

Коллоидный журнал, 2021, T. 83, № 1, стр. 75-85
Термодинамическое исследование солюбилизации краун-замещенного фталоцианината магния в водных растворах додецилсульфата натрия
Т. Г. Мовчан 1, *, А. И. Русанов 1, 2, Е. В. Плотникова 1
1 Институт физической химии и электрохимии им. А.Н. Фрумкина РАН
119071 Москва, Ленинский просп., 31, Россия
2 Менделеевский центр, Санкт-Петербургский государственный университет
199034 Санкт-Петербург, Университетская наб., 7/9, Россия
* E-mail: movchan_tamara@mail.ru
Поступила в редакцию 12.08.2020
После доработки 18.08.2020
Принята к публикации 20.08.2020
Аннотация
Проведено спектрофотометрическое исследование солюбилизации краун-замещенного фталоцианината магния(I) в водном растворе додецилсульфата натрия(II). Опыты проводились с насыщенными растворами I в условиях термодинамического равновесия раствора с осадком I. Более детально изучено известное явление перехода от димеров I к мономерам в процессе солюбилизации. Установлено, что мономеризация I начинается при концентрациях гораздо ниже критической концентрации мицеллообразования (ККМ) II, а при еще меньших концентрациях ПАВ образуются специфические мицеллы II с участием димеров I. Присутствие димеров характерно и при растворении I в чистой воде; растворимость I составила 7.38 мкМ (химики обычно считают, что I в воде не растворим). В методической части работы найдены коэффициенты экстинкции мономеров и димеров I. На основе экспериментальных данных определены термодинамические характеристики солюбилизации: солюбилизационная емкость мицелл, коэффициент распределения солюбилизата между мицеллами и окружающим раствором, а также стандартное сродство солюбилизации I. В расчете на одну молекулу I в мицелле найденное значение солюбилизационной емкости приводит к аномально большому числу агрегации (309). Среди объяснений этого явления – возможность возникновения бимодального распределения мицелл, в котором мицеллы с солюбилизатом сосуществуют с “пустыми” мицеллами, благодаря чему среднее число молекул солюбилизата в мицелле может оказаться меньше единицы.
ВВЕДЕНИЕ
Фталоцианины и их производные широко применяются в самых разных областях, включая молекулярную электронику, химию и медицину. И так случилось, что для более эффективного использования их можно применять в сочетании с ПАВ. В частности, речь идет о коллоидных ПАВ, способных к мицеллизации, и о таком явлении коллоидной химии как солюбилизация. В классической интерпретации солюбилизация – вхождение в мицеллу ПАВ постороннего вещества (солюбилизата) – это метод повышения растворимости при помощи мицелл. Для малорастворимых в воде фталоцианинов это тоже имеет значение, но здесь обнаруживается и другой, весьма примечательный эффект. Хотя растворимость фталоцианинов в воде очень мала (речь идет не о милли-, а о микромолях на литр), они часто находятся в растворе в агрегированном состоянии (обычно пишут о димерах, но распределение агрегатов по размерам пока еще не изучено). В то же время, по целому ряду причин (например, для проявления хромофорных свойств молекул) желательно иметь растворенные фталоцианины в мономерной форме. Именно это и достигается с помощью солюбилизации. К настоящему времени накопился целый ряд исследований в этом направлении [1–10], но все они чисто химического плана и опираются, в основном, на различные спектроскопические данные. Было бы полезно дополнить их термодинамическим анализом. Именно с этой целью и предпринята данная работа.
Основы термодинамики солюбилизации можно найти, например, в [11–13], где термодинамика мицеллообразования вообще строится на законе действия масс (современное развитие дано в [14–18]) в противовес модели фиктивной псевдофазы, заменяющей совокупность реальных мицелл [19]. В теории же солюбилизации альтернатива немного другая: рассматривать распределение солюбилизата между раствором и либо псевдофазой, либо реальными мицеллами. Надо сказать, что в термодинамике солюбилизации модель псевдофазы, будучи несостоятельной, все еще достаточно популярна (для примера можно взять работы [20, 21]) ввиду своей простоты. Из опыта определяется коэффициент распределения солюбилизата (в общем случае любого вещества i) между мицеллами (область внутри мицеллы обозначается символом фазы α) и раствором (фаза β)
где ${{x}_{i}}$ – мольная доля солюбилизата, индекс x показывает, что коэффициент распределения выражен в формализме мольных долей (в отличие, скажем, от коэффициента распределения ${{K}_{c}}$ как отношения концентраций). В формуле (1) кто-то считает область α псевдофазой, но в действительности это внутренняя область мицеллы, где что-то похожее на фазу (например, углеводородное ядро прямой мицеллы) действительно может быть. Сформулированная недавно [22] термодинамическая теория солюбилизации с участием реальных мицелл связывает коэффициент распределения со стандартным сродством солюбилизации ${{A}_{{i\operatorname{s} }}}$ соотношением(2)
$kT\ln {{K}_{x}} = \mu _{{i\operatorname{s} }}^{\beta } - \mu _{{i\operatorname{s} }}^{\alpha } \equiv {{A}_{{i\operatorname{s} }}},$(3)
$\Delta p \equiv {{p}^{\alpha }} - {{p}^{\beta }} = \gamma \left( {\frac{1}{{{{R}_{1}}}} + \frac{1}{{{{R}_{2}}}}} \right),$В работе [22] также решена задача о влиянии солюбилизации на критическую концентрацию мицеллообразования (ККМ) ПАВ. При этом использовался не только закон действия масс, но и определение ККМ через его константу и другие сопутствующие постоянные. Показано, что солюбилизация как из насыщенного, так и ненасыщенного раствора солюбилизата приводит к снижению величины ККМ. Это видно из формулы
(4)
${{c}_{{1\operatorname{m} }}} = {{\left( {\frac{1}{{nKc_{{21}}^{z}}}} \right)}^{{\frac{1}{{n - 1}}}}},$В данной работе изучалась солюбилизация краун-замещенного фталоцианината магния (I) в водном растворе додецилсульфата натрия (II) – одного из самых известных анионных коллоидных ПАВ.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Реактивы
Вещество I представляет собой довольно сложное комплексное соединение металла с плоской молекулярной структурой (рис. 1). Синтез и очистка I, произведенные в Институте физиологически активных веществ РАН совместно с Институтом физической химии и электрохимии им. А.Н. Фрумкина РАН, описаны в работе [1]. К нам препарат I поступил в виде кристаллического порошка, что свидетельствует в пользу его оценки как препарата высокой чистоты.
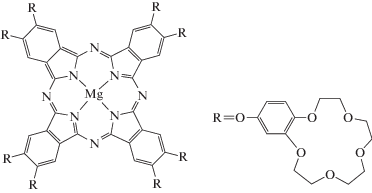
ПАВ II (Acros Organics, содержание основного вещества 99.8%) использовались без дальнейшей очистки. Вода бралась в виде тридистиллята с удельной электропроводностью не выше 4 × × 10–4 См/м при 27°C. Наконец, в работе с чисто вспомогательными целями (как растворитель I для калибровки) использовался еще один реактив – диметилсульфоксид (“х. ч.” производства Компонент-Реактив, Россия).
Методика измерений и результаты
Главной задачей эксперимента было определение равновесной концентрации I в мицеллярных растворах. Для этого был привлечен спектрофотометрический метод, подразумевающий измерение оптической плотности A раствора для характерной длины волны. Нахождение последней и было первой методической проблемой. Как уже отмечалось выше, фталоцианины могут находиться в растворах в агрегированном состоянии, и, чтобы отличать, например, димеры I от мономеров, требуются разные характерные длины волн. Для нас особый интерес представляют мономеры, но идеальный растворитель, в котором обнаруживались бы только мономеры, пока не найден. Однако есть растворители, где фталоцианины находятся преимущественно в мономерной форме. В [1] и последующих работах в качестве такого растворителя для I использовался дихлорметан.
Среди известных растворителей имеется также диметилсульфоксид [23, 24], который хорошо растворяет соединение I, а электронные спектры поглощения указывают на преимущественное содержание мономеров. На рис. 2 приведены фрагменты электронных спектров поглощения для I в насыщенном водном растворе (кривая 1) и при некоторой концентрации в диметилсульфоксиде (кривая 2). Видно, что водный раствор практически не содержит мономеров фталоцианина, тогда как в диметилсульфоксиде представлено значительное количество мономеров и лишь немного вещества I в другой форме. Мономерам I отвечает узкий интенсивный пик на длине волны 682 нм в видимой части спектра. Сопутствующий небольшой максимум при длине волны 616 нм было бы отнести к димерам соблазнительно, но опрометчиво, ибо по положению он не совпадает с максимумом для водного раствора. В конце концов выяснилось, что характерная длина волны 634 нм, четко позиционирующая себя на рис. 2, как раз и соответствует димерам I [6, 23].
Рис. 2.
Электронные спектры поглощения I в насыщенном водном растворе (1) и в диметилсульфоксиде при концентрации 3.7 мкМ (2) в диапазоне длин волн λ = 520–850 нм (длина оптического пути l = 1 см).

Известно также, что хорошими растворителями для фталоцианинов в мономерной форме являются водные растворы коллоидных ПАВ (эффект достигается с помощью солюбилизации, именно это и является предметом нашего исследования). В связи с этим необходимо взглянуть и на электронные спектры поглощения таких систем. В качестве примера на рис. 3 приведены спектры I в водном растворе II при разных значениях концентрации I (здесь и в дальнейшем условимся обозначать концентрацию солюбилизатора как ${{с}_{1}},$ а концентрацию солюбилизата как ${{с}_{2}}$). Видно, что при всех значениях ${{с}_{2}}$ мономерам I соответствует хорошо выраженный пик при λ = = 682 нм. На рис. 3 просматривается и небольшой пик при λ = 616 нм, его следует толковать как спутник пика мономеров. Мы теперь можем с уверенностью сказать, что характерные длины волн для мономеров (682 нм) и димеров (634 нм) I установлены.
Рис. 3.
Электронные спектры поглощения I в водных растворах при концентрации ПАВ с1 = 0.03 М и концентрации солюбилизата с2, равной (снизу вверх) 3.2, 4.2, 5.4, 6.7 и 8.1 мкМ, в диапазоне длин волн 580–720 нм (длина оптического пути l = 1 см).

Связь оптической плотности (экстинкции) A с концентрацией поглощающего свет вещества c дается законом Ламберта–Бера
где ε – коэффициент экстинкции и l – длина пути светового луча. Из (6) следует, что для определения концентрации по оптической плотности необходимо знать коэффициент экстинкции. Эта важная характеристика вещества должна быть найдена из независимого эксперимента, что составляло вторую методическую проблему данной работы. Дело в том, что нам нужен коэффициент экстинкции для мономеров фталоцианина, но, как уже отмечалось выше, пока не обнаружено растворителей, содержащих исключительно только мономеры. Мы проводили калибровку оптической плотности по концентрации I на примере диметилсульфоксида в качестве растворителя (рис. 4). Экспериментальная зависимость A(c) хорошо отображается прямой линией и приводит к значению молярного коэффициента экстинкции ε = 2.90 × 105 М–1 см–1. Нужно отметить, что в литературе уже имеются значения коэффициента экстинкции для I, полученные в дихлорметане с диэлектрической постоянной 8.9 [3] против 48.9 [23] для диметилсульфоксида. Такое отличие в полярности может сказаться и на значениях коэффициента экстинкции, хотя и между литературными значениями, полученными только с использованием дихлорметана, имеется разнобой: в единицах 105 М–1 см–1 это 1.64 [1] и 2.09 [2] (на длине волны λ = 690 нм). Мы же, проведя калибровку в дихлорметане по всем правилам, получили 4.43 для I, что не меньше, а уже значительно больше указанного значения 2.90. В такой ситуации использование органических растворителей для калибровки вряд ли целесообразно.Рис. 4.
Калибровочные зависимости оптической плотности растворов I в диметилсульфоксиде (1), в растворе II при с1 = 0.03 М (2) и при концентрациях с1 = 0.12–1.7 мМ, включающих димерную часть домицеллярного интервала (3).
Если коэффициент экстинкции вещества зависит от природы среды, возникает естественное желание найти его при использовании такой среды, в которой и будут проводиться эксперименты. Для I это водный раствор коллоидного ПАВ заданной концентрации. Растворив в нем какое-то количество солюбилизата и задав таким путем концентрацию c в формуле (6), а затем, измерив оптическую плотность, мы тут же находим коэффициент экстинкции по формуле (6). Этот простой прием весьма популярен (см., например, [25, 26]). С его помощью были определены значения коэффициента экстинкции I в мицеллярных растворах II и додецилбензолсульфоната натрия как 1.7 [1] и 1.89 [2] (при λ = 686 нм) соответственно (в единицах 105 М–1 см–1). Такой подход, однако, располагает к комментариям.
Во-первых, указанным способом определяется только “точечное” значение коэффициента экстинкции для заданной ситуации, но нахождение истинного коэффициента экстинкции как константы закона Ламберта–Бера требует построения калибровочной кривой, показывающей (своей прямолинейностью), что этот закон действительно выполняется. Во-вторых, в данном случае задание концентрации ПАВ определяет среду. Изменение концентрации ПАВ означает изменение среды. С учетом обсуждавшегося выше влияния среды на значение коэффициента экстинкции закономерно спросить: не требует ли это отдельной калибровки для каждой новой концентрации ПАВ? Можно ответить, что для коллоидных ПАВ здесь имеется одно важное смягчающее обстоятельство – мицеллообразование. Как уже отмечалось во введении, после прохождения ККМ свойства мицеллярной системы изменяются незначительно. В соответствии с требованиями термодинамики химический потенциал ПАВ продолжает возрастать с концентрацией, но настолько медленно, что ситуация напоминает фазовый переход, когда химический потенциал не изменяется совсем (отсюда и образ “псевдофазы”). Очень слабо изменяется и средняя активность ионного ПАВ, а потому можно надеяться, что калибровка, проведенная при некоторой концентрации ПАВ выше ККМ, будет работать в довольно широком интервале концентраций мицеллярного раствора. Что касается домицеллярного раствора ПАВ, то для него, действительно, калибровку в отношении солюбилизата нужно проводить для каждой концентрации ПАВ раздельно. Результатом такой кропотливой работы должна быть функция $\varepsilon ({{с}_{1}}),$ описывающая зависимость коэффициента экстинкции от концентрации ПАВ в диапазоне от чистой воды до ККМ. Выше же ККМ коэффициент экстинкции принимает практически постоянное значение.
В соответствии со сказанным выше, калибровку оптической плотности I в растворе II проводили при постоянной концентрации II, значение которой (естественно, выше ККМ) выбирали из условия полного растворения взятого количества солюбилизата. На рис. 4 показана калибровочная кривая 2 для I в растворах II при с1 = 0.03 М. Видно, что экспериментальная зависимость A(c2) при λ = 682 нм в интервале ${{с}_{2}}$ 1–12 мкМ хорошо (с квадратом смешанной корреляции около R2 = = 0.997) отображается прямой линией, наклон которой определяет значение молярного коэффициента экстинкции как ε = 2.62 × 105 М–1 см–1. Это значение и использовалось для определения концентрации мономеров I во всех наших опытах при λ = 682 нм. Заметим, что такое значение коэффициента экстинкции для мономеров I не сильно отличается от величины ε = 2.90 × 105 М–1 см–1, полученной с привлечением диметилсульфоксида в качестве растворителя.
Отыскание коэффициента экстинкции для димеров I при λ = 634 нм представляет более сложную задачу. Теперь нам требуется среда, где бы, наоборот, были практически только димеры, а мономеры отсутствовали или их концентрация была бы пренебрежимо малой. Такая среда известна – это вода. Но ведь нам нужно, чтобы среда сохраняла свои свойства при прибавлении ПАВ, а последнее, как известно, как раз вызывает образование мономеров. И все же оказалось, что мономеры возникают не сразу. Существует малый концентрационный зазор, внутри которого молекулы II уже появились, а мономеров I еще нет. В этом-то зазоре и нужно работать.
Возникшая ситуация хорошо иллюстрируется электронными спектрами поглощения I на рис. 5. Данные на рис. 5а свидетельствуют о том, что при добавлении II к чистой воде до концентрации 1.7 мМ в ней удается растворить без остатка вещество I до концентрации 8.2 мкМ (кривая 2) и что данная концентрация выше растворимости I в чистой воде (кривая 1). В то же время в спектре еще отсутствует характерный пик мономеров I. Это означает, что сначала присутствие ПАВ в воде повышает растворимость I в виде димеров, т.е. первичная стадии солюбилизации происходит на уровне димеров. Из рис. 5б следует, что при повышении содержания ПАВ ситуация меняется: при той же концентрации I ${{с}_{2}} = \;8.2$ мкМ уже при ${{с}_{1}} = 4.3$ мМ димеры почти не видны, но заметен небольшой пик мономеров I (кривая 1), интенсивность которого при ${{с}_{1}} = 5.7$ мМ резко возрастает (кривая 2). И все это, заметим, происходит ниже ККМ II (составляющей около 8 мМ).
Рис. 5.
Электронные спектры поглощения I в домицеллярных водных растворах II: (а) в области преобладания димеров I при с1 = 0 (кривая 1 – насыщенный раствор I в отсутствие ПАВ) и с1 = 1.7 мМ, с2 = 8.2 мкМ (кривая 2); (б) в области преобладания мономеров I при с2 = 8.2 мкМ и с1 = 4.3 мМ (кривая 1) или с1 = 5.7 мМ (кривая 2). Длина оптического пути l = 1 см.

Обратимся теперь к малому концентрационному зазору, изображенному на рис. 5а. Внутри него и на его верхней границе у нас имеются лишь две экспериментальных точки: A = 0.039 при с2 = = 1.1 мкМ (с1 = 0.12 мМ) и A = 0.3578 при с2 = = 8.2 мкМ (с1 = 1.7 мМ). Но для построения полноценной калибровочной кривой к ним можно прибавить и третью точку – начало координат. Если поставить условие, что калибровочная кривая должна проходить через начало координат, то указанные три точки прекрасно (с квадратом смешенной корреляции 0.999) ложатся на одну прямую (рис. 4, кривая 3), наклон которой и определяет коэффициент экстинкции димеров I как ε = 4.35 × 104 М–1 см–1. Зная коэффициент экстинкции димеров и величину A = 0.321 для точки максимума на кривой 1 рис. 5а, мы теперь можем рассчитать величину растворимости I в воде (часто считается, что в воде I не растворим). Это дает $с_{2}^{*}$ = 7.38 мкМ (звездочка означает растворимость, т.е. равновесную концентрацию раствора в контакте с твердым I). Результаты калибровки суммированы в табл. 1.
Таблица 1.
Значения молярного коэффициента экстинкции ε для I в разных средах
| Среда | λ, нм | ε, 105 М–1 см–1 |
|---|---|---|
| Диметилсульфоксид | 683 | 2.90 |
| Водный раствор II при c1 = 0.03 М | 682 | 2.62 |
| Водный раствор II при c1 = 0.0012 М | 634 | 0.435 |
После проведения калибровки можно было начать эксперимент по исследованию солюбилизации. Он проводился следующим образом. Сначала в воду помещался твердый солюбилизат (I), и система выдерживалась в закрытой емкости при комнатной температуре в защищенном от света месте в течение длительного времени (около месяца) вплоть до установления равновесия. Периодически отбирали пробу раствора над осадком и регистрировали спектр поглощения, после чего пробу возвращали в исходную систему. Далее, в насыщенный водный раствор I добавляли навеску сухого ПАВ II и снова дожидались равновесия (теперь уже в течение двух−трех суток), после чего измеряли оптическую плотность раствора и с помощью коэффициента экстинкции рассчитывали концентрацию солюбилизата. Процедура повторялась несколько раз, в результате чего строилась изотерма солюбилизации. Интервал задаваемых концентраций ПАВ составлял 0.2–10 ККМ (при указанном во введении определении ККМ через константу закона действия масс сама ККМ становится единицей концентрации [14–16]).
Измерения проводились с помощью однолучевого сканирующего спектрофотометра UNICO UV-2800 (США) в диапазоне длин волн 190− 900 нм с использованием стандартных кварцевых кювет с длиной l оптического пути 1 мм и 1 см. Нужно подчеркнуть, что все измерения относились к насыщенным растворам, в условиях, когда раствор все время находился в равновесии с твердой фазой I на дне сосуда. Это значит, что все наши исследования проводились в условиях постоянства химического потенциала I. Из данных по оптической плотности с использованием значений коэффициента экстинкции из табл. 1 вычислялась концентрация I в растворах II. Полученные данные приведены в табл. 2. Ее первые четыре строки отвечают концентрациям II ниже ККМ. Из них первые три строки были обсчитаны со значением коэффициента экстинкции 4.35 × × 104 М–1 см–1 для димеров. Такое значение с гарантией действует в первой и второй строках табл. 2, но полученное в третьей строке явно завышенное значение ${{с}_{2}} = 8.51$ мкМ свидетельствует о том, что при ${{с}_{1}} = 1.7$ мМ в растворе уже появляются мономеры. Действительно, если приглядеться к рис. 5а, можно увидеть искажение кривой 2 (в сравнении с кривой 1) как раз на длине волны, характерной для мономеров. Поскольку коэффициент экстинкции для мономеров в шесть раз больше, чем для димеров, то даже незначительное присутствие мономеров при ${{с}_{1}} = 1.7$ мМ может при расчете снизить концентрацию ${{с}_{2}} = 8.51$ мкМ до разумных значений. Фактически при ${{с}_{1}} = 1.7$ мМ и выше в растворе имеется смесь димеров и мономеров, и коэффициент экстинкции должен иметь эффективное значение между двумя значениями, указанными в нижней части табл. 1. Это значение нам неизвестно, а потому в четвертой строке табл. 2 расчет ${{с}_{2}}$ не проводился. Начиная же с пятой строки табл. 2, расчет относится к концентрациям ПАВ выше ККМ, где с гарантией работает значение ε = = 2.62 × 105 М–1 см–1 для мономеров, которые присутствуют в системе с большим преобладанием.
Таблица 2.
Оптическая плотность (А) и концентрация солюбилизата I (c2) как функции концентрации ПАВ II (c1) в водном растворе при заданном коэффициенте экстинкции (ε)
| c1, мМ | А | ε, 105 М–1 см–1 | c2, мкМ |
|---|---|---|---|
| 0 | 0.321 | 0.435 | 7.38 |
| 0.12 | 0.324 | 7.46 | |
| 1.7 | 0.37 | 8.51 | |
| 4.6 | 0.56 | ||
| 9 | 2.14 | 2.62 | 8.17 |
| 20 | 10.9 | 41.6 | |
| 30 | 20.9 | 79.8 | |
| 40 | 28.4 | 108.5 | |
| 40 | 27.9 | 106.5 |
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Анионное ПАВ II наиболее популярно и наиболее хорошо исследовано, включая даже использование его водного раствора для солюбилизации I [1], что является и нашим предметом изучения. Разница состоит в следующем. В [1] определяли зависимость оптической плотности водного раствора от концентрации II при заданной брутто-концентрации I. При этом, очевидно, в процессе прибавления ПАВ идет мицеллизация II и мономеризация I до тех пор, пока весь I не перейдет в мономерную форму. Как только это происходит, возрастание оптической плотности прекращается, и при дальнейшем прибавлении ПАВ она остается практически постоянной, как это видно на графике рис. 3 в [1]. Мы же всегда работали с насыщенными (по отношению к I) растворами, находящимися (в промежутке между измерениями) в постоянном контакте с равновесной твердой фазой I. Иными словами, мы работали в условиях постоянства химического потенциала солюбилизата, а осадок на дне сосуда играл роль резервуара вещества. По мере поступления ПАВ и солюбилизации свободного I в растворе химический потенциал I уменьшался, что вызывало поток новой порции I из осадка в раствор для восстановления прежнего значения химического потенциала. Таким образом, у нас поступление ПАВ в раствор всегда сопровождалось поступлением в жидкую фазу новой порции I. Это создавало стационарные условия для продолжения такого процесса как угодно долго, а зависимость A или ${{с}_{2}}$ от ${{с}_{1}}$ была всегда возрастающей (напомним, что номером 1 мы обозначили солюбилизатор, а номером 2 – солюбилизат, в данном случае это, соответственно, II и I).
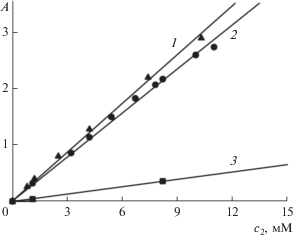
Поскольку расчет разных участков солюбилизационной кривой ${{с}_{2}}({{с}_{1}})$ требует разных и порой неизвестных нам значений коэффициента экстинкции I, мы не можем изобразить эту кривую сплошной линией. Но ее сплошность подтверждается сплошностью зависимости оптической плотности от концентрации ПАВ на рис. 6. Солюбилизационная же кривая ${{с}_{2}}({{с}_{1}})$ частично изображена точками на рис. 7. Их расположение отличается от классической солюбилизационной кривой, прежде всего, тем, что берет начало от нулевой точки системы координат, а не от значения ККМ (для II примерно 8 мМ) на оси абсцисс. Это означает, что для солюбилизации и мономеризации I совсем не обязательно наличие мицелл II, достаточно присутствия в растворе свободных ионов этого ПАВ. Правда, мы знаем, что мицеллы ПАВ присутствуют в растворе и ниже ККМ. Однако их количество настолько мало, что, если бы именно от них зависели мономеризация I и рост оптической плотности, то последний вряд ли был бы заметен.
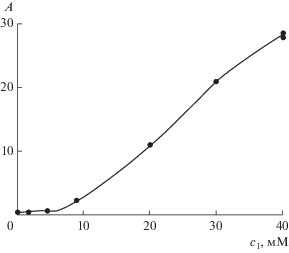
Рис. 7.
Расположение экспериментальных точек для зависимости ${{с}_{2}}$ от ${{с}_{1}}$ в системе I–II–вода.
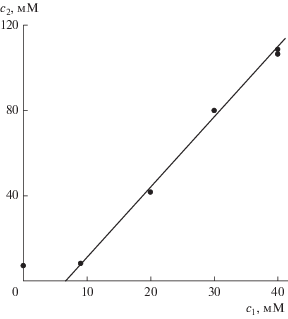
На рис. 7 также видно, что при малых концентрациях солюбилизационная кривая, по крайней мере, имеет другой наклон, чем при больших. Для них пять последних точек хорошо (с квадратом смешанной корреляции 0.997) ложатся на прямую линию, для которой компьютер дает уравнение (обе концентрации в мМ)
Из уравнения (7) следует, что пересечение прямой с осью абсцисс происходит при ${{с}_{1}}$ ≈ 6.5 мМ, и это была бы ККМ II, определенная классическим методом солюбилизации (причем весьма правдоподобная: немного меньше известного значения 8 мМ, а ведь установлено, что солюбилизат всегда понижает значение ККМ ПАВ [17, 18, 22]).
Следуя дальше классическим канонам коллоидной науки, мы должны положить наклон солюбилизационной прямой (т.е. коэффициент при ${{с}_{1}}$ в уравнении (7)) равным солюбилизационной емкости s мицелл II, что является отношением чисел агрегации солюбилизата z и солюбилизатора n (см. формулу (5)):
Отношение двух величин еще не определяет их по отдельности. Но в данном случае мы заранее знали, что в каждую мицеллу II входит лишь одна молекула I (в этом суть явления мономеризации). Положив z = 1, из (8) находим $n \approx 309,$ что более чем в четыре раза, превышает обычное число агрегации II $(n \approx 65).$ Из теории мицеллообразования [11–13] известно, что в процессе солюбилизации число агрегации ПАВ несколько возрастает. Но такой скачок от 65 к 309! Реально ли это?
Чтобы ответить на этот вопрос, прикинем, сколько поверхностно-активных ионов II может частоколом поместиться на поверхности одной молекулы I. По данным [3] диаметр плоской структуры I (рис. 1) составляет около 3 нм, и остается оценить ее толщину. Макроцикл металлофталоцианинов имеет плоско-квадратную координацию центрального иона металла [27] с радиусом внутренней полости макроцикла 0.135 нм [28]. В ней может свободно разместиться ион магния с радиусом 0.066 нм, не выходя за пределы макроцикла. Радиусы атомов углерода и азота, входящие в состав макроцикла соединения I, не превышают радиуса иона магния. Можно предположить, что толщина его макроцикла равна примерно 0.066 нм. Но в соединении I за счет кислородного мостика между бензо-краун-группой и периферией фталоцианина существует конформационная лабильность краун-групп и их подвижность относительно макроциклического лиганда. Поэтому атомы краун-эфирных колец в большей степени выходят из плоскости, проходящей через атомы азота пиррольных колец, чем остальные атомы фталоцианинового остова [27]. Согласно [27, 29], диаметр полости 15-краун-5 составляет 0.17–0.22 нм. При возможности поворота краун-фрагментов молекулы I перпендикулярно плоскости ее макроцикла в разные стороны максимальная толщина структуры может составлять 0.22 × 2 + 0.066 = 0.506 нм, а минимальная (при одностороннем повороте краун-фрагментов) – 0.286 нм.
Таким образом, если аппроксимировать форму молекулы I диском с диаметром 3 нм и максимальной толщиной 0.506 нм, то суммарная площадь его торцевой и боковой поверхности составит 23.06 нм2. Согласно расчету по методике Тэнфорда [30], объем углеводородного хвоста иона II (т.е. додецильного радикала) составляет 0.35 нм3, а его длина – 1.67 нм [11, с. 129]. Делением первого на второе получаем площадь поперечного сечения радикала около 0.21 нм2. Если теперь поделить на эту величину найденную выше площадь диска 23.06 нм2, то получим, что при самом плотном заполнении поверхности диска углеводородными хвостами II на ней уместится не более 110 ионов II. И это без учета двух важнейших факторов. Первый состоит в том, что на плоских участках поверхности диска реальная посадочная площадь иона II определяется размером полярной группы и потому больше, чем поперечная площадь углеводородного радикала (это неоспоримо следует из того факта, что II образует сферические мицеллы). Как следствие, и максимальное число ионов II на поверхности диска будет меньше. Второй фактор состоит в том, что если моделировать молекулу I диском, то не всю поверхность диска, а только ее гидрофобную часть (где работает гидрофобный эффект) следует считать доступной для углеводородных хвостов. Этот фактор также приводит к снижению числа агрегации II на диске. Справедливости ради нужно сказать, что имеется и третий фактор, работающий в противоположном направлении по отношению к первым двум. Это сам факт, что мы заменяем гладким диском сложную извилистую структуру с гораздо большей реальной поверхностью. Вопрос о правдоподобности числа агрегации 309 будет решен лишь после кропотливой работы по расчету истинной поверхности молекулы I и, самое главное, ее гидрофобной части.
Из сказанного видно, что обоснование большого числа агрегации II в процессе солюбилизации I не достигнуто, а выдвинутые контрдоводы выглядят весомо. Если бы все-таки число агрегации 309 оказалось артефактом, то можно ли было объяснить это? Легко. Как уже указывалось выше, образование смешанных мицелл I и II происходит независимо от классических мицелл II. Наш метод рассмотрения подразумевает наличие только одного вида мицелл. Фактически же распределение агрегатов по размерам имеет два максимума, т.е. является бимодальным. Наступает равновесие, и обычные “пустые” мицеллы II сосуществуют с “заполненными”. Как же так, скажет читатель, ведь у нас есть большой резервуар молекул I, и если прибавляются пустые мицеллы, они должны заполняться этими молекулами? Дело в том, что при заданных температуре и давлении состояние системы полностью определяется химическими потенциалами. Химический потенциал I, мы знаем, был закреплен с самого начала. Химический потенциал II менялся до ККМ, но выше ККМ меняется настолько слабо, что этим часто пренебрегают. А в трехкомпонентной системе (у нас I–II–вода) при постоянстве двух химических потенциалов третий будет также постоянным в силу термодинамического уравнения Гиббса–Дюгема. Иными словами, выше ККМ состояние системы становится практически постоянным. Любые пропорции (мы это хорошо видели на примере солюбилизационной емкости), в том числе и соотношение пустых и заполненных мицелл, меняться уже почти не могут. Наконец, если вернуться к одномодальному рассмотрению мицелл, то при одновременном наличии пустых и заполненных мицелл среднее число молекул I в одной мицелле будет меньше единицы. А при подстановке в (8) значений z < 1 мы можем получать любые разумные средние значения числа агрегации II. Число же 309 приобретает другой смысл – это среднее соотношение чисел молекул II и I в системе.
Приведенные соображения – лишь один из вариантов объяснения результатов, и пока существование мицеллы II, содержащей молекулу I, с числом агрегации 309 нельзя считать опровергнутой. В поддержку такой точки зрения говорят, например, результаты работы [31], где исследовалась солюбилизация мономеров замещенного фталоцианината цинка (структура II с заменой Mg на Zn при R = SO3H на рис. 1). При использовании в качестве солюбилизатора бромида додецилтриметиламмония (с такой же длиной углеводородной цепи как у II) методом динамического рассеяния света было установлено, что при солюбилизации диаметр мицелл возрастает от 3.12 до 21.04 нм, т.е. примерно в 7 раз!
Числа агрегации и детали кривой распределения мицелл не касаются общих термодинамических характеристик мицеллярной системы с солюбилизатом – коэффициента распределения солюбилизата и стандартного сродства солюбилизации (формулы (1) и (2)). Полученные нами экспериментальные данные позволяют рассчитать эти величины. Начнем с оценки мольных долей и сначала – средней мольной доли I в мицеллах $x_{2}^{\alpha }.$ Наша система состоит из трех компонентов, но если считать, что вода входит в мицеллы только в виде гидратных оболочек ионов II, то число компонентов мицелл сокращается до двух (I и II). Ввиду того, что мольная доля I в мицеллах очень мала, она практически равна солюбилизационной емкости (8), и мы можем оценить $x_{2}^{\alpha }$ примерно как 0.003237. Что касается мольной доли I в растворе (за вычетом мицелл) $x_{2}^{\beta },$ опять же очень малой, то здесь нужно учесть, что концентрация II, хоть и не настолько, но все ж достаточно мала, чтобы ее вкладом в $x_{2}^{\beta }$ можно было пренебречь. Тогда мольную долю можно оценить по растворимости I в воде, которую мы определили как 7.38 мкМ. Делением этого числа на 55.56 М находим $x_{2}^{\beta }$ ≈ 0.13284 × 10–6. По формуле (1) определяем коэффициент распределения как
ЗАКЛЮЧЕНИЕ
Можно условно представить, что процесс солюбилизации состоит в том, что мицелла ПАВ при столкновении с молекулой солюбилизата в растворе поглощает ее подобно тому, как хищная рыба заглатывает встречную живность. Но это лишь конечная схема процесса. В действительности мицелла ПАВ – весьма короткоживущая (1–3 с) и сугубо динамическая (с пребыванием в ней одной молекулы ПАВ до 10–7 с) структура. В сравнении с концентрацией свободных молекул или ионов ПАВ концентрация мицелл вблизи ККМ ничтожно мала, и пока произойдет столкновение солюбилизата с мицеллой случатся мириады его столкновений со свободными частицами ПАВ. Поэтому существующие в растворе структуры типично не трансформируются друг в друга, а формируются заново. На основе проведенного в настоящей работе исследования мы можем представить процесс солюбилизации I в растворе ПАВ следующим образом.
Растворимость I в воде очень мала (по нашим данным 7.38 мкМ при комнатной температуре) по той простой причине, что в молекуле I много органических фрагментов: действует гидрофобный эффект. Он же в растворе “припечатывает” одну плоскую молекулу I к другой, чтобы уменьшить площадь гидрофобного контакта. Поэтому основная форма растворенного в чистой воде I – димеры. Но у каждого димера остается еще много свободной гидрофобной поверхности, а потому, как только появляются ионы ПАВ, они начинают ее заполнять (функцию якорей выполняют прижимаемые водой углеводородные хвосты ионов). Если это происходит в насыщенном растворе I (например, при наличии осадка, как в наших опытах), то химический потенциал I закреплен. В [22] мы обсуждали выражение для химического потенциала солюбилизата. С его номером 2 в наших обозначениях и для малых концентраций I оно имеет вид
где $\mu _{2}^{0}$ – химический потенциал молекулы I в вакууме, ${{w}_{2}}$ – работа ее переноса в растворитель и ${{\Lambda }_{2}}$ – ее средняя длина волны де-Бройля. При заданной температуре величины $\mu _{2}^{0},$ kT и ${{\Lambda }_{2}}$ играют роль постоянных, и для регулирования химического потенциала можно использовать только ${{w}_{2}}$ и ${{c}_{2}}.$ После добавления ПАВ к воде уже не чистая вода, а раствор ПАВ играет роль растворителя для I. При этом работа ${{w}_{2}}$ существенно уменьшается благодаря гидрофобному эффекту. Тогда, согласно (9), для сохранения значения химического потенциала I, которое было в чистой воде, необходимо, чтобы растворимость I ${{c}_{2}}$ увеличилась. Это и есть солюбилизация, и роль солюбилизатора выполняет ПАВ, но не в мицеллярной, а молекулярной или ионной форме.По мере роста концентрации ПАВ, его молекулы все чаще сталкиваются с поверхностью димера I и задерживаются на ее гидрофобной части, которую, в конце концов, полностью покрывают. При взгляде на такую структуру логично спросить: а чем это не мицелла ПАВ с солюбилизированным димером I? Утвердительный ответ очевиден. Однако судьба таких мицелл недолговечна. Мы знаем, что все они заменятся мицеллами с мономерным солюбилизатом. Такие предварительные и недолговечные формы мицелл можно назвать протомицеллами. Что же происходит при дальнейшем увеличении концентрации ПАВ в растворе? Если ККМ не достигнута, то уменьшение числа гидрофобных контактов углеводородных хвостов путем массового образования обычных мицелл еще невозможно. Но есть и другой путь: развалить димеры I на мономеры, увеличив при этом свободную площадь молекул I и предоставив углеводородным хвостам дополнительную “парковку”. Но как развалить димеры? Здесь нет проблем. Как и все вокруг, димеры представляют собой динамические структуры и имеют среднюю продолжительность жизни. Как только димер разрушается тепловым движением и мономеры какое-то время существуют в растворе индивидуально, молекулы или ионы ПАВ спешат с ними связаться. Однако успех этого дела зависит от их концентрации. С ее ростом успех становится гарантированным, и все мицеллы переходят в состояние с солюбилизатом в мономерной форме, причем это может случиться еще до ККМ.
В описанной картине эволюции системы I–II–вода на каждом этапе особую роль играет концентрация ПАВ. Эта роль очевидна и в процессе мономеризации I, а потому вполне естественно ввести (подобно ККМ) такую физико-химическую характеристику как концентрация мономеризации солюбилизата. В наших опытах это примерно или условно была концентрация II 1.7 мМ. Это значение концентрации мономеризации может быть уточнено в последующих исследованиях.
Список литературы
Гольдшлегер Н.Ф., Черняк А.В., Калашникова И.П., Баулин В.Е., Цивадзе А.Ю. // Журн. общей химии. 2012. Т. 82. С. 856.
Гольдшлегер Н.Ф., Лобач А.С., Гак В.Ю., Калашникова И.П., Баулин В.Е., Цивадзе А.Ю. // Физикохимия поверхности и защита материалов. 2014. Т. 50. С. 496.
Гольдшлегер Н.Ф., Черняк А.В., Лобач А.С., Калашникова И.П., Баулин В.Е., Цивадзе А.Ю. // Физикохимия поверхности и защита материалов. 2015. Т. 51. С. 160.
Гольдшлегер Н.Ф., Гак В.Ю., Калашникова И.П., Баулин В.Е., Иванчихина А.В., Смирнов В.А., Ширяев А.А., Цивадзе А.Ю. // Физикохимия поверхности и защита материалов. 2018. Т. 54. № 4. Приложение. С. S3.
Гольдшлегер Н.Ф., Калашникова И.П., Горбунова Ю.Г., Мартынов А.Г., Баулин В.Е., Цивадзе А.Ю. // Физикохимия поверхности и защита материалов. 2018. Т. 54. С. 38.
Гольдшлегер Н.Ф., Гак В.Ю., Лапшина М.А., Баулин В.Е., Ширяев А.А., Цивадзе А.Ю. // Изв. АН. Сер. хим. 2018. С. 2205.
Мовчан Т.Г., Аверин А.А., Баулин Д.В., Плотникова Е.В., Баулин В.Е., Цивадзе А.Ю. // Коллоид. журн. 2018. Т. 80. С. 528.
Мовчан Т.Г., Чернядьев А.Ю., Плотникова Е.В., Аверин А.А., Цивадзе А.Ю., Баулин В.Е. // Коллоид. журн. 2018. Т. 80. С. 694.
Мовчан Т.Г., Чернядьев А.Ю., Плотникова Е.В., Цивадзе А.Ю., Баулин В.Е. // Коллоид. журн. 2019. Т. 81. С. 750.
Мовчан Т.Г., Чернядьев А.Ю., Плотникова Е.В., Цивадзе А.Ю., Баулин В.Е. // Коллоид. журн. 2020. Т. 82. С. 26.
Русанов А.И. Мицеллообразование в растворах поверхностно-активных веществ. СПб.: Химия, 1992.
Rusanov A.I. Micellization in Surfactant Solutions. Chemistry Reviews. Vol’pin, M.E., Ed. V. 22, Part 1. Reading: Harwood Academic Publ., 1996. ISBN 90-5702-297-4.
Русанов А.И., Щёкин А.К. Мицеллообразование в растворах поверхностно-активных веществ, 2-е изд., доп. СПб.: Лань, 2016.
Rusanov A.I. // Langmuir. 2014. V. 30. P. 14 443.
Русанов А.И. // Коллоид. журн. 2016. Т. 78. С. 358.
Русанов А.И. // Коллоид. журн. 2016. Т. 78. С. 613.
Русанов А.И. // Коллоид. журн. 2020. Т. 82. С. 463.
Русанов А.И. // Коллоид. журн. 2020. Т. 82. С. 604.
Shinoda K., Hutchinson E. // J. Phys. Chem. 1962. V. 66. P. 577.
Задымова Н.М., Иванова Н.И. // Коллоид. журн. 2013. Т. 75. С. 179.
Наумова К.А., Дементьева О.В., Зайцева А.В., Рудой В.М. // Коллоид. журн. 2019. Т. 81. С. 478.
Русанов А.И. // Коллоид. журн. 2021. Т. 83. С. 98.
Овсянникова Е.В., Ширяев А.А., Калашникова И.П., Баулин В.Е., Цивадзе А.Ю., Андреев В.Н., Алпатова Н.М. // Макрогетероциклы. 2013. Т. 6. С. 274.
Bıyıklıoğlu Z., Çakır V., Çakır D., Kantekin H. // J. Organomet. Chem. 2014. V. 749. P. 18.
Кушназарова Р.А., Бекмухаметова А.М., Гайнанова Г.А., Миргородская А.Б., Захарова Л.Я. // Структура и динамика молекулярных систем. М.: ИФХЭ РАН. 2016. Вып. 23. С. 307.
Миргородская А.Б., Кушназарова Р.А., Бекмухаметова А.М., Гайнанова Г.А., Захарова Л.Я. // Вестн. технол. ун-та. 2016. Т. 19. С. 8.
Цивадзе А.Ю. // Успехи химии. 2004. Т. 73. С. 6.
Венкатараман К. // Химия синтетических красителей. Т. II. Л.: Госхимиздат, 1957. С. 1279.
Овсянникова Е.В., Гольдшлегер Н.Ф., Курочкина Н.М., Баулин В.Е., Цивадзе А.Ю., Алпатова Н.М. // Макрогетероциклы. 2010. Т. 3. С. 125.
Tanford Ch. The Hydrophobic Effect: Formation of Micelles and Biological Membranes. N.Y.: Wiley, 1973.
Jadhao M., Mukherjee S., Joshi R., Kumar H., Ghosh S.K. // RSC Adv. 2016. V. 6. P. 77 161.
Дополнительные материалы отсутствуют.
Инструменты
Коллоидный журнал