

Кинетика и катализ, 2023, T. 64, № 2, стр. 153-172
Влияние добавок СО2 на некаталитическую конверсию природного газа в синтез-газ и водород
А. Р. Ахуньянов a, А. В. Арутюнов a, П. А. Власов a, *, В. Н. Смирнов a, В. С. Арутюнов a
a Федеральный исследовательский центр химической физики им. Н.Н. Семёнова РАН
119991 Москва, ул. Косыгина, 4, Россия
* E-mail: shocktube@yandex.ru
Поступила в редакцию 07.10.2022
После доработки 02.11.2022
Принята к публикации 03.11.2022
- EDN: GMLYME
- DOI: 10.31857/S0453881123020016
Аннотация
Проведен кинетический анализ некаталитического углекислотного риформинга СН4 в диапазоне температур 1500–1800 К в условиях непостоянной температуры за фронтом отраженной ударной волны. Установлены стадии превращения метана в синтез-газ, соответствующие этим стадиям характерные интервалы времени и наиболее важные элементарные реакции. На первой стадии молекулы метана в результате термического пиролиза последовательно превращаются в этан, этилен, а затем самый стабильный в данном интервале температур ацетилен. На второй стадии ацетилен превращается в СО и Н2, а для богатых смесей – и в частицы сажи. Конверсия СО2 протекает на второй и третьей стадиях, когда практически завершается конверсия СН4. Она происходит в результате взаимодействия молекул СО2 с появившимися в реагирующей системе атомами Н● и приводит к образованию молекул СО и радикалов ОН●. Ацетилен расходуется преимущественно в реакции с радикалами ОН●. Высокая концентрация ацетилена при риформинге метана способствует интенсивному формированию зародышей сажи, для которых ацетилен дает максимальный вклад в скорость их поверхностного роста. При этом сам ацетилен не является предшественником зародышей частиц сажи, которые преимущественно возникают из радикалов ${{{\text{С}}}_{{\text{3}}}}{\text{Н}}_{3}^{\centerdot }$.
ВВЕДЕНИЕ
Природный газ широко используется в энергетике и для производства разнообразных продуктов нефтехимии, в том числе синтетического жидкого топлива [1–3]. В большинстве случаев для этого необходима его предварительная конверсия в синтез-газ, то есть в смесь Н2 и СО с различным соотношением компонентов, из которого получают в промышленных масштабах водород, метанол, аммиак, синтетические жидкие углеводороды и другие продукты [4, 5]. Стадия конверсии природного газа в синтез-газ является наиболее энергоемкой стадией всего процесса синтеза нефтехимических продуктов из природного газа [5], что сдерживает развитие газохимии и водородной энергетики.
В настоящее время синтез-газ и чистый водород получают главным образом методами парового, окислительного и автотермического риформинга природного газа [5–10]. Это сложные и энергоемкие процессы, поэтому их совершенствование и создание более экономичных и гибких технологий производства синтез-газа и чистого водорода очень актуальны.
В качестве перспективного направления конверсии природного газа в синтез-газ и водород был предложен матричный риформинг углеводородных газов, в основе которого лежит некаталитическое газофазное парциальное окисление метана кислородом или воздухом [11–15]:
(I)
$\begin{gathered} {\text{С}}{{{\text{Н}}}_{{\text{4}}}}{\text{ + 1/2}}{{{\text{О}}}_{2}} \to {\text{СО + 2}}{{{\text{Н}}}_{2}}, \\ \Delta {{H}_{{298}}} = --36{\text{ кДж/моль}}. \\ \end{gathered} $Привлекательность парциального окисления метана в синтез-газ по реакции (I) по сравнению с традиционным паровым риформингом,
(II)
$\begin{gathered} {\text{С}}{{{\text{Н}}}_{{\text{4}}}}{\text{ + 2}}{{{\text{H}}}_{{\text{2}}}}{\text{O}} \to {\text{СО + 3}}{{{\text{Н}}}_{2}}, \\ \Delta {{H}_{{298}}} = + 206{\text{ кДж/моль}}, \\ \end{gathered} $состоит в том, что реакция (I) экзотермическая и в принципе может протекать за счет собственного тепловыделения, что делает процесс менее энергоемким по сравнению с паровым риформингом. Но в реакции (I) при обычных условиях выделяется слишком мало тепла для поддержания стабильного газофазного процесса окисления. При стехиометрическом соотношении реагентов, соответствующем этой реакции, коэффициент избытка окислителя a = [O2]/2[CH4] = 0.25 примерно в два раза ниже предела горения метановоздушных смесей при нормальных условиях [16, 17]. Поэтому для практической реализации парциального окисления метана необходима дополнительная энергия, которую обеспечивает параллельно протекающая реакция полного окисления метана
(III)
$\begin{gathered} {\text{С}}{{{\text{Н}}}_{{\text{4}}}}{\text{ + 2}}{{{\text{О}}}_{2}} \to {\text{С}}{{{\text{О}}}_{{\text{2}}}}{\text{ + 2}}{{{\text{Н}}}_{{\text{2}}}}{\text{О}}, \\ \Delta {{H}_{{298}}} = --802{\text{ кДж/моль}}, \\ \end{gathered} $При матричном риформинге парциальное окисление газа протекает в газовой фазе вблизи нагретой внутренней поверхности матрицы, изготовленной из проницаемого для газа термостойкого материала. В результате интенсивного конвективного и радиационного теплообмена фронта пламени с поверхностью матрицы ее внутренняя поверхность разогревается до 1300−1400 К. Почти до такой же температуры разогревается и свежая газовая смесь, поступающая через матрицу во фронт пламени. При этом температура продуктов конверсии снижается до 1500−1600 К. В замкнутом объеме риформера практически отсутствуют радиационные потери. Это позволяет реализовать “идеальное горение”. При определенных условиях возможно его протекание в так называемом “сверхадиабатическом” режиме как в каталитическом [18], так и некаталитическом [19, 20] вариантах. Температура продуктов конверсии может превышать адиабатическую температуру горения данного состава реагентов. Наряду со значительным снижением радиационных потерь в замкнутой полости матричного конвертора это позволяет существенно расширить пределы горения и тем самым поддерживать устойчивое окисление очень богатых смесей, невозможное в других типах горелочных устройств [21–23].
Достоинства матричного риформинга делают его потенциально привлекательным, в первую очередь, для малотоннажных процессов переработки углеводородных газов нетрадиционных источников, например, попутного нефтяного газа или биогаза [24]. Матричный риформинг открывает также возможность конверсии природного газа непосредственно в местах его добычи [25] или получения водорода в местах его потребления, что позволяет обойти нерешенные пока проблемы хранения и транспортировки водорода.
Разработка матричного риформинга метана потребовала решения ряда проблем, связанных с обеспечением устойчивости горения природного газа вблизи поверхности проницаемых объемных матриц, изготовленных из различных материалов, в том числе с нанесенными керамическими покрытиями [26–29]. Однако в формировании конечного состава продуктов конверсии большую роль играют и процессы, происходящие уже за фронтом пламени, в послепламенной зоне риформера. При достигаемой высокой температуре газа 1400–1600 K в отсутствие кислорода протекают относительно медленные процессы пиролиза оставшихся реагентов и получающихся продуктов, а также их паровая и углекислотная конверсия [30–32] под воздействием Н2О и СО2, образовавшихся на стадии окислительной конверсии продуктов. Кроме того, Н2О и СО2 можно дополнительно вводить в процесс риформинга для повышения его стабильности, увеличения выхода синтез-газа и снижения образования сажи.
На основании результатов кинетического и термодинамического моделирования ранее были выделены характерные стадии на пути достижения конечного состава продуктов парциального окисления метана в матричном риформере [33–38]. Первая стадия быстрых реакций с участием кислорода (зона пламени) завершается за времена 10–2−10–3 с практически полной конверсией метана и образованием в условиях недостатка кислорода, помимо СО2 и Н2О, таких продуктов, как СО, Н2, С2Н4, С2Н2 и ряда других, но в концентрациях, далеких от равновесных значений. Затем в послепламенной зоне при достигнутой высокой температуре протекают более медленные эндотермические процессы пиролиза метана и других присутствующих в газе углеводородов с последовательным образованием этана, этилена, и затем ацетилена, который в данном температурном диапазоне имеет наиболее низкую свободную энергию [39] и поэтому является основным промежуточным продуктом пиролиза остальных углеводородов.
В послепламенной зоне концентрация ацетилена достигает максимального значения. В связи с появлением существенной концентрации Н2О в этой зоне большую роль начинает играть экзотермическая реакция водяного газа (IV), приводящая к снижению концентрации Н2О и росту концентрации Н2 и СО2.
(IV)
$\begin{gathered} {\text{СО + }}{{{\text{Н}}}_{{\text{2}}}}{\text{О}} \to {\text{С}}{{{\text{О}}}_{{\text{2}}}}{\text{ + }}{{{\text{Н}}}_{2}}, \\ \Delta {{H}_{{298}}} = - 41{\text{ кДж/моль}}. \\ \end{gathered} $Также в указанной зоне со значительной скоростью идут процессы паровой и углекислотной конверсии ацетилена, ведущие к снижению его концентрации, а также конверсии остатков метана и других углеводородов.
В ходе наиболее продолжительной по времени стадии, соответствующей зоне III, протяженность которой обычно значительно превышает реальные размеры реактора, в результате медленных процессов паровой и углекислотной конверсии формируется конечный состав продуктов конверсии метана.
Кинетический анализ показал, что матричный риформинг гомологов метана и реальных углеводородных газов происходит аналогично риформингу метана. Различие лишь в том, что в послепламенной зоне пиролиз гомологов метана идет быстрее пиролиза метана, а в числе образующихся продуктов появляются пропилен, этилен и метан, которые затем уже пиролизуются в ацетилен [36, 38].
Для оптимальной работы матричного риформера необходимо обеспечить максимально полную конверсию ацетилена. Для этого требуется достаточно продолжительное (~1 с) время пребывания продуктов в послепламенной зоне, что может приводить к увеличению выхода сажи [40]. Если быстрые процессы парциального окисления углеводородов, протекающие на первой стадии в присутствии кислорода (в зоне пламени), исследованы достаточно хорошо, то процессы в последующих послепламенных зонах II и III требуют дополнительного анализа. В частности, необходим анализ роли образующихся при горении Н2О и СО2, которые также могут подаваться дополнительно для стабилизации процесса и снижения вероятности сажеобразования. Они практически не принимают участия в пиролизе углеводородов, и их заметная конверсия начинается существенно позже, уже под воздействием радикалов, возникших при пиролизе углеводородов.
В результате пиролиза углеводородов в этих условиях образуются в основном ацетилен и молекулярный водород [34–38]. В тоже время появляющиеся атомы Н● вовлекают в дальнейшее превращение термодинамически более стабильные Н2О и СО2, приводя к появлению крайне активных радикалов ОН●:
(V)
${{{\text{Н}}}_{{\text{2}}}}{\text{О + }}{{{\text{Н}}}^{\centerdot }} \to {{{\text{Н}}}_{{\text{2}}}}{\text{ + О}}{{{\text{Н}}}^{\centerdot }},$(VI)
${\text{C}}{{{\text{О}}}_{{\text{2}}}}{\text{ + }}{{{\text{Н}}}^{\centerdot }} \to {\text{CО + О}}{{{\text{Н}}}^{\centerdot }}.$Появление в результате взаимодействия Н2О и СО2 с атомами Н● радикалов ОН● ведет к конверсии ацетилена по реакциям:
(VII)
${{{\text{C}}}_{{\text{2}}}}{{{\text{Н}}}_{{\text{2}}}}{\text{ + О}}{{{\text{Н}}}^{\centerdot }} \to {\text{C}}{{{\text{Н}}}_{{\text{2}}}}{\text{CО + }}{{{\text{Н}}}^{\centerdot }},$(VIII)
${\text{C}}{{{\text{Н}}}_{{\text{2}}}}{\text{CО + }}{{{\text{Н}}}^{\centerdot }} \to {\text{ CН}}_{3}^{\centerdot }{\text{ + CО}},$Ацетилен, основной промежуточный продукт пиролиза углеводородов, является наиболее важным компонентом, участвующим в поверхностном росте образующихся частиц сажи. К сожалению, широко использовавшиеся ранее для моделирования процессов окисления углеводородов кинетические модели [41–43] не содержат блока реакций, который описывает процесс образования сажи. Тем не менее очевидно, что для повышения выхода основных компонентов синтез-газа Н2 и СО необходимо на послепламенной стадии создавать условия, максимально повышающие скорость конверсии ацетилена в эти продукты и подавляющие скорость реакций, ведущих к появлению продуктов конденсации (частиц сажи), в том числе путем введения в процесс риформинга дополнительного количества Н2О и СО2.
Существуют различные процессы преобразования метана в синтез-газ: паровая конверсия, углекислотная конверсия, парциальное окисление метана и их различные комбинации. В настоящей работе исследованы кинетические закономерности некаталитического углекислотного риформинга метана. Углекислый газ СО2 является парниковым газом, выбросы которого в атмосферу ведут к изменению климата. Поскольку процесс некаталитического углекислотного риформинга метана использует СО2 в качестве исходного компонента, то он способствует снижению выбросов СО2. С другой стороны, он требует большого количества энергии и идет при высоких температурах. Поэтому для разработки технологических установок некаталитического углекислотного риформинга нужна информация о высокотемпературной кинетике этого процесса, то есть необходимо знание об основных его стадиях, их характерных временах и о реакциях, доминирующих на каждой стадии.
Основной целью настоящей работы является анализ кинетики образования синтез-газа при некаталитическом окислительном риформинге метана с добавками СО2 в неизотермических условиях путем детального кинетического моделирования этого процесса.
ОПИСАНИЕ КИНЕТИЧЕСКОЙ МОДЕЛИ
В настоящее время имеется большое количество детальных кинетических механизмов, описывающих пиролиз и окисление как простых, так и достаточно сложных углеводородов. Современные детальные кинетические механизмы окисления даже относительно простых углеводородов насчитывают тысячи элементарных реакций с сотнями участвующих в них химических компонентов. Однако надежные экспериментальные данные, позволяющие установить адекватность всего механизма, имеются только для небольшого числа таких компонентов, которые обычно и используются для тестирования рассматриваемых кинетических механизмов.
Особенно большие сложности связаны с экспериментальной регистрацией свободных радикалов и электронно-, колебательно- или вращательно возбужденных компонентов. Экспериментальная регистрация возбужденных компонентов и их учет в кинетической схеме позволяют описать ряд тонких эффектов, таких как задержка воспламенения, период индукции, хемилюминесцентное излучение и химическая ионизация. В последнее время с развитием экспериментальной базы в такие сложные кинетические механизмы стали включать различные изомеры, имеющие одинаковый компонентный состав, но различное пространственное строение. При наличии заряженных частиц (положительных и отрицательных ионов и свободных электронов) и внешних источников энергии, как например, источников высокого напряжения или высокочастотных электромагнитных полей, непосредственно воздействующих на свободные электроны, в кинетические модели вводятся дополнительные уравнения, описывающие нестационарную функцию распределения свободных электронов по энергии. Такие современные детальные кинетические механизмы пиролиза и окисления углеводородов были успешно применены для прогнозирования различных эффектов при их горении, в том числе для анализа парциального газофазного окисления метана при высоких температурах [44].
Наиболее популярными кинетическими моделями для описания парциального газофазного окисления простейших углеводородов, в частности метана, являются GRI–Mech 3.0 (53 компонента, 325 элементарных стадий) [45] и Natural Gas III (NUI Galway) (289 компонентов, 1580 элементарных стадий) [46]. Механизм GRI–Mech 3.0 принципиально разрабатывался для описания высокотемпературной кинетики и не может претендовать на правильное описание ряда низкотемпературных эффектов. Также применяются механизмы Rasmussen 2008 (22 компонента и 67 элементарных стадий) [47], механизм Konnov (100 компонентов, 1140 элементарных стадий) [48], San–Diego (46 компонентов, 335 элементарных стадий) [49], USC–Mech-II (111 компонентов, 884 элементарные стадии) [50].
В работе [51] был выполнен подробный анализ существующих детальных кинетических механизмов, описывающих парциальное окисление метана. Основное внимание уделялось описательной способности этих механизмов для низкотемпературной области (ниже 1000 К). Ни один из 15 проанализированных механизмов не показал удовлетворительное соответствие результатов расчетов имеющимся экспериментальным данным, что говорит о необходимости совершенствования низкотемпературной части всех существующих механизмов.
В работе [52] сравнили механизмы Rasmussen 2008 и GRI–Mech 3.0 с результатами экспериментов по парциальному окислению метана в разбавленных азотом смесях при температурах 1270–1767 K и отношении СН4/О2 = 1.0–2.0. При проведении кинетического анализа использовался температурный профиль, полученный CFD-моделированием. Было показано, что экспериментальные результаты согласуются с расчетными данными. Для механизма Rasmussen 2008 наблюдается лучшее соответствие результатов кинетических расчетов результатам экспериментов, чем для GRI–Mech 3.0. Единственным исключением из всех трендов для обеих моделей была температурная зависимость концентрации этилена, который, как отмечено выше, является интермедиатом при образовании ацетилена. Моделирование кинетики парциального окисления метана было выполнено в [53] для условий фильтрационного горения: коэффициент избытка топлива ϕ = = 2[CH4]/[O2] = 2.4–2.6, тепловая мощность W = = 1540 кВт/м2. На основе полученных экспериментальных результатов оценена работоспособность следующих кинетических моделей: модель Konnov, модифицированная в [54], GRI–Mech 3.0, Leeds, SanDiego, USC–Mech-2, USC C1–C3. Кинетическая схема [54] дает наиболее близкое согласие расчетных концентраций целевых компонентов синтез-газа (Н2 и СО) с экспериментальными результатами. Однако в случае концентрации ацетилена модель [54] показала завышение результатов примерно в 2.5 раза. Лучшее соответствие концентрации ацетилена удалось получить, с помощью механизма San–Diego (концентрация ацетилена была занижена примерно в 1.5 раза). Для условий матричной конверсии в [55] было проведено кинетическое моделирование газофазного парового риформинга метана с использованием различных моделей: Konnov, San–Diego и Natural Gas III.
В работах [56–58] представлено подробное обсуждение результатов применения моделей GRI–Mech 3.0 и NUI Galway для кинетического моделирования парциального окисления углеводородов в синтез-газ и этилен для условий, близких к условиям матричной конверсии (температура, отношение реагентов). Так, в [58] показано, что детальный кинетический механизм Natural Gas III (NUI Galway) демонстрирует наилучшее соответствие экспериментальным результатам по образованию целевых компонентов синтез-газа и расходованию ацетилена.
Таким образом, на основе имеющихся литературных данных можно сделать вывод, что механизм Natural Gas III (NUI Galway) лучше остальных подходит для моделирования кинетики окисления углеводородов в условиях матричной конверсии. Данный механизм был проверен с использованием экспериментальных данных по окислению легких углеводородов в ударных трубах и машинах быстрого сжатия для следующих условий: T = 770–1580 K, P = 101–5050 кПa, О2/СН4 = 1.0–4.0 [59].
В настоящей работе для детального кинетического моделирования углекислотной конверсии метана в синтез-газ применялась одна из версий механизма NUI Galway – механизм Aramco 1.3, который был разработан для описания окисления простейших и кислородсодержащих углеводородов [59]. Этот механизм успешно описывает такие параметры, как задержки воспламенения, скорость пламени, качественный и количественный состав вредных выбросов. Принятые константы скорости элементарных процессов были отобраны в результате анализа литературных данных с минимальной последующей оптимизацией. Механизм был проверен для широкого диапазона начальных условий и разных экспериментальных методик, включая проточный реактор, ударную трубу, реактор со струйным перемешиванием, для пламен различных углеводородов. Механизм Aramco 1.3 тестировали путем сравнения результатов кинетических расчетов с результатами экспериментов для насыщенных и ненасыщенных углеводородов, включая метан, этан, этилен и ацетилен, а также кислородсодержащих соединений: формальдегид, метанол, ацетальдегид и этанол.
Механизм ARAMCO 1.3 описывает поведение углеводородов С1–С5, но не включает реакции с участием ароматических и полиароматических углеводородов, таких как бензол, толуол и т.д. Как было показано в работе [60] формирование частиц сажи при пиролизе и окислении метана идет по полиароматическому пути, то есть зародыши частиц сажи образуются из полиароматических углеводородов, а рост массы частиц сажи идет по механизму HACA за счет присоединения к активным сайтам на поверхности частицы молекул ацетилена. Для выяснения влияния полиароматических углеводородов и частиц сажи на процесс некаталитического углекислотного риформинга метана часть расчетов проводили с использованием программы MACRON и единого кинетического механизма сажеобразования, представленного в работе [61].
Предварительные расчеты для богатых смесей метана с кислородом показали интенсивное формирование сажи для самых богатых смесей, исследованных в наших кинетических расчетах. Ввиду важности проблемы сажеобразования подробное изучение ее влияния на температуру и кинетические закономерности риформинга метана будет выполнено в отдельной работе.
Таким образом, кинетическое моделирование в настоящей работе проводили с использованием программного пакета Chemkin Pro [60] и механизма ARAMCO 1.3, а также программы MACRON и единого кинетического механизма сажеобразования [61, 62]. Кроме того, программу MACRON применяли для определения вклада основных реакций образования и расходования различных компонентов, наиболее важных на разных стадиях матричной конверсии метана.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Условиям в послепламенной зоне матричного риформера достаточно хорошо соответствуют условия гомогенного риформинга метана, реализующиеся в экспериментах на ударной трубе при переменной температуре за фронтом отраженной ударной волны в диапазоне 1500–1800 К, что позволяет использовать для анализа процессов в послепламенной зоне матричного рифомера опыт и условия их моделирования в отраженных ударных волнах. В послепламенной зоне достигается максимум концентрации ацетилена, который затем превращается в СО и Н2, а для очень богатых смесей – и в частицы сажи. После этого начинается конверсия СО2 при его взаимодействии с появляющимися в реагирующей системе атомами Н●, что приводит к образованию молекул СО и радикалов ОН●. Ацетилен расходуется в основном в реакции с радикалами ОН●.
Основные результаты нашего кинетического анализа гомогенного углекислотного риформинга метана в диапазоне температур 1500–1800 К при атмосферном давлении в неизотермических условиях представлены на рис. 1–8. Особое внимание уделялось влиянию коэффициента избытка горючего ϕ, величина которого определяет склонность реагирующей смеси к образованию конденсированных частиц сажи. Расчеты были проведены для смесей СН4/О2/СО2, в которых концентрация метана оставалась постоянной, а концентрации О2 и СО2 менялись. Соотношение СН4/О2 задавало величину коэффициента ϕ, а концентрация СО2 определялась условием, что сумма всех коэффициентов перед компонентами была строго равна 1. С ростом ϕ концентрация СО2 в исходной смеси увеличивалась.
Рис. 1.
Расчет изменения концентрации СН4, СО2 и С2Н2 для различных значений ϕ при Т50 = 1500 К и Р50 = 1 бар.
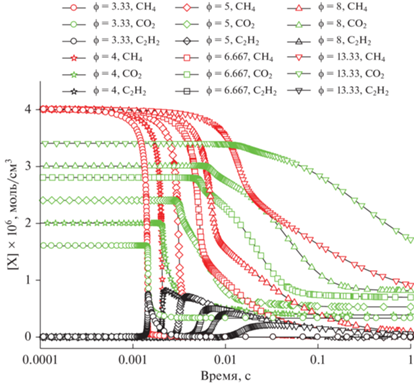
Рис. 2.
Расчет изменения концентраций СН4, СО2, СО, Н2, Н2О, С2Н2, О2 и температуры при Т50 = 1500 К, Р50 = 1 бар и ϕ = 3.33.
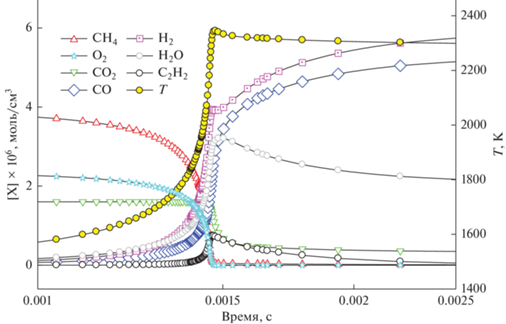
Рис. 3.
Расчет изменения концентрации СН4, СО2, СО, Н2, Н2О, С2Н2, О2 и температуры при Т50 = 1500 К, Р50 = 1 бар и ϕ = 8.
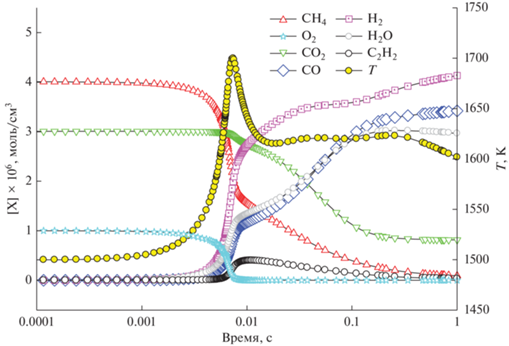
Рис. 4.
Расчет изменения концентрации СН4, СО2, СО, Н2, Н2О, С2Н2, О2 и температуры при Т50 = 1800 К, Р50 = 1 бар и ϕ = 3.33.
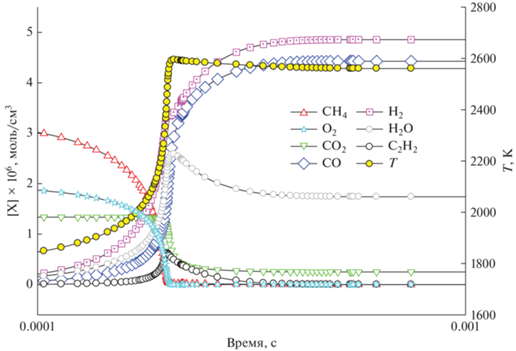
Рис. 5.
Расчет изменения концентрации СН4, СО2, СО, Н2, Н2О, С2Н2, О2 и температуры при Т50 = 1800 К, Р50 = 1 бар и ϕ = 8.
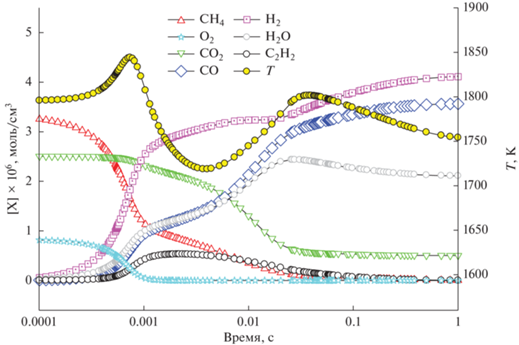
Рис. 6.
Расчет изменения концентрации СН4, СО2 и С2Н2 для смеси 0.5СН4 + 0.3О2 + 0.2СО2 (ϕ = 3.33) при Р50 = 1 бар и различных значениях начальной температуры Т50 = 1500, 1600, 1700, 1800 К.
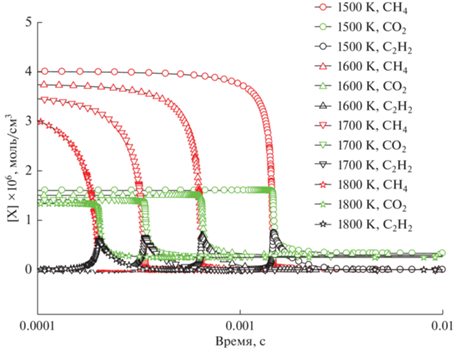
Рис. 7.
Расчет изменения концентрации СН4, СО2, и С2Н2 для смеси 0.5СН4 + 0.125О2 + 0.375СО2 (ϕ = 8) для Р50 = = 1 бар и различных значений начальной температуры Т50 = 1500, 1600, 1700, 1800 К.
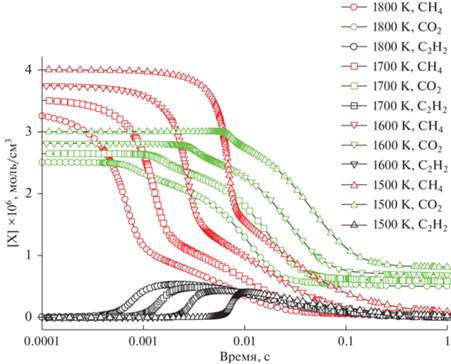
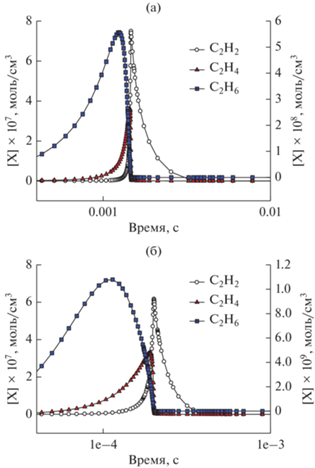
Рис. 8.
Кинетика изменения концентрации основных С2 углеводородов: С2Н2 (левая шкала), С2Н4 (левая шкала) и С2Н6 (правая шкала) для смеси 0.5СН4 + + 0.3О2 + 0.2СО2 (ϕ = 3.33) при Р50 = 1 бар и Т50 = 1500 (а) и 1800 К (б).
При кинетическом моделировании рассматривалось поведение во времени основных исходных и конечных компонентов реагирующей смеси (СН4, СО2 и С2Н2) для различных ϕ при начальной температуре за фронтом отраженной ударной волны Т50 = 1500, 1600, 1700 и 1800 К и давлении Р50 = 1 бар. Нижние индексы “50” при температуре и давлении указывают на значения параметров непосредственно за фронтом отраженной ударной волны. Именно они определяются по измеренной в эксперименте скорости падающей ударной волны на основании сигналов датчиков давления. В дальнейшем в ходе процесса риформинга величины Т50 и Р50 изменяются. Все расчеты проводили для условий постоянной плотности ρ = const. Были исследованы три состава смеси метана с кислородом и добавкой СО2 с различными значениями ϕ: 1) 0.5СН4 + 0.3О2 + 0.2СО2 (ϕ = 3.33); 2) 0.5СН4 + 0.125О2 + 0.375СО2 (ϕ = 8.0); 3) 0.5СН4 + + 0.075О2 + 0.425СО2 (ϕ = 13.33). Для того, чтобы отобразить все стадии процесса, на рисунках использована логарифмическая шкала времени.
При минимальных значениях ϕ, когда смесь ближе всего к стехиометрическому составу, конверсия метана протекает наиболее быстро, за время порядка 10–3 с. Из-за высокой температуры максимум скорости расходования СО2 и максимум концентрации С2Н2 наблюдаются на временах порядка 10–3 с. При увеличении ϕ, когда концентрация О2 падает, а концентрация СО2 растет, характерное время конверсии метана повышается, а характерная скорость расходования СН4 и СО2 уменьшается. Также возрастает время, когда концентрация С2Н2 проходит максимум (рис. 1). Для всех значений ϕ кроме наибольшего из рассматриваемых ϕ = 13.33 метан расходуется полностью, а концентрация СО2 достигает равновесного значения. Наши расчеты показали, что концентрация ацетилена возрастает до максимального значения для ϕ = 4, и с повышением ϕ можно было бы ожидать дальнейшего ее увеличения, однако этого не происходит, видимо, из-за снижения максимальной температуры, достигаемой в ходе процесса.
Результаты предварительного расчета процесса сажеобразования показали, что при ϕ = 3.33 частицы сажи формируются в очень низких концентрациях. Практически это граница сажеобразования. При бóльших значениях ϕ этот процесс интенсифицируется, и для ϕ = 8 расчетный выход сажи составляет порядка 50–70% в зависимости от начальной температуры. При этом начальная концентрация метана в реагирующей смеси при расчетах строго сохранялась, и изменялись только концентрация кислорода О2 и, соответственно СО2. Результаты детального расчета сажеобразования в богатых смесях метана с добавками СО2 будут представлены в отдельной работе.
Как видно из рис. 2, процесс окисления метана происходит вполне традиционно. После определенного периода индукции, в течение которого в реагирующей системе нарабатываются активные частицы, вступающие в экзотермические реакции, интенсивно повышается температура, резко возрастает скорость расходования метана и кислорода, увеличивается скорость выхода водорода и воды, а также СО. Несколько позже вступает в реакцию и расходуется СО2. К этому моменту концентрация ацетилена достигает наибольшего значения, а концентрация воды после прохождения локального максимума снижается. При данном составе смеси образование сажи не оказывает заметного влияния на профили температуры и концентраций рассматриваемых компонентов.
Для более богатой смеси с ϕ = 8 в неизотермическом случае ρ = const наблюдается сложный профиль изменения температуры (рис. 3), которая проходит выраженный максимум при времени порядка 0.01 с, а затем быстро снижается до квазипостоянного уровня с небольшим спадом в конце. К моменту полного расходования кислорода О2 достигаются максимум температуры и максимальная скорость расхода метана, начинается заметный расход СО2, а также отмечается максимальная скорость образования Н2, СО и Н2О. Несколько позднее доходит до максимального значения и концентрация С2Н2. При временах порядка 1 с концентрации всех компонентов стремятся к стационарным значениям. Такие компоненты, как метан и ацетилен, к этому времени расходуются полностью. Поскольку рассматривается очень богатая смесь с ϕ = 8, по нашим предварительным расчетам происходит интенсивное сажеобразование, которое начинается на временах порядка 10–2 с, и ко времени 1 с наблюдается очень высокий расчетный выход сажи, порядка 65%. Следует отметить, что это сугубо расчетные результаты, не наблюдаемые экспериментально.
Из рис. 3 видно, что первый максимум температуры достигается, когда кислород полностью израсходован, после чего скорость убыли метана заметно снижается, начинает медленно расходоваться СО2, и замедляется скорость образования Н2О и СО. Одновременно концентрация ацетилена проходит через максимум, температура спадает до локального минимума (на ~100 К), а затем снова растет. Скорее всего, этот повторный рост связан с началом образования частиц сажи. При повышении начальной температуры до Т50 = 1800 К представленная на рис. 2 картина практически сохраняется, лишь сокращается период задержки воспламенения, что хорошо видно на рис. 4.
Профили температуры и концентрации СН4, СО2, СО, Н2, Н2О, С2Н2, О2 (рис. 4 и 5) для одних и тех же начальных значений температуры и давления, но различных ϕ (3.33 и 8.0 соответственно), демонстрируют резкое изменение. По-видимому, это обусловлено повышением ϕ от 3.33 до 8.0. Столь существенное увеличение параметра ϕ (переход к очень богатым смесям) должно приводить к росту концентрации частиц сажи. Поэтому эти изменения температуры и концентраций естественно связать с процессом сажеобразования.
Для более высокой начальной температуры Т50 = 1800 К наблюдаемые профили концентрации метана, диоксида углерода и ацетилена аналогичны представленным на рис. 1. Однако при данной температуре все концентрации выходят на плато к моменту времени 1 с, а метан и ацетилен полностью расходуется. Задержка воспламенения метана резко сокращается по сравнению с характерными временами, представленными на рис. 1 для Т50 = 1500 K. Концентрация ацетилена проходит через максимум, величина которого немонотонно зависит от ϕ. С увеличением ϕ полуширина максимума сильно возрастает. Заметной корреляции между скоростью расходования СН4 и СО2 не наблюдается, поэтому можно заключить, что их расходование происходит различными путями.
Рис. 6 демонстрирует изменение задержки воспламенения богатой смеси 0.5СН4 + 0.3О2 + + 0.2СО2 (ϕ = 3) при повышении температуры Т50 от 1500 до 1800 K. На рисунке также показан профиль концентрации образующегося ацетилена, максимум концентрации которого практически совпадает с моментом достижения наибольшей скорости расходования метана. С возрастанием температуры задержка воспламенения сокращается почти на порядок, а максимум концентрации ацетилена снижается незначительно, в то время как максимальные скорости расходования СН4 и СО2 практически совпадают.
Рис. 7 демонстрирует изменение задержки воспламенения с повышением температуры Т50 от 1500 до 1800 K для еще более богатой смеси 0.5СН4 + 0.125О2 + 0.375СО2 (ϕ = 8). Концентрация СО2 начинает заметно падать в момент времени, соответствующий наибольшей скорости расходования метана. Максимум концентрации ацетилена наблюдается при временах, при которых график скорости расходования метана претерпевает излом, и скорость заметно снижается. K моменту времени 1 с концентрация СО2 выходит на плато, а метан и ацетилен полностью расходуются. С повышением начальной температуры максимальная концентрация ацетилена несколько возрастает. Она достигается в области резкого замедления скорости расходования метана, и затем метан расходуется в течение продолжительного времени одновременно с ацетиленом.
Кинетические расчеты показывают определенную последовательность появления молекул С2-углеводородов при окислительном пиролизе метана с добавками СО2: сначала происходит образование С2Н6, далее С2Н4 и затем С2Н2. Рис. 8 демонстрирует эту последовательность для смеси 0.5СН4 + 0.3О2 + 0.2СО2 (ϕ = 3.33) для Т50 = 1500 и 1800 K. Для обеих температур первой достигает максимума концентрация С2Н6, затем концентрация С2Н4 и только после этого наблюдается максимум концентрации С2Н2. Отметим, что концентрация С2Н6 на порядок меньше концентраций С2Н4 и С2Н2.
Повышение начальной температуры сокращает характерные времена образования продуктов, но не влияет на характер изменения концентрации основных С2-углеводородов (рис. 8).
Для того чтобы установить основные реакции образования и расходования какого-либо компонента, исследовали интегральные скорости его образования и расхода. Как показали кинетические расчеты, сначала появляются молекулы этана и этилена, а затем ацетилен. Поэтому далее приводятся результаты для этана, этилена и ацетилена. Все результаты получены для расчета по полному времени реакции 1 с.
Рис. 9 демонстрирует результаты анализа основных реакций образования и расходования С2Н6 для смеси 0.5СН4 + 0.3О2 + 0.2СО2 (ϕ = 3.33) при начальной температуре Т50 = 1500 K и давлении 1 бар. При этих условиях главным источником этана является реакция
Рис. 9.
Основные реакции образования и расхода этана С2Н6 для смеси 0.5СН4 + 0.3О2 + 0.2СО2 (ϕ = 3.33), Т50 = 1500 К и Р50 = 1 бар.
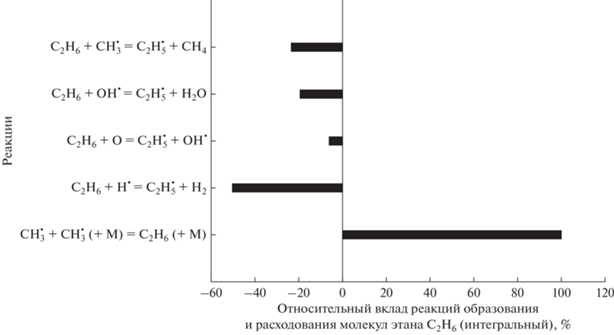
Основные реакции расхода молекул С2Н6:
Основные реакции образования и расходования С2Н4 для смеси 0.5СН4 + 0.3О2 + 0.2СО2 (ϕ = = 3.33) при Т50 = 1500 K и Р50 = 1 бар представлены на рис. 10. Этилен производится главным образом в реакциях
Рис. 10.
Основные реакции образования и расхода этилена С2Н4 для смеси 0.5СН4 + 0.3О2 + 0.2СО2 (ϕ = 3.33), Т50 = = 1500 К и Р50 = 1 бар.
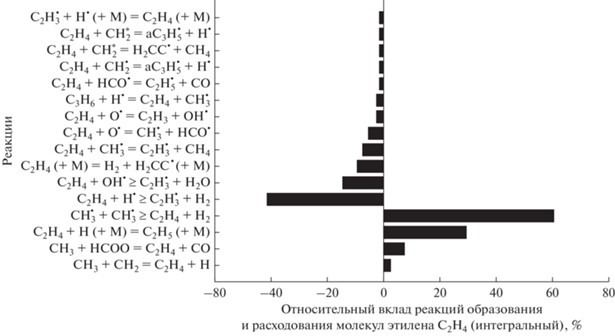
Основные реакции расходования молекул С2Н4:
Основные реакции образования и расходования С2Н2 для смеси 0.5СН4 + 0.3О2 + 0.2СО2 (ϕ = 3.33) для Т50 = 1500 K и Р50 = 1 бар представлены на рис. 11. Ацетилен производится главным образом в реакции
Рис. 11.
Основные реакции образования и расхода С2Н2 для смеси 0.5СН4 + 0.3О2 + 0.2СО2 (ϕ = 3.33), Т50 = 1500 К и Р50 = 1 бар.
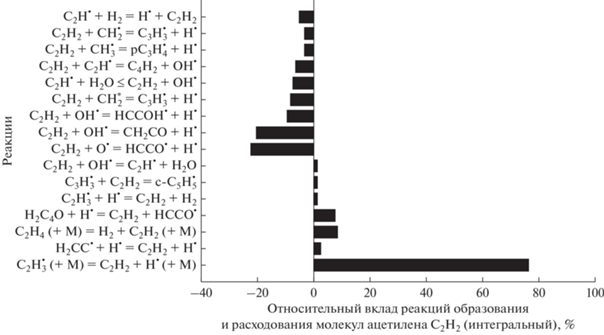
Основные реакции расходования С2Н2:
Важный аспект некаталитического углекислотного риформинга метана – появление зародышей и конденсированных частиц сажи. К основным параметрам, определяющим склонность реагирующей смеси к сажеобразованию, можно отнести коэффициент избытка горючего ϕ и температуру. Формирование высокой концентрации ацетилена в процессе некаталитического углекислотного риформинга метана ведет к появлению большого количества сажи. Однако по существующим представлениям о механизме сажеобразования молекулы ацетилена являются “строительным” материалом растущих частиц сажи, но не являются непосредственно зародышами этих частиц [63].
Основной предвестник зародышей частиц сажи – радикалы ${{{\text{С}}}_{{\text{3}}}}{\text{Н}}_{3}^{\centerdot }$. Наиболее важные реакции с их участием показаны на рис. 12 и 13 для давления 1 бар и температур Т50 =1500 и 1800 K. Для обеих температур радикалы С3Н3 появляются главным образом в реакции
Рис. 12.
Основные реакции образования и расхода радикалов ${{{\text{C}}}_{{\text{3}}}}{\text{H}}_{3}^{\centerdot }$ для смеси 0.5СН4 + 0.3О2 + 0.2СО2 (ϕ = 3.33), Т50 = 1500 К и Р50 = 1 бар.
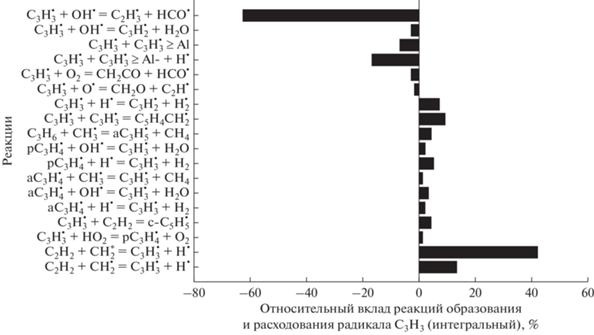
Рис. 13.
Основные реакции образования и расходования радикалов ${{{\text{C}}}_{{\text{3}}}}{\text{H}}_{3}^{\centerdot }$ для смеси 0.5СН4 + 0.3О2 + 0.2СО2 ( ϕ = = 3.33), Т50 = 1800 К и Р50 = 1 бар.
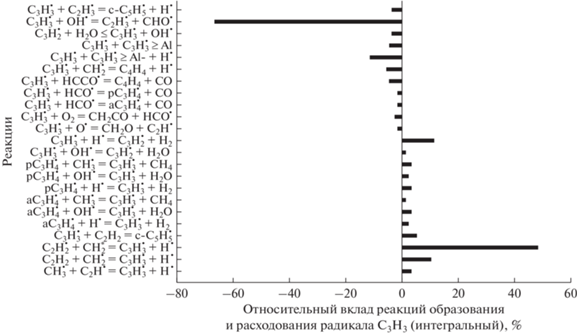
Для обеих температур основные реакции появления радикалов ${{{\text{С}}}_{{\text{3}}}}{\text{Н}}_{3}^{\centerdot }$ практически одинаковы, а вот число реакций их расходования с ростом температуры заметно увеличивается. Анализ показывает, что основная роль в образовании ${{{\text{С}}}_{{\text{3}}}}{\text{Н}}_{3}^{\centerdot }$ принадлежит реакциям молекул ацетилена с радикалами ${\text{СН}}_{2}^{\centerdot }$ в синглетном и триплетном состоянии. Основные реакции гибели радикалов ${{{\text{С}}}_{{\text{3}}}}{\text{Н}}_{3}^{\centerdot }$ – это их взаимодействие с гидроксильным радикалом ОН●. При этом появляются радикалы ${{{\text{С}}}_{{\text{2}}}}{\text{Н}}_{3}^{\centerdot }$, которые являются предвестником молекул С2Н4. Радикалы ${{{\text{С}}}_{{\text{3}}}}{\text{Н}}_{3}^{\centerdot }$ играют ключевую роль в формировании финильных радикалов и молекул бензола. Именно с этой стадии начинается процесс образования зародышей частиц сажи.
На рис. 14 показаны наиболее важные реакции, влияющие на расходование молекул добавки СО2 к смеси метана с кислородом и появление молекул СО.
Рис. 14.
Основные реакции образования и расхода молекул СО2 для смеси 0.5СН4 + 0.3О2 + 0.2СО2 (ϕ = 3.33), Т50 = = 1800 К и Р50 = 1.
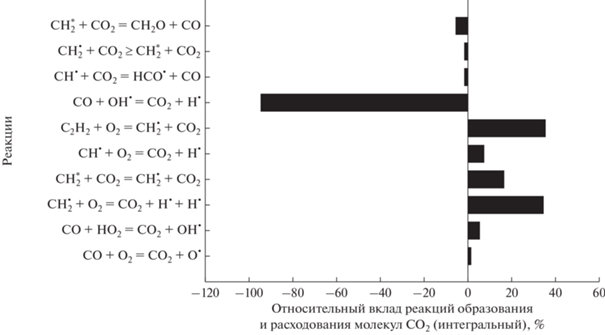
Как видно из рис. 14, основной реакцией является реакция взаимодействия СО2 с атомом водорода, в результате которой образуются молекулы СО:
Концентрация молекул СО2 восстанавливается в реакциях
Поскольку главный продукт углекислотной конверсии метана – синтез-газ, были определены реакции, дающие основной вклад в образование и расходование составляющих его молекул. Как видно из рис. 15, молекулы СО, получающиеся из молекул СО2, вступают в реакции, основными из которых являются:
Рис. 15.
Основные реакции образования и расхода СО для смеси 0.5СН4 + 0.3О2 + 0.2СО2 (ϕ = 3.33), Т50 = 1800 К и Р50 = 1 бар.
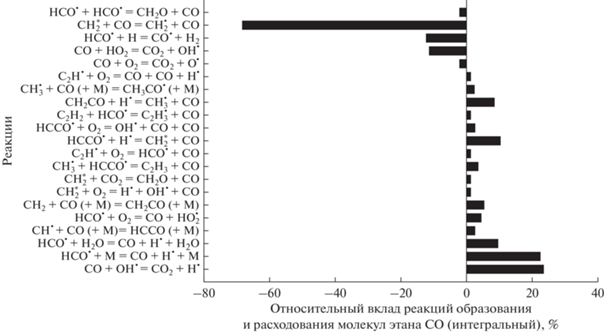
Их концентрация восстанавливается в реакциях:
Молекулы водорода Н2 образуются в следующих реакциях (рис. 16):
Рис. 16.
Основные реакции образования и расхода молекул Н2 для смеси 0.5СН4 + 0.3О2 + 0.2СО2 (ϕ = 3.33), Т50 = 1800 К и Р50 = 1 бар.
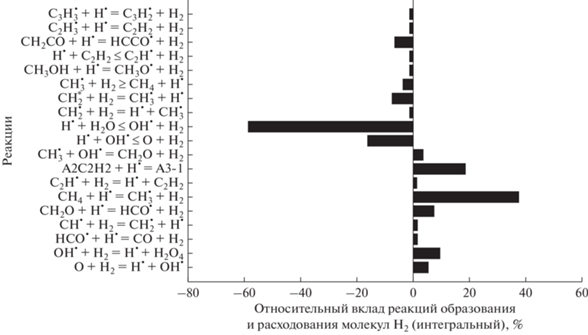
Расходуются молекулы Н2, главным образом, в реакциях:
Молекулы кислорода расходуются в реакциях (рис. 17):
Рис. 17.
Основные реакции образования и расхода молекул O2 для смеси 0.5СН4 + 0.3О2 + 0.2СО2 (ϕ = 3.33), Т50 = 1800 К и Р50 = 1 бар.
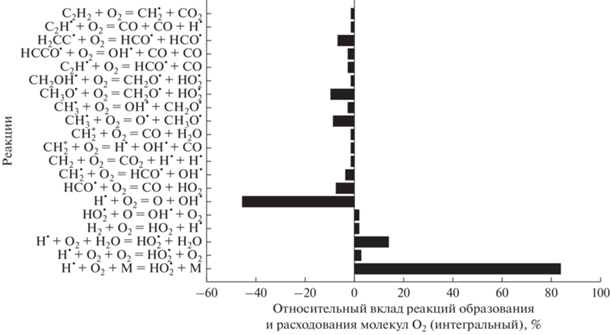
Восполняется концентрация кислорода в реакции:
Молекулы метана изначально при столкновениях с третьим телом М дают радикал ${\text{CH}}_{3}^{\centerdot }$ и атом Н●. Эта реакция, по сути, инициирует весь последующий процесс превращений исходных компонентов реагирующей смеси. В дальнейшем, после наработки пула активных радикалов, молекулы метана расходуются главным образом в реакциях:
Концентрация метана восстанавливается в реакциях (рис. 18):
Рис. 18.
Основные реакции образования и расхода молекул CН4 для смеси 0.5СН4 + 0.3О2 + 0.2СО2 (ϕ = 3.33), Т50 = = 1800 К и Р50 = 1 бар.
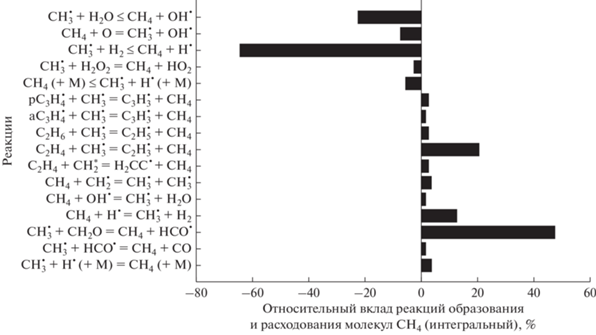
Проведенный анализ основных реакций образования и расхода различных компонентов процесса позволяет проследить путь от компонентов исходной смеси (СН4, О2, СО2) до конечных продуктов. Процесс начинается с термического пиролиза молекулы метана с формированием радикала ${\text{CH}}_{3}^{\centerdot }$ и атома Н● (инициирование). Затем атом Н● реагирует с молекулой кислорода с появлением двух радикалов О● и ОН● (разветвление цепи). Атомы Н● также реагируют с молекулой метана с образованием радикала ${\text{CH}}_{3}^{\centerdot }$ и молекулы водорода. Основной реакцией расходования молекул Н2 является их реакция с гидроксильным радикалом ОН●
Основной канал расхода молекул СО2 – реакция
Молекула СО вступает в основном в реакции:
и формируется ансамбль разнообразных активных радикалов. Таков механизм действия добавки СО2 к смеси метана с кислородом при получении синтез-газа.
Проведенный кинетический анализ показал явную стадийность образования различных С2-углеводородов в процессе конверсии метана. Первым по времени появляется С2Н6, затем С2Н4 и, наконец, С2Н2. Одновременно с достижением максимума концентрации С2Н2 идет процесс ускоренного расходования молекул добавки СО2 и увеличивается концентрация молекул СО, одного из основных компонентов синтез-газа.
ЗАКЛЮЧЕНИЕ
Кинетический анализ некаталитического углекислотного (сухого) риформинга СН4 в диапазоне температур 1500–1800 K в условиях переменной температуры за фронтом отраженной ударной волны позволил установить стадии превращения метана в синтез-газ и соответствующие этим стадиям характерные интервалы времени. На первой стадии молекулы метана в процессе термического пиролиза последовательно превращаются в этан, этилен, а затем наиболее стабильный при высоких температурах ацетилен. На второй стадии ацетилен превращается в СО и Н2, а для наиболее богатых смесей – и в частицы сажи. Kонверсия СО2 начинается на третьей стадии, когда практически завершается конверсия СН4, и происходит взаимодействие молекул СО2 с появившимися в реагирующей системе атомами Н●, что приводит к образованию молекул СО и радикалов ОН●. Молекулы ацетилена расходуются преимущественно в реакциях с радикалами ОН●.
Необходимо отметить, что в процессе некаталитического углекислотного риформинга метана с появлением свободных радикалов в реагирующей системе образуется большое количество молекул Н2О, влияние которых проявляется главным образом на поздних стадиях риформинга. Исследование влияния воды на конверсию метана в синтез-газ на ранних стадиях процесса риформинга метана требует отдельного анализа.
Важной проблемой некаталитического углекислотного риформинга метана является образование конденсированных частиц сажи. Высокая концентрация ацетилена на ранних стадиях риформинга метана приводит к высокой концентрации зародышей сажи, для которых ацетилен служит основным компонентом их поверхностного роста. Следует отметить, что сам ацетилен не является предшественником образования зародышей частиц сажи, которые формируются главным образом из радикалов ${{{\text{С}}}_{{\text{3}}}}{\text{Н}}_{3}^{\centerdot }$. В большинстве случаев появление частиц сажи в реагирующей системе – крайне нежелательный процесс, который ведет к целому ряду экологических и технологических проблем. Поэтому в процессах конверсии метана в синтез-газ, особенно в процессах, использующих катализаторы, снижение концентрации частиц сажи до минимально возможного уровня представляет собой важную и очень сложную проблему, которая требует дальнейшего анализа.
Список литературы
Брагинский О.Б. Мировая нефтехимическая промышленность. Москва: Наука, 2003. 566 с.
Брагинский О.Б. Нефтегазовый комплекс мира. Москва: Изд-во “Нефть и газ” РГУ нефти и газа им. Губкина, 2006. 640 с.
Савченко В.И., Макарян И.А., Арутюнов В.С. // Мир нефтепродуктов. Вестник нефтяных компаний. 2013. № 11. С. 3.
Rostrup-Nielsen J.R. // Catal. Today. 2002. V. 71. P. 243.
Арутюнов В.С., Голубева И.А., Елисеев О.Л., Жагфаров Ф.Г. Технология переработки углеводородных газов. Москва: Изд-во Юрайт, 2020. 723 с.
Gregoire Padro C.E., Lau F. // Adv. Hydrogen Energy. 2002.
Bockris J.O’.M. // Int. J. Hydrogen Energy. 2013. V. 38. P. 2579.
Kalamaras C.M., Efstathiou A.M. / Hindawi Publishing Corporation. Conference Papers in Energy, 2013. https://doi.org/10.1155/2013/690627
Aasberg-Petersen K., Hansen J.-H.B., Christensen T.S., Dybkjaer I., Christensen P.S., Nielsen C.S., Madsen S.E.L.W., Rostrup-Nielsen J.R. // Appl. Catal. 2001. V. 221. P. 379.
Aasberg-Petersen K., Dybkjær I., Ovesen C.V., Schjødt N.C., Sehested J., Thomsen S.G. // J. Natur. Gas Sci. Eng. 2011. V. 3. P. 423. https://doi.org/10.1016/j.jngse.2011.03.004
Арутюнов В.С., Шмелев В.М., Лобанов И.Н., Политенкова Г.Г. // Теоретические основы химической технологии. 2010. Т. 44. №. 1. С. 21. https://doi.org/10.1134/S0040579510010033
Arutyunov V.S., Shmelev V.M., Sinev M.Yu., Shapovalova O.V. // Chem. Eng. J. 2011. V. 291. P. 176. https://doi.org/10.1016/j.cej.2011.03.084
Арутюнов В.С., Савченко В.И., Седов И.В., Шмелев В.М., Никитин А.В., Фокин И.Г., Эксанов С.А., Шаповалова О.В., Тимофеев К.А. // Журнал прикладной химии. 2016. Т. 89. № 11. С. 1450.
Алдошин С.М., Арутюнов В.С., Савченко В.И., Седов И.В., Никитин А.В., Фокин И.Г. // Химическая физика. 2021. Т. 40. № 5. С. 46. https://doi.org/10.31857/S0207401X21050034
Арутюнов В.С., Никитин А.В., Стрекова Л.Н., Сав-ченко В.И., Седов И.В., Озерский А.В., Зимин Я.С. // Журнал технической физики. 2021. Т. 91. № 5. С. 713. https://doi.org/10.21883/JTF.2021.05.50681.265-20
Van den Schoor F., Verplaetsen F. // J. Hazard. Mater. 2006. V. 128. I. 1. P. 1. https://doi.org/10.1016/j.jhazmat.2005.06.043
Wan X., Zhang Qi, Lian Z. // Ind. Eng. Chem. Res. 2016. V. 55. I. 30. P. 8472. https://doi.org/10.1021/acs.iecr.6b01012
Максимов Ю.М., Кирдяшкин А.И., Аркатова Л.А. // Катализ в промышленности. 2013. Т. 2. С. 45.
Dorofeenko S.O., Polianczyk E.V. // Chem. Eng. J. 2016. V. 292. P. 183. https://doi.org/10.1016/j.cej.2016.02.013
Dorofeenko S.O., Polianczyk E.V. // Int. J. Hydrogen Energy. 2019. V. 44. P. 30039. https://doi.org/10.1016/j.ijhydene.2019.09.208
Arutyunov V.S., Shmelev V.M., Rakhmetov A.N., Shapovalova O.V. // Ind. Eng. Chem. Res. 2014. V. 53. I. 5. P. 1754. http://pubs.acs.org/doi/ipdf/10.1021/ie4022489
Арутюнов В.С., Савченко В.И., Седов И.В., Шмелев В.М., Никитин А.В., Фокин И.Г., Эксанов С.А., Шаповалова О.В., Тимофеев К.А. // Журнал прикладной химии. 2016. Т. 89. № 11. С. 1450.
Nikitin A., Ozersky A, Savchenko V., Sedov I., Shmelev V., Arutyunov V. // Chem. Eng. J. 2019. V. 377. ISCRE 25 Special Issue: Bridging Science and Technology. 2019. Article 120883. https://doi.org/10.1016/j.cej.2019.01.162
Arutyunov V., Nikitin A., Strekova L., Savchenko V., Sedov I. // Catal. Today. 2021. V. 379. P. 23. https://doi.org/10.1016/j.cattod.2020.06.057
Арутюнов В.С., Стрекова Л.Н., Савченко В.И., Седов И.В., Никитин А.В., Елисеев О.Л., Крючков М.В., Лапидус А.Л. // Нефтехимия. 2019. Т. 59. № 3. С. 246. https://doi.org/10.1134/S002824211903002X
Шмелев В.М. // Химическая физика. 2010. Т. 29. № 7. С. 27.
Shmelev V.M. // Combust. Sci. Technol. 2014. V. 186. I. 7. P. 943. https://doi.org/10.1080/00102202.2014.890601
Шмелев В.М., Николаев В.М. // Химическая физика. 2016. Т. 35. № 3. С. 56.
Shmelev V. // Energy Power Eng. 2017. V. 9. P. 366. http://www.scirp.org/journal/epe
Wang L., Liu Z., Chen S., Zheng C., Li. J. // Energy Fuels. 2013. V. 27. I. 12. P. 7602. https://doi.org/10.1021/ef401559r
Lavoe. J.-M. // Front. Chem. 2014. V. 2(81). https://doi.org/10.3389/fchem.2014.00081
Shah Y.T., Gardner T.H. // Catal. Rev. 2014. V. 56. I. 4. P. 476. https://doi.org/10.1080/01614940.2014.946848
Wittich K., Schunk S.A., Kramer M., Bottke N. // Chem. Cat. Chem. 2020. V. 12. P. 2130. https://doi.org/10.1002/cctc.201902142
Савченко В.И., Шаповалова О.В., Никитин А.В., Ару-тюнов В.С., Седов И.В. // Журнал прикладной химии. 2018. Т. 91. № 9. С. 1332. https://doi.org/10.1134/S0044461818090128
Савченко В.И., Никитин А.В., Озерский А.В., Седов И.В., Арутюнов В.С. // Нефтехимия. 2020. Т. 60. № 4. С. 538. https://doi.org/10.31857/S0028242120040139
Савченко В.И., Зимин Я.С., Никитин А.В., Седов И.В., Арутюнов В.С. // Нефтехимия. 2021. Т. 61. № 4. С. 520. https://doi.org/10.1134/S0965544121070021
Бузилло Э., Савченко В.И., Арутюнов В.С. // Нефтехимия. 2021. Т. 61. № 6. С. 820. https://doi.org/10.1134/S0965544121110037
Савченко В.И., Зимин Я.С., Бузилло Э., Никитин А.В., Седов И.В., Арутюнов В.С. // Нефтехимия. 2022. Т. 62. № 3. С. 375. https://doi.org/10.1134/S0965544122050048
Duff R.E., Bauer S.H. // J. Chem. Phys. 1962. V. 36. P. 1754.
Warnatz J.,·Maas U., Dibble R.W. Combustion: Physical and Chemical Fundamentals, Modeling and Simulation, Experiments, Pollutant Formation. Springer, 2006. 378 p.
Healy D., Kalitan D.M., Aul C.J., Petersen E.L., Bourque G., Curran H.J. // Energy Fuels. 2010. V. 24. I. 3. P. 1521.
Chemical-Kinetic Mechanisms for Combustion Applications. https://web.eng.ucsd.edu/mae/groups/combustion/mechanism.html) Date of access 30.11.2022.
Konnov A.A. // 28-th Symposium (Int.) on Combustion. Edinburgh. Abstr. Symp. Pap. 2000. P. 317.
Zhang Q., Liu Y., Chen T., Yu X., Wang J., Wang T. // Chem. Eng. Sci. 2016. V. 142. P. 126.
Smith G.P., Golden D.M., Frenklach M., Moriarty N.W., Eiteneer B., Goldenberg M., Bowman C.T., Hanson R.K., Song S., Gardiner W.C.Jr.; Lissianski V., Qin Z. Available at http://combustion.berkeley.edu/gri-mech/version30/text30.html.
Healy D., Kalitan D.M., Aul C.J., Petersen E.L., Bourque G., Curran H.J. // Energy Fuels. 2010. V. 24. № 3. P. 1521.
Rasmussen C.L., Jakobsen J.G., Glarborg P. // Int. J. Chem. Kinet. 2008. V. 40. № 12. P. 778.
Fomin A., Zavleva T., Alekseev V., Rahinov I., Cheskis S., Konnov A. // Combustion and Flame. 2016. V. 171. P. 198.
San Diego Mechanism. http://web.eng.ucsd.edu/mae/ groups/combustion/mechanism.html
Wang H., You X., Joshi A. V., Davis S. G., Laskin A., Egolfopoulos F., Law C. K. USCMech Version II. High-Temperature Combustion Re-action Model of H2/CO/C1-C4 Compounds. http://ignis.usc.edu/ Mechanisms/USC-Mech%20II/USC_Mech%20II.htm, May 2007.
Беляев А.А., Никитин А.В., Токталиев П.Д., Власов П.А., Озерский А.В., Дмитрук А.С., Арутюнов А.В., Арутюнов В.С. // Горение и взрыв. 2018. Т. 11. № 1. С. 19.
Li C., Kuan B., Lee W.J., Burke N., Patel J. // Chem. Eng. Sci. 2018. V. 187. P. 189.
Fotovat F., Rahimpour M. // Int. J. Hydrogen Energy. 2021. V. 46. № 37. P. 19312.
Capriolo C., Alekseev V.A., Konnov A.A. // Combustion and Flame. 2018. V. 197. P. 11.
Savchenko V.I., Nikitin A.N., Zimin Y.S., Ozerskii A.V., Sedov I.V., Arutyunov V.S. // Chem. Eng. Res. Des. 2021. V. 175. P. 250.
Zhang Q., Wang J., Wang T. // Ind. Eng. Chem. Res. 2016. V. 55. P. 8383.
Wang T., Liu Y., Wang D. // Chem. Eng. J. 2012. V. 207–208. P. 235.
Liu Y., Zhang Q., Wang T. // Combust. Sci. Technol. 2017. V. 189. № 5. P. 908.
Metcalfe W.K., Burke S.M., Ahmed S.S., Curran H.J. // J. Chem. Kinet. 2013. V. 45. P. 638.
CHEMKIN-PRO Release 15101. Reaction Design:San Diego, 2010.
Агафонов Г.Л., Билера И.В., Власов П.А., Жильцова И.В., Колбановский Ю.А., Смирнов В.Н., Тереза А.М. // Кинетика и катализ. 2016. Т. 57. № 5. С. 571.
Власов П.А., Ахуньянов А.Р., Смирнов В.Н. // Кинетика и катализ. 2022. Т. 63. № 2. С. 160.
Frenklach M., Mebel A.M. // Phys. Chem. Chem. Phys. 2020. V. 22. P. 5314.
Дополнительные материалы отсутствуют.
Инструменты
Кинетика и катализ