

Коллоидный журнал, 2019, T. 81, № 5, стр. 617-624
Механохимия Bi2O3. 1. Дефектная структура и реакционная способность механически активированного Bi2O3
А. Н. Стрелецкий 1, *, И. В. Колбанев 1, Г. А. Воробьева 1, А. В. Леонов 2, А. Б. Борунова 1, А. А. Дубинский 1
1 Институт химической физики им. Н.Н. Семенова РАН
119991 Москва, ул. Косыгина, 4, Россия
2 Московский государственный университет им. М.В. Ломоносова,
химический факультет
119991 Москва, Ленинские горы, 1, стр. 3, Россия
* E-mail: str1945@yandex.ru
Поступила в редакцию 10.04.2019
После доработки 22.04.2019
Принята к публикации 25.04.2019
Аннотация
Методами рентгеновской дифракции, измерения удельной поверхности и синхронного термоанализа, совмещенного с масс-спектроскопией, проанализированы закономерности механической активации α-Bi2O3, природа и термическая устойчивость образующихся при этом дефектов и увеличение реакционной способности оксида. Механическую активацию Bi2O3 можно разделить на две стадии. На стадии раскола частиц происходит рост удельной поверхности до S = 3.2 м2/г и уменьшение размера частиц до 100 нм, а размера L областей когерентного рассеяния до 40 нм. На стадии трения S немного уменьшается, а L не изменяется. После помола на воздухе наряду с основной фазой моноклинного α-Bi2O3 обнаружена фаза висмутита Bi2O2CO3, возникающего в результате сорбции CO2 из воздуха. При прогреве активированного образца висмутит разлагается с выделением CO2 в широком температурном диапазоне. Для активированного образца наноразмерного оксида поглощение тепла при фазовом переходе α-Bi2O3 → δ-Bi2O3 начинается при температуре на 10°C ниже обычной. Реакционная способность активированного Bi2O3 проверена на примере его восстановления в атмосфере CO. Механическая активация повышает глубину восстановления Bi2O3 при 600°C в 2.5 раза и снижает температуру начала восстановления примерно на 100°C.
1. ВВЕДЕНИЕ
Оксид висмута Bi2O3 существует в четырех кристаллических модификациях. Две из них – α-Bi2O3(моноклинная, пространственная группа P21/c, параметры ячейки a = 0.585, b = 0.8166, c = 1.3827 нм, β = 90°) и δ-Bi2O3 (высокотемпературная, кубическая гранецентрированная, Fm3m) стабильны, а β-Bi2O3 (тетрагональная, P4b2) и γ-Bi2O3 (кубическая, объемноцентрированная, I23) метастабильны. α-Bi2O3 при нагревании до 727°C переходит в δ-Bi2O3 –x с частичной потерей кислорода. Метастабильная фаза β-Bi2O3 образуется при охлаждении δ-Bi2O3 до 646°C, а при дальнейшем охлаждении до 620–605°C переходит в α-Bi2O3. Фазу γ-Bi2O3 удается получить при охлаждении δ-Bi2O3 в среде кислорода до 635°C. Температура плавления α-Bi2O3 составляет 825°C [1].
Достаточно большое количество работ посвящено изучению систем на основе Bi3+, обладающих сегнетоэлектрическими, сверхпроводящими, сцинтилляционными и магнитными свойствами [2], за счет различных нецентросиметричных структур типа перовскита, флюорита, пирохлора и т.д. Это связано с высокой поляризуемостью висмута и наличием у Bi3+ неподеленной 6s 2 электронной пары. Все это обеспечивает широкое использование Bi2O3 как одного из исходных веществ при создании газовых сенсоров, антиотражающих покрытий, детекторов радиации, фотоэлектрических батарей, оптоэлектронных устройств [3]. Кроме того, широкое применение соединений висмута в различных областях промышленности связано с его безопасностью, нетоксичностью. Большое количество работ посвящено использованию катализаторов на основе оксида висмута в процессах окислительной димеризации метана [4, 5], окисления пропилена [6]. В последние годы широкое применение Bi2O3 нашел в синтезе катализаторов, используемых при фотокатализе. Фотокатализаторы, приготовленные на основе оксида висмута, проявляют активность при поглощении видимого излучения, при этом возрастает каталитическая эффективность этих систем. Известно, что α-Bi2O3 используется при фотоминерализации хлорфенола, циануровой кислоты, дихлоруксусной кислоты [7] и формальдегида при облучении светом в видимом диапазоне [8]. Фотокаталитическую активность в реакциях разложения органических красителей демонстрируют полиморфные модификации α-, δ- и γ-Bi2O3 [9–11].
Опубликован ряд работ по механической активации (МА) Bi2O3. В [12] рассмотрены свойства аморфной фазы, получаемой методом МА этого оксида. МА двух- и трехкомпонентных смесей, содержащих Bi2O3, применяется для приготовления висмутсодержащих высокотемпературных пьезоэлектриков [13], различной керамики для СВЧ-печей [14–16], механохимической стабилизации фазы γ-Bi2O3, приготовления нанокомпозитов Bi–Fe2O3 [17] и т.д.
В последнее время стало очевидным, что Bi2O3 является одним из наиболее перспективных материалов для приготовления твердооксидных топливных элементов. Высокотемпературная фаза δ-Bi2O3 считается одним из лучших кислород-ионных проводников и обладает высокодефектной структурой флюорита [18–20]. δ-Bi2O3 характеризуется уникальными свойствами: высокими поляризуемостью катионной решетки и степенью разупорядочения анионной подрешетки, присутствием до 25% анионных вакансий и сильно ассиметричным окружением ионов висмута. Все это приводит к очень высокой ионной проводимости δ-Bi2O3 – на 2 порядка большей, чем у стабилизированного ZrO2: при 730°C она равна 1 Ом–1 см–1.
Подчеркнем, что для δ-Bi2O3, так же как и для ZrO2, возникает необходимость стабилизации этой высокотемпературной модификации. Для того чтобы сохранить высокотемпературную модификацию δ-Bi2O3 при комнатной температуре, необходимо провести замещение части катионов Bi3+ на Mn+ (M – щелочноземельный элемент, Ln, Y, Pb, Mo, W, Sn) [18–20]. Выполнены первые попытки применить для этого методы механохимии [21].
Еще одна возможность применения α-Bi2O3 – его использование в составе энергонасыщенных нанокомпозитов ряда Me/X (X – твердый окислитель). Одним из перспективных способов приготовления нанокомпозитов с рекордными свойствами (например, с большой скоростью горения) является метод МА [22–24]. Повышение химической активности в процессе МА может быть связано как с созданием большой поверхности контакта компонентов, так и с образованием разнообразных точечных и линейных дефектов, в том числе, на поверхности [25].
Настоящий цикл работ посвящен возможностям использования методов механохимии для улучшения свойств композитов, содержащих Bi2O3. В первой работе проанализированы образование и свойства дефектной структуры α-Bi2O3 при его МА.
2. ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
В работе использовался α-Bi2O3 марки “х. ч.”. В связи с тем, что в исходном оксиде висмута оказалось значительное количество карбоната висмута, перед проведением экспериментов образцы прогревали на воздухе при температуре 650–670°C. При этом происходит разложение карбоната с выделением CO2 в газовую фазу. Дифракционные данные подтвердили исчезновение линий карбоната и однофазность Bi2O3 после такой термообработки.
Механоактивацию основного количества образцов проводили в мельнице SPEX-8000 на воздухе. Загрузка оксида – 13 г. Масса шаровой загрузки – 43 г (4 шара диаметром 10 мм и 10 шаров диаметром 5 мм). В отдельных опытах была использована существенно более энергонапряженная планетарная мельница Активатор-2SL.
Фазовый состав образцов определяли с помощью рентгенофазового анализа (РФА) на дифрактометре Дрон-3М. Количественный фазовый состав определяли, пользуясь программой Phan% [26]. Размер L областей когерентного рассеяния (ОКР), величину микроискажений (с учетом геометрического уширения), а также параметры решетки рассчитывали с помощью программы Profile [26].
Удельную поверхность S образцов, предварительно прогретых в вакууме при 150°C, определяли из изотерм низкотемпературной адсорбции аргона по методу БЭТ.
Термогравиметрические исследования проводили на приборе синхронного термического анализа STA 449C (NETZSCH, Германия), сопряженного с масс-спектрометром Aeolos-32, в среде инертного газа He высокой чистоты (марка “55”, 99.9995%) при различных значениях температуры вплоть до 900°C. Скорость нагрева образца составляла 10 град/мин.
Восстановление α-Bi2O3 проводили в среде CO при температуре до 600°C.
3. РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
3.1. Механическая активация Bi2O3
На рис. 1 приведена зависимость величины удельной поверхности α-Bi2O3 от времени t его МА. Видно, что значение S увеличивается к t = 90 мин от 0.1 до 3.3 м2/г, а затем уменьшается. По аналогии с [27] начальный участок роста удельной поверхности и уменьшения размера частиц мы назовем стадией раскола, а участок активации материала на этапе уменьшения удельной поверхности – стадией трения. Из максимальной величины S в предположении сферической формы монодисперсных частиц можно оценить их диаметр D по соотношению S = 3/(ρD), где ρ – плотность Bi2O3. Подставляя значения S = 3.3 м2/г и ρ = 9.36 г/см3, получаем D ~ 100 нм.

На рис. 2 представлены дифрактограммы исходного и механически активированных образцов Bi2O3. На дифрактограмме исходного образца зарегистрированы линии только одной фазы – моноклинного α-Bi2O3 с параметрами a = 5.8622 ± ± 0.0017, b = 8.1516 ± 0.0039, c = 7.5292 ± 0.0009 Å, β = 113.057 ± 0.018°, что близко к табличным значениям. При МА на стадии раскола образец остается однофазным, а при временах активации более 90 мин появляются линии, соответствующие фазе висмутита Bi2O2CO3 (рис. 2 и табл. 1). При максимальной степени активации количество висмутита достигает 18 мас. %. Так как МА проводили на воздухе, то можно полагать, что образование висмутита обусловлено хемосорбцией CO2 из воздуха.
Рис. 2.
Дифрактограммы исходного (1), активированных (2–4) и прогретого до 400°С после активации (5) образцов Bi2O3. Время активации: 2 – 20, 3 – 90, 4, 5 – 180 мин.

Таблица 1.
Фазовый состав, размер L областей когерентного рассеяния при разной продолжительности активации t α-Bi2O3 и потеря массы ΔM(T) при прогреве образцов в He до 800 и 900°C
| № образца | t, мин | Фазовый состав, мас. % | L, нм | ΔМ(800) | ΔМ(900) |
|---|---|---|---|---|---|
| 1 | 0 | α-Bi2O3 | – | 0.22% | 0.42% |
| 2 | 20 | α-Bi2O3 | 52 ± 4 | – | – |
| 3 | 90 | α-Bi2O3 + следы Bi2O2CO3 | 38 ± 3 | – | – |
| 4 | 180 | 82 α-Bi2O3 + 18 Bi2O2CO3 | 40 ± 3 | 1.63% | 1.80% |
При МА параметры фазы α-Bi2O3 в пределах точности измерений не изменяются, однако ширина дифракционных линий увеличивается. Анализ, проведенный с учетом геометрического уширения, показал, что уширение линий является чисто блочным, а размеры L ОКР приведены в табл. 1. Видно, что на стадии раскола значение L уменьшается до примерно 40 нм, а на стадии трения оно не изменяется.
В результате МА в образце возникают парамагнитные дефекты, регистрируемые в спектрах ЭПР. На стадии раскола спектры ЭПР Х-диапазона имеют вид широких линий, расположенных от очень слабых полей (большие значения g-фактора) до области центральных полей (вблизи 0.3 мТл для g = 2). С увеличением продолжительности активации интенсивность этих линий возрастает. На стадии трения, при временах МА t > 90 мин на фоне этого спектра в той же протяженной области регистрации ЭПР проявляются парамагнитные дефекты с множественными разрешенными линиями тонкой структуры. На качественном уровне высокая мультиплетность и большие значения g-фактора свидетельствуют о локализации дефектов вблизи ядер висмута (спин IBi = 9/2 формирует 10 линий сверхтонкой структуры для каждой ориентации дефекта в порошковом образце; сильная спин-орбитальная связь приводит к значительным расщеплениям магнитных подуровней электрона в малых полях спектрометра). Определение состава и структуры дефектов требует количественного анализа полученных спектров, выходящего за рамки данной работы.
3.2. Термическая устойчивость дефектов
Данные о поведении МА-образцов при их прогреве приведены в табл. 2 и на рис. 3 и 4. В табл. 2 сопоставлены данные по изменению удельной поверхности образцов при их хранении при комнатной температуре или прогреве до разных значений температуры. Образцы 1 и 2 в табл. 2 соответствуют максимуму удельной поверхности (рис. 1), а образец 3 получен при максимальной продолжительности МА. Оказалось, что хранение активированных образцов на воздухе в течение года приводит к уменьшению величины удельной поверхности, причем для образца 1 этот эффект выражен ярче, чем для образца 3 с максимальной продолжительностью активации. Последующий прогрев до 400°C сопровождается дальнейшим уменьшением величины S. Для образца 1 к 400°C удельная поверхность уменьшается более чем в два раза, а для образца 3 значение S после прогрева до 400°C составляет 71% от исходного. Образец 2 был приготовлен недавно, и для него измерения S проведены сразу после получения. В этом случае уменьшение удельной поверхности после прогрева до 400°C оказалось более существенным, чем в предыдущих случаях, а после прогрева до 700°C величина S уменьшилась почти на порядок.
Таблица 2.
Относительное уменьшение удельной поверхности S/S0 образцов α-Bi2O3, активированных в течение времени t, при их хранении при комнатной температуре и прогреве. S0 – удельная поверхность сразу после активации, S(1 год) – значение S после хранения МА-образца в течение 1 года, S(400) и S(700) – значения S после прогрева МА-образцов до 400 и 700°C
| № образца | t, мин | S(1 год)/S0 | S(400)/S0 | S(700)/S0 |
|---|---|---|---|---|
| 1 | 90 | 0.71 | 0.45 | – |
| 2 | 90 | – | 0.38 | 0.15 |
| 3 | 180 | 0.87 | 0.71 | – |
Рис. 3.
Данные ДСК (1, 2), ТГ (1 ', 2 ') и измерений ионного тока O2 (1 '', 2 '') для исходного (1, 1 ', 1 '') и активированного 180 мин (2, 2 ', 2 '') образцов Bi2O3; 2 ''' –ионный ток CO2 для активированного образца.
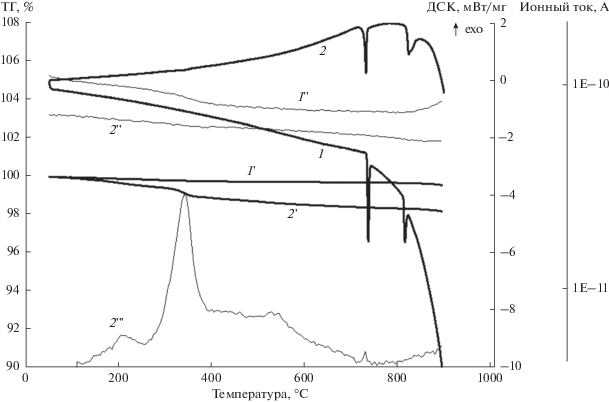
Рис. 4.
Тепловые эффекты для неактивированного (1) и активированного в течение 180 мин (2) образцов Bi2O3.

Одновременно со спеканием порошка при прогреве происходит уменьшение ширины дифракционных линий, обусловленное ростом размеров ОКР. Спектр 5 на рис. 2 измерен после прогрева МА-образца 4 до 400°C. Видно, что ширина линий этого спектра несколько уменьшилась по сравнению со спектром 4. Оценки по формуле Шеррера показали, что после прогрева до 400°C значение L увеличилось до 75 нм.
На рис. 3 сопоставлены данные калориметрических, термогравиметрических и масс-спектральных измерений при прогреве исходного и механоактивированного Bi2O3. При прогреве исходного оксида происходит уменьшение массы (кривая 1 ' рис. 3, табл. 1) в диапазоне 100–800°C на 0.22% (выделение остатков CO2 и соединения с массой 30). При температуре 800–900°C наблюдается увеличение ионного тока O2 (1") и дополнительное уменьшение массы образца (табл. 1). Можно полагать, что в этом температурном диапазоне происходит разложение оксида с выделением кислорода. На ДСК-кривой (1) наблюдаются эндоэффекты при 733 и 812°C, связанные с фазовым переходом α-Bi2O3 → δ-Bi2O3 и плавлением Bi2O3 соответственно. При повторном прогреве исходного образца (данные не приведены) потери массы до 800°C практически нет, а в диапазоне 800–900°C снова наблюдается выделение кислорода, зафиксированное методами масс-спектрометрии и термогравиметрии (ТГ). Оба эндотермических процесса (фазовый переход и плавление) при повторном прогреве также наблюдаются, однако величины тепловых эффектов заметно меньше. Оказалось, что жидкий оксид висмута растворяется в стенках кюветы ДСК, что ограничивает возможности количественных высокотемпературных измерений.
Активированному образцу Bi2O3 на рис. 3 соответствуют кривые 2, 2 ', 2 '' и 2 '''. В этом случае при низких температурах наблюдается более существенное уменьшение массы (кривая 2 '), обусловленное выделением CO2 – максимальным при 340°C (кривая 2 ''') и продолжающимся при более высоких значениях температуры. Выделение CO2 вызвано разложением висмутита Bi2O2CO3 (дифрактограмма 4 рис. 2 и табл. 1), который образовался при помоле в результате сорбции углекислого газа из воздуха. После прогрева фаза висмутита из образца исчезла (спектр 5 рис. 2). Если исходить из дифракционных данных по содержанию висмутита в образце (табл. 1), уменьшение массы при реакции
(1)
${\text{B}}{{{\text{i}}}_{{\text{2}}}}{{{\text{O}}}_{{\text{2}}}}{\text{C}}{{{\text{O}}}_{3}} \to {\text{B}}{{{\text{i}}}_{{\text{2}}}}{{{\text{O}}}_{{\text{3}}}} + {\text{C}}{{{\text{O}}}_{2}}$Положение и форма двух высокотемпературных эндотермических пиков для максимально активированного образца заметно изменены по сравнению с исходным образцом Bi2O3 (рис. 4). Следует отметить, что для исходного образца температура фазового перехода α-Bi2O3 → δ-Bi2O3 примерно соответствует литературным данным, а температура плавления ниже стандартного значения примерно на 10°C [1]. Для активированного образца температура фазового перехода сдвинута в низкотемпературную область примерно на 10°C, а кривая плавления – на 10°C в сторону более высоких температур. Сдвиг фазового перехода α-Bi2O3 → δ-Bi2O3 в низкотемпературную область может быть обусловлен образованием при МА наноразмерных частиц оксида, причина сдвига температуры плавления требуют дополнительного исследования. Не исключено, что в форму кривых плавления вносит вклад растворение жидкого висмута в стенках кюветы.
Форма спектра ЭПР и концентрация парамагнитных центров, согласно предварительным данным, не изменяется при прогреве до 400°C.
3.3. Реакционная способность активированного Bi2O3
Реакционная способность исходного и механически активированных образцов α-Bi2O3 была сопоставлена на примере реакции восстановления Bi2O3 при прогреве в атмосфере CO. Для этого помимо исходного Bi2O3 были использованы образцы, полученные его помолом в течение 2 и 4 мин в энергонасыщенной планетарной мельнице Активатор-2SL. Величины удельной поверхности этих образцов приведены в первом столбце табл. 3. Образец 1 являлся исходным порошком α-Bi2O3, образец 2 соответствовал максимуму удельной поверхности (см. рис. 1), а образец 3 был близок к образцу с максимальной дозой активации, приготовленному уже на стадии трения. Были проведены опыты по восстановлению всех этих образцов оксида при их прогреве в ДСК-ячейке в атмосфере CO (рис. 5). Схема опытов была следующей: прогрев в CO до 600°C, охлаждение в He и повторный прогрев в He до 350°C (выше температуры плавления висмута). После этого измеряли рентгеновские дифрактограммы.
Таблица 3.
Концентрация Bi, образовавшегося при восстановлении Bi2O3 в атмосфере CO, по данным о потере массы (ΔМ), количественного фазового анализа и о его теплоте плавления Q. S – удельная поверхность, ΔMexp и ΔMb – значения экспериментальной и фоновой потерь массы
| № образца | S, м2/г | ΔM, % | РФА | ДСК | |||
|---|---|---|---|---|---|---|---|
| ΔMexp | ΔMexp – ΔMb | [Bi], мас. % | [Bi], мас. % | Q, Дж/г | [Bi], мас. % | ||
| 1 | 0.41 | 1.91 | 1.26 | 12.2 | 9.3 | 4.29 | 9.1 |
| 2 | 4.5 | 7.49 | 6.52 | 63.3 | 52.8 | 26.36 | 55.9 |
| 3 | 2.4 | 6.13 | 5.16 | 50 | 38.6 | 20.11 | 42.6 |
Рис. 5.
Уменьшение массы (кривые 1–3), снижение парциального давления кислорода (кривые 1 '–3 ') и выделение CO2 (кривые 1 ''–3 '') при прогреве исходного (1–1 '') и активированных образцов № 2 (2–2 '') и № 3 (3–3 '') в атмосфере CO.

Из рис. 5 следует, что при прогреве в CO масса образца уменьшается одновременно с ростом выделения CO2. Естественно полагать, что уменьшение массы обусловлено, прежде всего, отрывом атома кислорода от оксида для образования CO2 из CO.
Дифракционные данные (рис. 6) подтверждают, что после прогрева образцов в CO образуется фаза Bi. Следует отметить, во всех образцах присутствуют только фазы Bi2O3 и Bi, а в образце 2 обнаружены еще следы карбоната.
Рис. 6.
Дифрактограммы активированного образца Bi2O3 (№ 3 в табл. 3) до (1) и после (2) прогрева в CO.

Таким образом, протекает реакция
Как видно на рис. 5, восстановление исходного образца 1 начинается при 350°C. Уменьшение массы к 600°C составляет 1.91%. Вычитая фоновое изменение массы и учитывая, что убыль массы обусловлена отрывом кислорода, получаем, что в этом опыте образовалось 12 мас. % висмута. В активированных образцах 2 и 3 реакция начинается примерно на 100°C ниже, а степень превращения заметно выше. Следует отметить, что максимальное превращение характерно для образца 2, т.е. для образца с наибольшей удельной поверхностью.
Из рис. 5 следует также, что одновременно с выделением CO2 происходит уменьшение парциального давления O2. Это указывает на определенное участие газофазного кислорода в образовании CO2.
На рис. 7 приведены термограммы образцов при их прогреве в He после восстановления в CO. Эндотермические пики соответствуют плавлению висмута, образовавшегося при восстановлении Bi2O3 в CO. Там же приведена кривая плавления “эталонного” висмута. Видно, что температура плавления “восстановленного” висмута примерно на 3°C ниже, чем эталонного образца. Из площади пиков можно оценить количество образовавшегося висмута.
Рис. 7.
Термограммы плавления висмута при повторном прогреве образцов в He после восстановления в CO. Номера кривых 1–3 соответствуют номерам образцов в табл. 3; кривая 4 относится к эталонному образцу Bi.

В табл. 3 сопоставлены данные трех методов (РФА, ТГ и ДСК) определения количества образовавшегося висмута. Видно, что результаты, полученные с помощью РФА и измерения теплоты плавления, хорошо совпадают. В то же время при расчете содержания Bi по потере массы надо учитывать выделение из образца CO2. Такая поправка сделана, однако очевидно, что точность этого метода ниже.
ЗАКЛЮЧЕНИЕ
Таким образом, механическую активацию α-Bi2O3 можно разделить на две стадии – раскола и трения.
На стадии раскола уменьшается средний размер частиц до 100 нм и растет удельная поверхность S до 3.2 м2/г, уширяются дифракционные линии, вследствие уменьшения размеров ОКР до 40 нм, возникают парамагнитные дефекты. Сформировавшаяся на стадии раскола дефектная структура достаточно легко релаксирует. При длительном хранении на воздухе значение S активированного образца оксида уменьшается на 30%. Прогрев активированного Bi2O3 до 400°C сопровождается уменьшением S более чем в два раза с одновременным ростом размеров ОКР, а прогрев до 700°C приводит к падению значения S почти на порядок величины.
При увеличении дозы активации удельная поверхность перестает расти и происходит агрегирование частиц материала, сопровождающееся уменьшением величины S в 1.5–2 раза при сохранении размеров ОКР. Эту стадию мы называем стадией трения. В таких агрегатах поверхность обладает большей термической устойчивостью: при прогреве до 400°C образца α-Bi2O3, активированного с максимальной дозой, S уменьшается только на 30%. На стадии трения образуются парамагнитные дефекты, имеющие множественные линии тонкой структуры и расположенные вблизи ядер висмута. Однако строение и состав этих парамагнитных дефектов пока не удалось расшифровать.
После помола на воздухе на стадии трения наряду с основной фазой моноклинного α-Bi2O3 обнаружена фаза висмутита Bi2O2CO3, возникающая в результате сорбции оксидом CO2 из воздуха. При прогреве активированного образца висмутит разлагается с выделением CO2 в широком температурном диапазоне. Для активированного образца наноразмерного оксида поглощение тепла при фазовом переходе α-Bi2O3 → δ-Bi2O3 начинается при температуре на 10°C ниже обычной.
Реакционная способность активированного Bi2O3 проверена на примере его восстановления в атмосфере CO. Механическая активация повышает степень восстановления Bi2O3 при 600°C в 2.5 раза и снижает температуру начала восстановления примерно на 100°C.
Список литературы
Medernach J.W., Snyder R.L. // J. Am. Ceram. Soc. 1978. V. 61. P. 494.
Орлов В.Г., Буш А.А., Иванов С.А., Журов В.В. // Физика твердого тела. 1997. Т. 39. С. 865.
Юхтин Ю.М., Михайлов Ю.И. Химия висмутовых соединений и материалов. Новосибирск: Издательство СО РАН, 2001.
Воскресенская Е.Н., Куртеева Л.И., Аншиц А.Г. // Нефтехимия. 1991. Т. 31. С. 416.
Воскресенская Е.Н., Куртеева Л.И., Цыганкова С.И., Аншиц А.Г. // Химия твердого топлива. 1993. № 2. С. 79.
Lei Y.-H., Chen Z.-X. // Sci. China Chem. 2015. V. 58. P. 593.
Eberl J., Kisch H. // Photochem. Photobiol. Sci. 2008. V. 7. P. 1400.
Ai Z., Huang Y., Lee S., Zhang L. // J. Alloys Compd. 2011. V. 509. P. 2044.
Schlesinger M., Schulze S., Hietschold M., Mehring M. // Dalton Trans. 2013. V. 42. P. 1047
Iyyapushpam S., Nishanthi S.T., Padiyan D.P. // J. Alloys Compd. 2014. V. 601. P. 85.
Wu Y., Lu G. // Phys. Chem. Chem. Phys. 2014. V. 16. P. 4165.
Рыков А.И., Павлюхин Ю.Т., Болдырев В.В., Сиротина Н.И., Колышев А.Н. // Изв. СО АН СССР. Сер. хим. наук. 1986. Вып. 2. С. 36.
Ng S.H., Xue J.M., Wang J. // Mater. Chem. Phys. 2002. V. 75. P. 131.
Zhou D., Wang H., Yao X., Pang L.-X., Zhou H.-F. // J. Am. Ceram. Soc. 2008. V. 91. P. 139.
Petkov V., Selbach S.M., Einarsrud M.-A., Grande T., Shastri S.D. // Phys. Rev. Lett. 2010. V. 105. P. 185501.
Poleti D., Karanovic L., Zdu M., Jovalekic C., Brankovic Z. // Solid State Sci. 2004. V. 6. P. 239.
Jiang N., Wachsman E.D., Jung S.-H. // Solid State Ion. 2002. V. 150. P. 347.
Obbade S., Huve M., Suard E., Drache M., Conflant P. // J. Solid State Chem. 2002. V. 168. P. 91.
Battle P.D., Catlow C.R.A., Heap J.M., Moroney L.M. // J. Solid State Chem. 1986. V. 63. P. 8.
Battle P.D., Catlow C.R.A., Moroney L.M. // J. Solid State Chem. 1987. V. 67. P. 42.
Poleti D., Karanovic L., Zdujic M., Jovalekic C., Brankovic Z. // Solid State Sci. 2004. V. 6. P. 239.
Dreizin E.L. // Prog. Energy Combust. Sci. 2009. V. 35. P. 141.
Baláž P., Achimovičová M., Baláž M., Billik P., Cherkezova-Zheleva Z., Criado J.M., Delogu F., Dutková E., Gaffet E., Gotor F.J., Kumar R., Mitov I., Rojac T., Senna M., Streletskii A., Wieczorek-Ciurowa K. // Chem. Soc. Rev. 2013 V. 42. P. 7571.
Dolgoborodov A.Yu. // Combust. Explos. Shock Waves. 2015. V. 51. P. 86.
Streletskii A.N., Sivak M.V., Dolgoborodov A.Yu. // J. Mater Sci. 2017. V. 52. P. 11810.
Shelekhov E.V., Sviridova T.A. // Met. Sci. Heat Treat. 2000. V. 42. P. 309.
Стрелецкий А.Н., Бутягин П.Ю. // Кинетика и катализ. 1980. Т. 21. С. 770.
Дополнительные материалы отсутствуют.
Инструменты
Коллоидный журнал