль/л [24, 39], табл. 2. Рассчитывая по данным этой публикации kнабл_изм = wS(VI)/${{H}_{{{\text{S}}{{{\text{O}}}_{{\text{2}}}}}}}$ × ${{P}_{{{\text{S}}{{{\text{O}}}_{{\text{2}}}}}}}$ × × [Mn(II)] ≈ 300 л моль–1 с–1 при рН ∼1, μ ≈ ≈ 5 моль/л, [Mn(II)] = 0.2 моль/л и ${{P}_{{{\text{S}}{{{\text{O}}}_{{\text{2}}}}}}}$ = 150 ppb, для численного значения этой константы находим:
С учетом отмеченного в [23] слабого влияния µ на kнабл при невысоких рН, в настоящих расчетах принималось, что kXIa(µ ≈ 0) ≈ kXIa(µ ≈ 5 моль/л). Повторяя вычисления для иных ${{P}_{{{\text{S}}{{{\text{O}}}_{{\text{2}}}}}}}$, [Mn(II)] и t в [28] и усредняя эти результаты, приходим к δ(μ ≈ 5 моль/л) = kXIb/kXIa ≈ 3, где t – время наработки сульфатов в аэрозольных частицах в опытах в smog chamber, с. Найденная нами усредненная величина комбинации констант скорости
Полученное выражение (1) для скорости реакции в ВР-режиме находится в согласии с 1-ыми порядками по ионам металла и S(IV), определенными опытным путем в [23, 24]. В тоже время в [28] при рН ≤ 1.5 (α > αкр, μ ≤ 5 моль/л) сообщалось о более высоком порядке по S(IV):
В рамках ВР-механизма этот “прирост” в порядке по сульфиту (0.3) [28] является следствием выявленного в цитируемой работе замедления реакции по мере закисления влаги в аэрозольных частицах. Во избежание этого в [23, 24] использовали добавки буфера или в воздушную смесь вводили примесь аммиака. Сравнивая выражение [28] с уравнением (1), приходим к:
При [${\text{H}}_{{{\text{aq}}}}^{ + }$] ≈ 0.1 моль/л и ${{P}_{{{\text{S}}{{{\text{O}}}_{{\text{2}}}}}}}$ = 150 ppb (K(μ) ≈ 6 × × 10–3 моль/л [39]), как в опытах [28], будем иметь kнабл_изм ≈ 200 л моль–1 с–1, что не слишком отклонятся от рассчитанных нами kнабл_изм и говорит о 1-ом в действительности по S(IV) порядке реакции и в [28].
О СПЕЦИФИКЕ КАТАЛИТИЧЕСКОЙ РЕАКЦИИ В АЭРОЗОЛЕ В ВР-РЕЖИМЕ
Неоспоримым свидетельством этой специфики выступают различия динамики каталитической реакции в bulk условиях и в аэрозольных частицах, которые присущи, однако, лишь ВР-режиму реакции, рис. 2. Переход реакции в ВР-режим – важная, но не единственная причина наблюдаемого на опыте усиления каталитической активности ионов марганца, что проявляется в бóльших wS(VI) в сравнении с теми, что были найдены в bulk условиях. Еще одной причиной ускорения наработки сульфатов в аэрозольных частицах в сравнении с bulk условиями служит близость к постоянству по ходу реакции концентраций сульфита и промежуточного продукта ${\text{HSO}}_{5}^{ - }$ в частицах. Их постоянство благодаря высокому соотношению поверхности к объему частиц (S/V ≥ 104 [23, 24, 28]) в опытах с аэрозолем поддерживает быстрый захват из газа SO2. При этом в виду высокой скорости разветвления цепей концентрация ${\text{HSO}}_{5}^{ - }$ в частицах может в таких условиях быть и больше уровня [S(IV)]. В сопряжении быстрых каталитической реакции и подпитки реагентом и видится физико-химический механизм ускорения каталитической реакции в аэрозольной фазе в ВР-режиме в сравнении с опытами в bulk условиях [23, 24, 28]. В bulk условиях подобное ускорение каталитической реакции в условиях подпитки сульфитом следует ожидать при барботаже растворов солей марганца воздушными смесями, содержащими микропримесь SO2, что представляет интерес для разработки новых подходов к сероочистке дымовых газов. О таких экспериментах, но лишь при α < αкр, сообщалось в [11, 12, 21].
В стационарных условиях для концентрации полупродукта ${\text{HSO}}_{5}^{ - }$ в аэрозольной фазе в ВР-режиме находим:
Отсюда:
При рН ∼1.5, [Mn(II)] = 0.2 моль/л и ${{P}_{{{\text{S}}{{{\text{O}}}_{{\text{2}}}}}}}$ = 150 ppb, например, приходим к [${\text{HSO}}_{5}^{ - }$]/[S(IV)] ≈ 4 × 103! В тоже время в щелочных растворах в bulk условиях, т.е. при непостоянстве концентраций сульфита и ${\text{HSO}}_{5}^{ - }$ и практически полном подавлении (VI), концентрация ${\text{HSO}}_{5}^{ - }$ не превысила и ∼10% от концентрации сульфита(!) [40]. О потенциально высоких концентрациях ${\text{HSO}}_{5}^{ - }$ в облачных каплях и важной роли этого полупродукта в атмосфере указывалось и в [41].
На рис. 2 показана рассчитанная нами кривая ϕкр = [${\text{HSO}}_{5}^{ - }$]/[S(IV)] = 1, демонстрирующая сочетание α и pH, обеспечивающих равенство концентраций ${\text{HSO}}_{5}^{ - }$ и сульфита в ВР-режиме реакции при μ ≈ 0. Выше этой кривой располагается область, в которой ϕ > ϕкр, а ниже – ϕ < ϕкр. Видно, что точки на рис. 2, отвечающие концентрационным условиям опытов в smog chambers и контролю содержания сульфатов в частицах атмосферной дымки, располагаются над кривой ϕкр, подтверждая, таким образом, наличие значительных концентраций ${\text{HSO}}_{5}^{ - }$ в частицах и намного бóльшие потому wS(VI), чем в bulk условиях. Отсюда явствует также, что ВР-режим при низких рН возможен лишь при самых высоких [Mn(II)]. Это объясняется высокой скоростью реакции (VI). Преобладание (XIV) над (VI) в таких условиях требует концентрированных по [Mn(II)] растворов. Бóльшую скорость образования сульфатов в аэрозольных частицах в сравнении с наблюдаемой в bulk условиях [18] обеспечивают высокие концентрации переносчиков цепи, обусловленные разветвлением цепи в (XIV). Напротив, высокие скорости реакции в аэрозоле в ВР режиме при бóльших рН достигаются уже при относительно низких [Mn(II)] [23, 24]. Причиной служит рост [S(IV)], вызванный увеличением растворимости SO2. Одновременно с этим нарастает скорость реакции (XI) и, как результат, возрастают [${\text{HSO}}_{5}^{ - }$] и wS(VI).
СРАВНЕНИЕ С ДАННЫМИ ЛАБОРАТОРНОГО МОДЕЛИРОВАНИЯ
На рис. 3 представлены найденные корреляции рассчитанных по ходу каталитической реакции концентраций сульфатов ([S(VI)]расч) в ВР-режиме и их измеренных величин по результатам smog chambers опытов ([S(VI)]изм) [23, 28]. Ввиду неопределенности концентрационных условий в опытах с аэрозолем [9, 42–44] эти данные, хотя и указывающие на достаточно высокий уровень wS(VI), в настоящей работе к рассмотрению не привлекались.
Рис. 3.
Соответствие рассчитанных в настоящей работе ([S(VI)]расч) и измеренных ([S(VI)]изм) в работах [23, 28] концентраций сульфатов в частицах. Сплошная линия – соответствие этих данных при коэффициенте корреляции равном единице. На вставке показана расчетная зависимость скорости реакции от рН (сплошная кривая); точкам соответствуют экспериментальные данные [23, 24, 28], см. комментарии в тексте.

При вычислениях [S(VI)]расч принимали во внимание различие ионной силы и кислотности растворов в опытах [23, 28]. Для исключения их влияния учитывали влияние ионной силы на ${{H}_{{{\text{S}}{{{\text{O}}}_{{\text{2}}}}}}}$(μ) и K(μ), см. табл. 2 в [39]:
Усредненное по ∼25 индивидуальным измерениям значение $k_{{{\text{набл}}}}^{*}$ не зависит ни от рН (∼0.3–3.7), ни от ионной силы растворов (∼2–10 моль/л), и при Т = 298 К оно составило 1.4 л моль–1 с–1. Это значение $k_{{{\text{набл}}}}^{*}$ было использовано ниже при расчетах [S(VI)]расч в smog chambers опытах.
Концентрации сульфатов вычисляли по выражению:
Здесь L – объемная доля частиц, см3 аэрозоль/см3 воздуха, а t – время экспозиции, с, в экспериментах. Величины L, [${\text{H}}_{{{\text{aq}}}}^{ + }$] и γH (безразмерный коэффициент активности протонов в расчете на мольную долю) применительно к условиям опытов [23, 28] рассчитывали с применением модели E-AIM [45] с учетом данных об относительной влажности газовых смесей в экспериментах, Т и концентрациях фоновых электролитов в частицах ((NH4)2SO4 [23] и NaNO3 [28]). По данным этих расчетов вычисляли также коэффициенты активности ${\text{H}}_{{{\text{aq}}}}^{ + }$ в расчете на молярную концентрацию ($\gamma _{{\text{H}}}^{*}$) и рассчитывали рН водной фазы в частицах [46 ] .
Несмотря на отличающиеся значения α, μ, рН, L и t в [23, 28], рис. 3 демонстрирует неплохую корреляцию между [S(VI)]расч и [S(VI)]изм и согласованный поэтому характер результатов цитированных работ. Их согласие, несмотря на различие в рассматриваемых работах рН на ∼2–3 единицы, с очевидностью указывает на рост wS(VI) с уменьшением кислотности частиц в [23]. В работе [28] вопреки этому сообщалось о слабом влиянии кислотности частиц на wS(VI). Сплошной линией на рис. 3 показана зависимость в координатах “[S(VI)]расч–[S(VI)]изм”, отвечающая коэффициенту корреляции равному 1.
Для иллюстрации влияния рН на скорость реакции в ВР-режиме на вставке рис. 3 в качестве примера приводятся рассчитанные для Т = 298 К, μ ≈ 5 моль/л и L = 100 и 300 мкг/м3 зависимости wS(VI)(моль л–1 с–1) от кислотности частиц (рН 0–5) при ${{P}_{{{\text{S}}{{{\text{O}}}_{{\text{2}}}}}}}$ = 40 ppb, концентрации растворимого марганца 40 нг/м3 и Т = 298 К, близких к натурным условиям [23]. Результаты показаны на рис. 3 сплошной кривой. Сведения о содержании в газовой фазе ионов металла и диоксида серы отвечают результатам мониторинга газового и аэрозольного состава атмосферы в Пекине (январь 2016 г.). Учитывая, что содержание марганца согласно [23] соответствует растворимой форме металла, рост L от 100 до 300 мкг/м3, влекущий за собой снижение концентрации ионов марганца в аэрозольной фазе, не сопровождается изменением wS(VI) ввиду одновременного увеличения объемной доли влаги, см. показанную сплошной линией расчетную кривую на рис. 3. При вычислениях принимали во внимание зависимость [S(IV)] от рН при заданных μ [23, 39]. Вид приведенной здесь зависимости от рН скорости каталитической реакции в ВР-режиме отличается от ее колоколообразного вида при протекании реакции в избытке ионов металла, но при α < αкр [16, 24]. На вставке рис. 3 значками показаны рассчитанные нами зависимости wS(VI) от рН по данным экспериментов [23, 24, 28]. Расчеты wS(VI) производили с учетом найденных по результатам этих опытов значений $k_{{{\text{набл\_изм}}}}^{*}$ и пересчета концентраций ионов марганца и сульфита в частицах к приводившимся концентрационным условиям. Видно, что точки, отражающие опытные данные, в целом следуют ходу расчетной кривой, указывая на корректность развитого здесь подхода к толкованию механизма каталитической реакции в ВР-режиме в аэрозоле.
Из вставки на рис. 3 следует, что при рН < 1.5 наблюдается близкая к насыщению wS(VI) ≈ ≈ 10–7 моль л–1 с–1. Из уравнения(1) для таких условий будем иметь:
ЗАКЛЮЧЕНИЕ
В работе показано, что определяющую роль в кажущемся ускорении образования сульфатов в аэрозольных частицах в опытах по каталитическому окислению сульфита в присутствии ионов марганца и в избытке ионов металла играют разветвление цепей по реакции ${\text{HSO}}_{5}^{ - }$ + Mn2+ и подпитка сульфитом за счет захвата из газа диоксида серы. При этом образование сульфатов происходит со скоростью разветвления цепи. Приводится разграничение каталитической реакции в избытке ионов металла на медленный и быстрый (ВР) режимы. При рН >1.5 каталитическую реакцию в ВР-режиме характеризует экспоненциальный рост скорости наработки сульфатов с увеличением рН: wS(VI) ≈ 1/10–2pH. Приводится определенная по данным лабораторных экспериментов наблюдаемая константа скорости реакции $k_{{{\text{набл}}}}^{*}$, пригодная для расчетов динамики накопления сульфатов в аэрозольной фазе, в том числе и применительно к атмосферным условиям. Проведенные с использованием $k_{{{\text{набл}}}}^{*}$ оценки динамики образования сульфатов находят удовлетворительное согласие с данными лабораторного моделирования и результатами натурных экспериментов. Рассматриваемый ВР-режим каталитической реакции представляет интерес для разработки нового процесса очистки дымовых газов от диоксида серы.
Список литературы
Семенов Н.Н. Цепные реакции. 2-е изд., испр. и доп. Москва: Наука, 1986. 535 с.
Brandt Ch., van Eldik R. // Chem. Rev. 1995. V. 95. № 1. P. 119.
Kuo D.T.F., Kirk D.W., Jia C.Q. // J. Sulfur. Chem. 2006. V. 27. № 5. P. 461.
Ermakov A.N., Purmal A.P. // Kinet. Catal. 2002. V. 43. № 2. P. 249.
Yermakov A.N., Purmal A.P. // Prog. React. Mech. 2003. V. 28. № 3. P. 189.
Hoather R.C., Goodeve C.F. //Trans. Faraday Soc. 1934. V. 30. P. 1149.
Johnstone H.F., Coughanowr D.R. // Ind. Engng. Chem. 1958. V. 50. № 8. P. 1169.
Coughanowr D.R., Krause F.E. // Ind. Eng. Chem. Fund. 1965. V. 4. № 1. P. 61.
Matterson M.J., Stober W., Luther H. // Ind. Eng. Chem. Fund. 1969. V. 8. № 4. P. 677.
Barrie L.A., Georgii H.W. // Atmos. Env. 1976. V. 10. № 9. P. 743.
Hudson J.L., Erwin J., Catiopovich N.M. Research Report. Kinetics of sulfur dioxide oxidation in aqueous solutions. The University of Illinois Urbana. Illinois 61801. US Environmental Protection Agency EPA-600/7-79-030, 1979, January. 82 p.
Pasiuk-Bronikowska W. Bronikowski T. // Chem. Engng. Sci. 1981. V. 36. № 1. P. 215.
Kaplan D.J., Himmelblau D.M., Kanaoka C. // Atmos. Env. 1981. V. 15. № 5. P. 763.
Huss A.Jr., Lim P.K., Eckert C.A. // J. Phys. Chem. 1982. V. 86. № 21. P. 4224.
Martin L.R., Hill M.W. // Atmos. Envir. 1987. V. 21. № 10. P. 2267.
Ibusuki T., Takeuchi K. // Atmos. Env. 1987. V. 21. № 7. P. 1555.
Grgić I., Hudnik V., Bizjak M., Levec J. // Atmos. Env. 1991. V. 25A. P. 1591.
Berglund J., Fronaeus S., Elding L.I. // Inorg. Chem. 1993. V. 32. № 21. P. 4527.
Grgić I., Berčič G. // J. Atmos. Chem. 2001. V. 39. № 2. P. 155.
Connick R.E., Zhang Y.-X. // J. Inorg. Chem. 1996. V. 35. № 16. P. 4613.
Ulrich R.K., Rochelle G.T., Prada R.E. // Chem. Engng. Sci. 1986. V. 41. № 8. P. 2183.
Seinfeld J.H., Pandis J.H. Atmospheric Chemistry and Physics: From Air Pollution to Climate Change. New York: John Wiley and Sons, 1998. 1326 p.
Wang G.H., Zhang R.Y., Gomes M.E., Song Y., Zhou L., Cao J.,Hu J., Tang G., Chen Z., Li Z., Xu Z., Peng C., Lian C., Chen Y., Pan Y. // Proc. Natl. Acad. Sci. U.S.A. 2016. V. 113. № 48. P. 13630.
Liu T., Clegg S.L., Abbatt J.P.D. // Proc. Natl. Acad. Sci. U.S.A. 2020. V. 117. № 3. P. 1354.
Liu P., Ye C., XueCh., Zhang Ch., Mu Yu., Sun X. // Atmos. Chem. Phys. 2020. V. 20. № 7. P. 4153.
Cheng Y.F., Zheng G., Way Ch., Mu Q. // Sci. Adv. 2016. V. 2. № 12. e1601530.
Alexander B., Park R.J., Jacob D.J., Gong S. // J. Geophys. Res. Atmos. 2009. V. 114. D02309.
Zhang H., Xu Y., Jia L. // Atmos. Env. 2021. V. 245. 118019.
Angle K.J., Neal E.E., Grassian V.H. // Env. Sci. Technol. 2021. V. 55. № 15. P. 10291.
Ермаков А.Н.//Кинетика и катализ. 2021. Т. 62. № 5. С. 518.
Berglund J., Elding L.I., Buxton G.V., McGowan S., Salmon G.A. // J. Chem. Soc. Faraday Trans. 1994. V. 90. № 21. P. 309.
Fronaeous S., Berglund J., Elding L.I. // J. Inorg. Chem. 1998. V. 37. № 19. P. 4939.
Herrmann H., Ervens B., Jacobi H.-W., Wolke R., Nowacki P., Zellner R. // J. Atmos. Chem. 2000. V. 36. № 3. P. 231.
Stanbury D.M. // Adv. Inorg. Chem. 1989. V. 33. P. 69.
Buxton G.V., Mulazzani Q., Ross A. // J. Phys. Chem. Ref. Data. 1995. V. 24. № 3. P. 1055.
Fischer M., Paydar M., Warneck P., Ziajka J. Final Report Contract № STEP - 0005 - C(MB). Report F. 1992. P. 75.
Liu M., Song Y., Zhou T., Zhou T., Xu Z., Yan C., Zheng M., Wu Z., Hu M., W Y., Zhu T. // Geophys. Res. Lett. 2017. V. 44. № 10. P. 5213.
Betterton E.A., Hoffman M.R. // J. Phys. Chem. 1988. V. 92. № 21. P. 5962.
Millero F.J., Hershey J.B., Johnson G., Zhang J.-Z. // J. Atmos. Chem. 1989. V. 8. № 4. P. 377.
Larson T.J., Horike N.R., Harrison H. // Atmos. Env. 1978. V. 12. № 8. P. 1597.
Jacob D.J. // J. Geophys. Res.1986. V. 91. D. 9. P. 9807.
Cains P.W., Carabine M. // J. Chem. Soc. Faraday Trans, 1. 1978. V. 74. № 0. P. 2689.
Berresheim H., Jaeschke W. // J. Atmos. Chem. 1986. V. 4. № 3. P. 311.
Crump J.G. Flagan R.C., Seinfeld J.H. // Atmos. Env. 1967. V. 17. № 7. P. 1277.
http://www.aim.env.uea.ac.uk/aim/aim.php
Робинсон Р., Стокс Р. Растворы электролитов. Москва: Из-во Иностранной литературы, 1963. 646 с. (Для англ. версии. Robinson R.A., Stokes R.H. Electrolite solutions. Second edition. London. Butterworth scienfic publications, 1959. 565 p.)
Дополнительные материалы отсутствуют.
Инструменты
Кинетика и катализ