

Кинетика и катализ, 2023, T. 64, № 1, стр. 31-38
Сравнительное исследование гидрирования 1,3,5-тринитробензола и 2,4,6-тринитротолуола на медно-алюминиевом оксидном катализаторе в проточном реакторе
А. Л. Нуждин a, *, И. А. Щурова a, b, М. В. Бухтиярова a, П. Е. Плюснин c, Н. А. Алексеева a, b, С. В. Сысолятин b, Г. А. Бухтиярова a
a ФБГУН ФИЦ Институт катализа им. Г.К. Борескова СО РАН
630090 Новосибирск,
просп. Акад. Лаврентьева, 5, Россия
b ФБГУН Институт проблем химико-энергетических технологий СО РАН
659322 Бийск, ул. Социалистическая, 1, Россия
c ФБГУН Институт неорганической химии им. А.В. Николаева СО РАН
630090 Новосибирск, пр-т Акад. Лаврентьева, 3, Россия
* E-mail: anuzhdin@catalysis.ru
Поступила в редакцию 08.08.2022
После доработки 27.09.2022
Принята к публикации 27.09.2022
- EDN: KHAEGN
- DOI: 10.31857/S0453881123010045
Аннотация
Исследованы каталитические свойства медно-алюминиевого оксидного катализатора, приготовленного на основе двойного слоистого гидроксида, в гидрировании 1,3,5-тринитробензола (ТНБ) и 2,4,6-тринитротолуола (ТНТ) в проточном реакторе. Реакцию проводили при температуре 120°C, общем давлении 30 бар и концентрации субстрата 0.10−0.15 М, используя метанол в качестве растворителя. 1,3,5-Триаминобензол (ТАБ) и 2,4,6-триаминотолуол (ТАТ) выделяли из реакционной смеси в виде двойных солей с серной кислотой ТАБ⋅2H2SO4 и ТАТ⋅2H2SO4, выход которых составил 92 и 98% соответственно. При исходной концентрации тринитроарена 0.10 М гидролиз солей триаминобензолов позволил синтезировать флороглюцин и метилфлороглюцин с выходом 78 и 91%. Увеличение концентрации до 0.15 М снижает выход до 71 и 88% соответственно. Согласно данным термического анализа наблюдаемые различия в выходах солей триаминобензолов и полифенолов объясняются образованием разного количества смолоподобных побочных продуктов на поверхности катализатора в ходе гидрирования тринитроарена. При гидрировании ТНТ количество смол меньше, что приводит к более высоким выходам ТАТ⋅2H2SO4 и метилфлороглюцина. Вероятно, это связано с наличием электронодонорного метильного заместителя, который замедляет поликонденсацию молекул ТАТ.
ВВЕДЕНИЕ
Флороглюцин (1,3,5-тригидроксибензол) и его структурные аналоги находят широкое применение в различных областях химической промышленности. Флороглюцин используется в качестве исходного компонента для синтеза лекарственных препаратов, обладающих широким спектром фармацевтической активности [1–6], эпоксидных смол, красителей и ряда других ценных продуктов тонкого органического синтеза [7–9]. Метилфлороглюцин (2,4,6-тригидрокситолуол) также представляет интерес для получения фармацевтических препаратов [2]; исследуется как основа для получения азокрасителей и пигментов [9].
Промышленный способ производства флороглюцина включает окисление 2,4,6-тринитротолуола (ТНТ) хромовой смесью до 2,4,6-тринитробензойной кислоты с последующим декарбоксилированием в 1,3,5-тринитробензол (ТНБ), который затем восстанавливают стехиометрическими количествами восстановительных реагентов (таких как Fe или Sn в соляной кислоте) до 1,3,5-триаминобензола (ТАБ). Гидролиз последнего в кислой среде приводит к целевому продукту [7, 9, 10]. Традиционный метод синтеза метилфлороглюцина также основан на восстановлении ТНТ до 2,4,6-триаминотолуола (ТАТ) с использованием Fe/HCl или Sn/HCl и последующем гидролизе [9, 10]. Однако применение данных восстановителей сопровождается образованием большого количества отходов [11–13]. Каталитическое гидрирование тринитробензолов газообразным водородом, несомненно, более эффективный и экологически безопасный способ получения 1,3,5-триаминобензолов [14–26]. Метилфлороглюцин также может быть синтезирован из флороглюцина по реакции Вильсмайера–Хаака с последующим восстановлением [2], однако такой подход является нерациональным и дорогим.
На сегодняшний день гидрирование тринитроароматических соединений (1,3,5-тринитробензола, 2,4,6-тринитротолуола и 2,4,6-тринитробензойной кислоты) как правило было реализовано в присутствии катализаторов на основе платиновых металлов, таких как Pd/C [14–22], Pt/С [14] и Pd/Al2O3 [22, 23] или никеля Ренея [24–26]. Однако большим недостатком Pd- и Pt-содержащих катализаторов является их высокая стоимость и низкая доступность [12], тогда как Ni Ренея пирофорен, что в сочетании c использованием легковоспламеняющихся жидкостей (растворителей) и тринитробензолов делает процесс потенциально опасным, особенно при масштабировании. Следовательно, поиск недорогих, безопасных и при этом эффективных катализаторов гидрирования тринитробензолов на основе неблагородных металлов представляет как научный, так и практический интерес. Следует также отметить, что в подавляющем большинстве работ гидрирование производных 1,3,5-тринитробензола проводили в периодическом режиме в автоклавных реакторах [14–20, 24–26]. Между тем, применение проточных систем повышает производительность, обеспечивает более точный контроль технологических параметров, позволяя минимизировать накопление побочных продуктов, исключает проблемы с отделением и рециркуляцией катализатора, облегчает масштабирование, снижает затраты на очистку оборудования, уменьшает контакт персонала с вредными веществами, что в совокупности делает такие процессы более безопасными и привлекательными для внедрения [27–29].
Гетерогенные медные катализаторы обеспечивают селективное гидрирование нитрогруппы до аминогруппы в различных нитроаренах [12, 30, 31]. В нашей предыдущей публикации было показано, что смешанные Cu–Al-оксиды, полученные на основе двойных слоистых гидроксидов (ДСГ) [31–33], являются эффективными катализаторами гидрирования тринитробензолов до соответствующих триаминобензолов в проточном режиме [31]. В настоящей работе изучена стабильность медно-алюминиевого оксидного катализатора оптимального состава в гидрировании ТНБ и ТНТ. Исследование образцов отработанных катализатора методом термического анализа позволило предложить возможный механизм их дезактивации.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Приготовление катализатора
ДСГ с мольным отношением Cu : Al = 1 : 1 был приготовлен соосаждением смеси нитрата меди(II) и нитрата алюминия при pH 9.0 и 70°C, с использованием раствора NaOH и Na2CO3 ([${\text{CO}}_{3}^{{2 - }}$]/[Al3+] = 0.86, [OH−] = 1.6([Al3+] + [Cu2+]) в качестве осаждающего агента. “Старение” полученной суспензии проводили при 70°C в течение 2 ч. Затем осадок отфильтровывали, промывали горячей водой и сушили 14 ч при 110°C. Синтезированный ДСГ прокаливали при 650°C в течение 4 ч, что приводит к образованию Cu–Al смешанного оксида (далее обозначен как CuAlOx) [31–33].
Физико-химические методы исследования
Химический анализ на содержание меди и алюминия выполняли с применением оптического эмиссионного спектрометра с индуктивно-связанной плазмой Optima 4300 DV (“Perkin Elmer”, США). Текстурные характеристики катализатора исследовали на установке ASAP 2400 (“Micromeritics”, США), основываясь на изотермах низкотемпературной (–196°C) адсорбции азота. Рентгенофазовый анализ (РФА) проводили на дифрактометре Bruker D8 Advance (“Bruker”, Германия), используя CuKα-излучение. Средний размер кристаллитов (области когерентного рассеяния, ОКР) металлической меди оценивали с помощью уравнения Шеррера. Термический анализ образцов катализатора выполняли на приборе STA 449F1 Jupiter, оборудованном масс-спектрометром QMS 403DAëolos (“NETZSCH”, Германия).
Каталитическое гидрирование
Каталитическое гидрирование тринитроаренов осуществляли в проточной установке H-Cube Pro™ (“Thalesnano”, Венгрия), оборудованной реакторами из нержавеющей стали CatCart®70 (толщина каталитического слоя − 64 мм, внутренний диаметр − 4 мм) [31–34]. До начала реакции катализатор (470 мг в виде гранул 0.25–0.50 мм) восстанавливали в смеси водород/метанол (99.8%, J.T. Baker) при температуре 120°C в течение часа (скорость потока CH3OH и H2 составляла 0.5 и 100 мл/мин). Затем, в реактор вводили реакционную смесь, представляющую собой 0.10 или 0.15 М раствор тринитроарена в метаноле. Реакцию проводили при температуре 120°C, общем давлении 30 бар и скоростях потоков реакционной смеси и водорода 0.5 и 100 мл/мин соответственно. Для предотвращения окисления продуктов реакции кислородом воздуха гидрогенизат на выходе из реактора направляли в колбу, содержащую 7%-ный раствор серной кислоты (H2SO4 : тринитроарен = 2.1 : 1) в метаноле, что приводило к образованию двойных солей серной кислоты с ТАБ и ТАТ [20, 31]. Отбор проб начинали спустя 30 мин с момента подачи реакционной смеси и продолжали в течение 8 ч, отбирая пробу каждые 2 ч. Далее пробы суспензии соли охлаждали, осадок отфильтровывали, промывали 20 мл холодного метанола и сушили при 70°C под вакуумом в течение 6 ч. Соли триаминобензолов легко окисляются в растворе, и их чистоту очень сложно оценить методом ЯМР, поэтому выделенные соли ТАБ и ТАТ далее подвергали гидролизу по ранее описанным методикам [31], что позволило получить полифенолы с точно определенным выходом.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Физико-химические свойства катализатора
По данным элементного анализа смешанный оксид CuAlOx содержал 46.5 мас. % Cu и 17.4 мас. % Al. Изучение текстурных характеристик показало, что удельная площадь поверхности и объем пор в данном образце составляют 107 м2/г и 0.85 см3/г соответственно. Рентгенограмма CuAlOx демонстрирует рефлексы, относящиеся к фазе CuO (тенорит, PDF 05-661) [31, 33], восстановление которой в потоке водорода при 120°C в течение 1 ч приводит к образованию фазы металлической меди (рис. 1) со средним размером кристаллитов (области когерентного рассеяния) 7 нм. Кроме того, на рентгенограмме присутствуют низкоинтенсивные пики при 2θ = 31.3°, 36.9°, 60.5° и 65.6°, вероятно, относящиеся к твердому раствору состава Cu1 – xAl2 + xO4 [31].

Каталитические свойства
Каталитические свойства CuAlOx изучали в гидрировании ТНБ и ТНТ в проточном реакторе. Непосредственно перед проведением реакции катализатор восстанавливали в токе водорода при 120°C. В качестве растворителя использовали метанол, в котором ТНБ и ТНТ хорошо растворяются в отличие от других спиртов (EtOH, 2-PrOH). Гидрирование осуществляли при температуре 120°C, общем давлении 30 бар в течение 8 ч, используя 0.10 и 0.15 М растворы тринитроаренов. Для предотвращения окисления триаминобензолов кислородом воздуха гидрогенизат непосредственно на выходе из реактора обрабатывали раствором разбавленной серной кислоты в метаноле, что приводило к образованию стабильных двойных солей:

Согласно полученным результатам, гидрирование ТНТ происходило с 98%-ным выходом двойной соли ТАТ (ТАТ⋅2H2SO4) при обеих использованных концентрациях, при этом не наблюдалось заметной дезактивации катализатора (рис. 2). В случаи ТНБ выход двойной соли ТАБ (ТАБ⋅2H2SO4) был ниже: в течение первых 4 ч реакции он находился на уровне 94%, а затем снижался до 90–92% (рис. 2). Увеличение концентрации ТНБ с 0.10 до 0.15 М не оказывало видимого влияния на выход изолированного продукта.
Рис. 2.
Зависимость от времени выходов солей ТАБ⋅2H2SO4 и ТАТ⋅2H2SO4 при концентрации субстрата 0.10 (а) и 0.15 М (б).

Выделенные порции солей ТАБ и ТАТ далее объединяли и гидролизовали в водном растворе [31] по реакции:
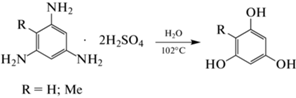
Гидролиз солей ТАБ⋅2H2SO4, синтезированных при использовании 0.10 и 0.15 М растворов ТНБ, привел к образованию флороглюцина с выходом 85 и 77% соответственно. В случае солей ТАТ⋅2H2SO4, полученных в экспериментах с 0.10 и 0.15 М растворами ТНТ, был синтезирован метилфлороглюцин с выходом 93 и 90%. Таким образом, при увеличении концентрации субстрата с 0.10 до 0.15 M происходит образование солей триаминобензолов, содержащих большее количество побочных продуктов синтеза, что ведет к снижению выхода соответствующих полифенолов на стадии гидролиза.
Термический анализ отработанных катализаторов
Для выявления влияния природы субстрата на выход целевого продукта и стабильность катализатора образцы отработанного катализатора, взятые из разных частей реактора, были исследованы методом термического анализа на наличие углеродных отложений (высокомолекулярных плохо растворимых в метаноле смолоподобных веществ) [35]. В качестве образца сравнения использовали предварительно восстановленный исходный катализатор CuAlOx. Следует отметить, что согласно полученным ранее результатам [32, 33] при гидрировании органических субстратов в метаноле в присутствии CuAlOx не происходит вымывания Cu и Al из состава катализатора.
Термогравиметрическая (ТГ) кривая восстановленного исходного катализатора содержит два пика потери массы: широкий пик с максимумом около 120°C и узкий сигнал при ~150°C, которые обусловлены десорбцией физически адсорбированной воды. Кроме того, на кривой ТГ имеются пики прироста массы, связанные с окислением металлической меди. Процесс окисления приводит к увеличению массы образца на 2.4% в диапазоне 150–350°C и сопровождается экзотермическими пиками на дифференциальной термической кривой (ДТА) при 160 и ~240°C, а также эффектами на дифференциальной термогравиметрической (ДТГ) кривой (рис. 3).

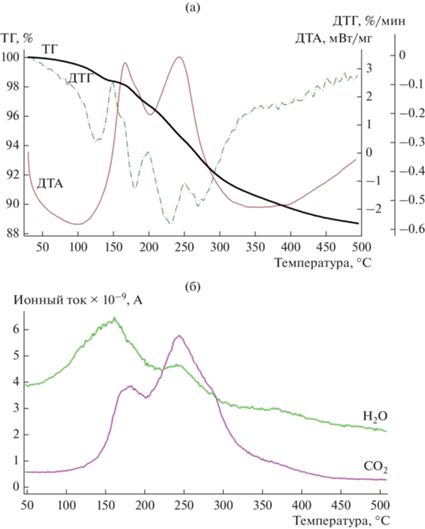
Типичные кривые ТГ/ДТГ/ДТА отработанного катализатора представлены на рис. 4a. После завершения реакции образец катализатора промывали чистым метанолом в течение 50 мин (скорость потока – 0.5 мл/мин), картридж с катализатором доставали из установки и оставляли на воздухе в течение ночи, а затем извлекали катализатор и сушили его под вакуумом при 70°C в течение 3 ч. ДТГ-кривые отработанных катализаторов демонстрируют две области потери массы: первая (~50–150°C) обусловлена удалением адсорбированной воды (потеря массы в этом интервале температур составляет 1–2%), вторая (~150–350°C) связана со сгоранием высокомолекулярных смолоподобных веществ на поверхности катализатора и окислением наночастиц металлической меди [32, 33, 35]. При этом процесс сгорания приводит к потере массы, а окисление Cu – к ее росту. Положение экзотермических пиков на ДТА-кривой и пиков CO2 и H2O на масс-спектрометрических (МС) кривых в диапазоне 150–350°C (рис. 4a, 4б) подтверждает сгорание высокомолекулярных смолоподобных веществ на поверхности катализатора, которые образуются в ходе гидрирования тринитробензолов. В продуктах сгорания не обнаружены оксиды азота (NO и др.), что, вероятно, объясняется замещением NH2/NH-групп в составе смол на ОН/O-фрагменты вследствие протекания гидролиза в ходе реакции [36] и/или при хранении на воздухе.
Рис. 4.
Кривые ТГ/ДТГ/ДТА (а) и МС (б) отработанного катализатора CuAlOx (из средней части реакторе) после гидрирования ТНБ (C = 0.15 M) в течение 8 ч.
На рис. 5 показано содержание смолоподобных веществ на поверхности образцов отработанного катализатора, отобранных на входе, выходе и в середине реактора, в зависимости от природы тринитроарена и его концентрации в реакционной смеси. Для определения содержания смол к потере массы в диапазоне 150–350°C (ТГ-кривые) прибавляли прирост массы восстановленного исходного катализатора в этом диапазоне вследствие окисления меди (2.4%). Хорошо видно, что количество смол в образцах за одно и то же время реакции возрастает как с увеличением исходной концентрации, так и при переходе от ТНТ к ТНБ. Для обоих субстратов содержание смолоподобных веществ на поверхности катализатора на входе в реактор выше, чем в средней части и на выходе из него.
Рис. 5.
Влияние природы тринитроарена и его концентрации на содержание смолоподобных веществ на поверхности отработанного катализатора CuAlOx в различных частях реактора.

Таким образом, в ходе гидрирования ТНБ происходит образование большего количества смол, чем в реакции с ТНТ. Вследствие этого выход ТАТ⋅2H2SO4 оказывается больше выхода ТАБ⋅2H2SO4 при одинаковых условиях реакции, и катализатор дольше сохраняет свою активность на первоначальном уровне (рис. 2).
Ранее в работе В.П. Талзи и др. методами 1H и 13C ЯМР-спектроскопии были идентифицированы ди- и тримеры молекул ТАБ, возникающие в процессе аквафазного гидрирования 2,4,6-тринитробензойной кислоты на катализаторе Pd/сибунит [19]. На основании этих результатов мы предполагаем следующий механизм образования высокомолекулярных побочных продуктов на поверхности катализатора, которые вызывают его дезактивацию:

Наличие электронодонорного метильного заместителя в ТАТ, вероятно, уменьшает величину частично положительного заряда на атомах углерода, связанных с аминогруппой, и приводит к снижению реакционной способности ТАТ в реакциях нуклеофильного замещения, предотвращая тем самым поликонденсацию. Другой возможный механизм дезактивации катализатора может быть обусловлен с образованием азокси-, азо- и гидразосоединений, получающихся в результате конденсации нитрозоаренов и арилгидроксиламинов (промежуточных продуктов восстановления нитроаренов) [11–13].
Следует отметить, что в ряде предыдущих исследований нами также было показано образование смолоподобных веществ на поверхности катализаторов CuAlOx в ходе проведения реакций гидрирования [32, 33], при этом “углеродные отложения” могли быть удалены прокаливанием образца на воздухе при 330°C с восстановлением его исходной активности [33].
ЗАКЛЮЧЕНИЕ
Проведено сравнительное исследование каталитических свойств смешанного Cu–Al-оксида (CuAlOx), приготовленного на основе двойного слоистого гидроксида, в гидрировании 1,3,5-тринитробензола (ТНБ) и 2,4,6-тринитротолуола (ТНТ) в проточном реакторе. При проведении реакции в метаноле в течение 8 ч из реакционной смеси были выделены 1,3,5-триаминобензол (ТАБ) и 2,4,6-триаминотолуол (ТАТ) в виде стабильных двойных солей ТАБ⋅2H2SO4 и ТАТ⋅2H2SO4 с выходом 92 и 98% соответственно. При исходной концентрации субстрата 0.10 М гидролиз солей триаминобензолов позволил синтезировать флороглюцин и метилфлороглюцин с выходом 78 и 91% в расчете на исходное нитроароматическое соединение. Увеличение концентрации тринитроарена до 0.15 М снижает выход полифенолов до 71 и 88% соответственно.
Согласно данным термического анализа, гидрирование ТНБ сопровождается образованием большего количества смолоподобных побочных продуктов на поверхности катализатора, чем гидрирование ТНТ, в результате чего в случае ТНБ получены существенно более низкие выходы соли триаминобензола и соответствующего полифенола. Кроме того, при гидрировании ТНБ наблюдается медленная дезактивация катализатора, которая не была зафиксирована в экспериментах с ТНТ. Отмечаемые различия, вероятно, объясняются электронным эффектом метильного заместителя в ТНТ. Электронодонорная CH3-группа в образующемся в ходе реакции ТАТ уменьшает величину частично положительного заряда на атомах углерода, связанных с NH2-группами, что предотвращает поликонденсацию молекул ТАТ и, тем самым, снижает скорость смолообразования.
Список литературы
Lee H., Park R.Y., Park K. // Bull. Korean Chem. Soc. 2021. V. 42. P. 66.
Teng X., Wang Y., Gu J., Shi P., Shen Zh., Ye L. // Molecules. 2018. V. 23. № 3116.
Mittal N., Tesfu H.H., Hogan A.M., Cardona S.T., Sorensen J.L. // J. Antibiot. 2019. V. 72. P. 253.
Verbanac D., Jain S.C., Jain N., Chand M., Paljetak H.C., Matijašic M., Peric M., Stepanic V., Saso L. // Bioorg. Med. Chem. 2012. V. 20. P. 3180.
Sun Q., Schmidt S., Tremmel M., Heilmann J., Konig B. // Eur. J. Med. Chem. 2014. V. 85. P. 621.
Li N., Khan S.I., Qiu S., Li X.-C. // Molecules. 2018. V. 23. № 3232.
Fiege H., Voges H.W., Hamamoto T., Umemura S., Iwata T., Miki H., Fujita Y., Buysch H.J., Garbe D., Paulus W. / In: Phenol derivatives, Ullmann’s Encyclopedia of Industrial Chemistry. Wiley-VCH Verlag GmbH & Co: Weinheim, 2012. V. 26. P. 557.
Abdel-Ghany S.E., Day I., Heuberger A.L., Broeckling C.D., Reddy A.S.N. // Sci. Rep. 2016. V. 6. P. 38483.
Ушкаров В.И., Кобраков К.И., Алафинов А.И., Шевелев С.А., Шахнес А.Х. // Химическая технология. 2006. Т. 8. С. 5.
Kastens M.L., Kaplan J.F. // Ind. Eng. Chem. 1950. V. 42. P. 402.
Blaser H.-U., Steiner H., Studer M. // ChemCatChem. 2009. V. 1. P. 210.
Formenti D., Ferretti F., Scharnagl F.K., Beller M. // Chem. Rev. 2019. V. 119. P. 2611.
Cardenas-Lizana F., Keane M.A. // J. Mater. Sci. 2013. V. 48. P. 543.
Kürschner U., Ehwald H., Alscher G. // Catal. Lett. 1995. V. 34. P. 191.
Belskaya O.B., Mironenko R.M., Talsi V.P., Rodionov V.A., Sysolyatin S.V., Likholobov V.A. // J. Mol. Catal. A: Chem. 2016. V. 420. P. 190.
Belskaya O.B., Mironenko R.M., Talsi V.P., Rodionov V.A., Gulyaeva T.I., Sysolyatin S.V., Likholobov V.A. // Catal. Today. 2018. V. 301. P. 258.
Мироненко Р.М., Бельская О.Б., Талзи В.П., Родионов В.А., Сысолятин С.В., Лихолобов В.А. // Изв. Акад. Наук. Сер. хим. 2016. № 6. С. 1535.
Бельская О.Б., Мироненко Р.М., Гуляева Т.И., Тренихин М.В., Лихолобова В.А. // Изв. Акад. Наук. Сер. хим. 2018. № 1. С. 71.
Talsi V.P., Belskaya O.B., Yurpalov V.L. // Magn. Reson. Chem. 2020. V. 58. P. 84.
Кашаев В.А., Хисамутдинов Г.X., Шевелев С.А., Валешний С.И., Шахнес А.X., Баврина А.П. // Химическая технология. 2007. Т. 8. С. 346.
Scholles H., Luebben M., Reichelt K., Kluger K., Niehoff W. // DE Patent 4131471, 1993.
van Gelder K.B., Damhof J.K., Kroijenga P.J., Westerterp K.R. // Chem. Eng. Sci. 1990. V. 45. P. 3159.
Halcour K., Schwerdtel W., Swodenk W., Thelen H. // DE Patent 2135154, 1973.
Temme O., Dickner T., Laschat S., Fröhlich R., Kotila S., Bergander K. // Eur. J. Org. Chem. 1998. P. 651.
Kim H.J., Lee W.S. // WO Patent 2007126262, 2007.
Schuster L. // DE Patent 3218665, 1981.
Yu T., Jiao J., Song P., Nie W., Yi C., Zhang Q., Li P. // ChemSusChem. 2020. V. 13. P. 2876.
Masuda K., Ichitsuka T., Koumura N., Sato K., Kobayashi S. // Tetrahedron. 2018. V. 74. P. 1705.
De Risi C., Bortolini O., Brandolese A., Di Carmine G., Ragno D., Massi A. // React. Chem. Eng. 2020. V. 5. P. 1017.
Kirichenko O., Kapustin G., Mishin I., Nissenbaum V., Shuvalova E., Redina E., Kustov L. // Molecules. 2022. V. 27. № 5147.
Nuzhdin A.L., Shchurova I.A., Bukhtiyarova M.V., Bulavchenko O.A., Alekseyeva N.A., Sysolyatin S.V., Bukhtiyarova G.A. // Synthesis. 2022. V. 54. P. 3605.
Nuzhdin A.L., Bukhtiyarova M.V., Bukhtiyarova G.A. // J. Chem. Technol. Biotechnol. 2020. V. 95. P. 3292.
Nuzhdin A.L., Bukhtiyarova M.V., Bulavchenko O.A., Bukhtiyarova G.A. // Mol. Catal. 2020. V. 494. № 111 132.
Нуждин А.Л., Симонов П.А., Бухтияров В.И. // Кинетика и катализ. 2021. Т. 62. № 4. С. 459.
Nuzhdin A.L., Reshetnikov S.I., Bukhtiyarova G.A., Moroz B.L.,·Gerasimov E.Yu., Pyrjaev P.A., Bukhtiyarov V.I. // Catal. Lett. 2017. V. 147. P. 572.
Рассолов А.В., Иванов Г.А., Брагина Г.О., Баева Г.Н., Смирнова Н.С., Казаков А.В., Усачев Н.Я., Стахеев А.Ю. // Кинетика и катализ. 2021. Т. 62. № 5. С. 591.
Дополнительные материалы отсутствуют.
Инструменты
Кинетика и катализ