

Коллоидный журнал, 2021, T. 83, № 5, стр. 540-547
“Нерезонансное” усиление оптического поглощения в органических пленках с плазмонными частицами
А. И. Звягина 1, А. А. Ежов 2, Н. В. Кузьмина 1, М. А. Калинина 1, *
1 Институт физической химии и электрохимии им. А. Н. Фрумкина РАН
119071 Москва, Ленинский проспект, 31, корп. 4, Россия
2 Физический факультет Московского государственного университета им. М.В. Ломоносова
119991 Москва, Ленинские горы, ГСП-1, Россия
* E-mail: kalinina@phyche.ac.ru
Поступила в редакцию 24.05.2021
После доработки 26.05.2021
Принята к публикации 28.05.2021
Аннотация
Усиление поглощения видимого света органическими хромофорами, обусловленное их взаимодействием с металлическими наночастицами, представляет большой интерес как способ повышения эффективности фотовольтаических элементов и фотокатализаторов. Поскольку в качестве органического компонента часто используют хромофоры, склонные к ароматическому стекингу, изучение условий усиления оптического поглощения в системах на их основе имеет принципиальное значение. В работе исследованы ультратонкие слоистые системы на основе изопентилдиимида перилен-3,4,9,10-тетракарбоновой кислоты и двумерных плазмонных ансамблей различного строения из наночастиц золота. Показано, что для системы, содержащей одиночные (неагрегированные) наночастицы, наблюдается аддитивное спектральное поведение, несмотря на совпадение спектров поглощения компонентов. В то же время усиление оптического поглощения на 35% реализуется в системе на основе ансамблей агрегатов наночастиц при отсутствии совпадения спектральных характеристик плазмонного и органического компонентов. Полученные результаты открывают новые возможности управления взаимодействием света с ультратонкими пленками красителей-хромофоров, использующихся для создания устройств органической оптики и оптоэлектроники.
ВВЕДЕНИЕ
Плазмонные ансамбли (ПА) на твердых поверхностях представляют собой латерально организованные наноструктуры, состоящие из плазмонных наночастиц, в большинстве случаев, золота и серебра [1–3]. Эти системы являются основой для целого ряда практических приложений, связанных с усилением оптических свойств за счет плазмонного резонанса, таких как усиленные поверхностью спектроскопия комбинационного рассеяния (SERS) [4–6] и инфракрасная спектроскопия [7, 8], флуоресценция и абсорбция, усиленные плазмоном [9–13], и ряда других.
ПА, интегрированные с тонкими органическими пленками, имеют особое значение для развития новых технологий, связанных со сборкой слоистых структур способом “снизу вверх”. Эта стратегия широко применяется в конструировании органических солнечных батарей, фотовольтаических и оптоэлектронных устройств [14–17]. В органических преобразователях солнечного света плазмонные частицы вводятся в слои полимеров или красителей для увеличения эффективности поглощения видимого света [18–23]. Как правило, конструкция этих элементов следует принципу максимально полного перекрывания спектров поглощения органических слоев и плазмонных наночастиц для достижения резонансного усиления поглощения светового излучения [11, 24–26]. В таких системах управление поглощением путем настройки характеристик коллективного плазмона в организованных агрегатах наночастиц [1, 27] используется значительно реже, чем настройка оптических характеристик системы путем варьирования размера или формы частиц [28–32]. Наиболее сложным аспектом использования агрегированных ансамблей частиц для реализации плазмонного усиления оптического поглощения в тонких органических слоях является управление их свойствами с высокой точностью без привлечения дорогостоящих литографических методов. Поскольку количество методов самосборки частиц в ансамбли с управляемой латеральной структурой весьма ограничено, взаимодействие плазмонных частиц с веществом на границах между слоями с различной диэлектрической проницаемостью остается в значительной степени неисследованным. В то же время, знания о взаимодействии поля плазмона, создаваемого организованными протяженными ансамблями наночастиц, с тонкослойными органическими структурами, необходимы для разработки новых подходов к миниатюризации органических преобразователей солнечного света без потери ими эффективности. Влияние таких взаимодействий на оптические характеристики особенно существенно для систем, в которых агрегацию плазмонных частиц при их введении в органические слои невозможно надежно контролировать.
Ранее нами был предложен новый способ управляемой сборки двумерных ПА за счет контролируемой латеральной агрегации наночастиц золота в смешанном монослое ионогенных ПАВ на межфазной границе [33, 34]. Такие ансамбли были использованы для изучения усиленного плазмонным резонансом оптического поглощения (plasmon resonance enhanced absorption, PREA) в ультратонких слоях ряда органических хромофоров [35]. Настройка оптических характеристик ПА осуществляется непосредственно при его сборке в слое ПАВ на поверхности гидрозоля золота, затем ансамбль с фиксированным спектральным положением полосы поглощения плазмона переносят на твердую подложку с предварительно нанесенным слоем органического хромофора. Поскольку положение полосы поглощения коллективного плазмона ансамбля может быть задано с точностью до нескольких нанометров в диапазоне 525–620 нм, метод позволяет обеспечить максимально полное перекрывание спектров поглощения хромофора и ПА для достижения эффекта резонансного усиления.
Было показано, что в органических пленках со сравнительно слабыми взаимодействиями между молекулами хромофоров достигается максимальный эффект усиления оптического поглощения, более чем на 200%, при максимальном перекрытии спектров ПА и слоев органических красителей. В то же время, это эмпирическое правило спектрального перекрывания как условие резонансного усиления оптических свойств, по-видимому, не распространяется на слои J-агрегатов и кристаллитов периленовых и порфириновых красителей. Было установлено, что системы на основе тонких слоев таких агрегатов с сильными межмолекулярными взаимодействиями за счет ароматического стекинга, интегрированных с ПА с соответствующими спектральными характеристиками, проявляют только аддитивное оптическое поведение. Анализ систем со слабым межмолекулярным взаимодействием с помощью методов компьютерного моделирования показал, что поле плазмона индивидуальной частицы, создающее эффект усиления, может распространяться внутри органического слоя только в том случае, если обеспечено ее погружение в органическую пленку. В том случае, когда частица находится на поверхности пленки, поле плазмона распространяется в фазе с меньшим показателем преломления (вакуум или воздух).
Эти данные позволяют выдвинуть гипотезу о том, что для инициирования эффекта плазмонного резонансного усиления оптического поглощения на границе фаз взаимное пространственное расположение компонентов системы не менее важно, чем совпадение их спектральных характеристик. Следовательно, существует вероятность того, что PREA-эффект может проявляться и при минимальном перекрытии спектров поглощения компонентов в таких слоях при условии, что поле плазмона проникает в органический слой. Это условие потенциально может выполняться для агрегированных плазмонных структур, так как известно, что пространственное распределение поля коллективных плазмонов отличается от такового для одиночных частиц. Это различие было выявлено для плазмонных частиц и ансамблей в вакууме и объемных фазах [36, 37], в то время как для межфазных систем оно не было подтверждено ни модельными расчетами, ни экспериментальными данными.
Для экспериментальной проверки этой гипотезы, в данной работе была создана двухслойная система слой хромофора/ПА, с заданным различным положением максимумов оптического поглощения компонентов. Ультратонкая пленка состояла из слоя агрегатов хромофора – изопентилдиимида перилен-3,4,9,10-тетракарбоновой кислоты, интегрированного со стабилизированной ПАВ двумерной сеткой агрегированных плазмонных частиц золота. Оптическое поведение такой структуры было изучено в сравнении с системой на основе тех же компонентов, в которой положения максимумов поглощения слоя хромофора и ПА совпадают.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Использовали изопентилдиимид перилен-3,4,9,10-тетракарбоновой кислоты (EP-PDI, см. рис. 1а), стеариновую кислоту (СК), октадециламин (ОДА), золотохлористоводородную кислоту и цитрат натрия (все – от Sigma-Aldrich). Хлороформ (Химмед, Россия), перегоняли над CaCl2 и фильтровали через Al2O3. Деионизированную воду с сопротивлением 16 MОм получали с помощью системы Водолей. Гидрозоль золота с размером частиц 18 ± 1 нм синтезировали по методу Туркевича [38].
Рис. 1.
Схема получения ультратонкой гибридной системы на основе хромофора и наночастиц золота, в которой реализуется нерезонансное усиление поглощения. (а) Структура EP-PDI. (б) Получение монослоя EP-PDI на межфазной границе воздух/вода. (в) Перенос плотноупакованного монослоя EP-PDI на твердую подложку. (г) Формирование гибридной системы: твердая подложка с осажденным слоем EP-PDI погружается в гидрозоль золота, на поверхность которого наносится смесь ПАВ. После формирования ПА с требуемыми спектральными характеристиками он переносится на подложку.

Для формирования монослоев EP-PDI 530 мкл раствора в хлороформе с концентрацией 1.4 × × 10–4 M наносили на поверхность деионизированной воды площадью 243 см2. Изотермы сжатия монослоев получали с помощью автоматизированных поверхностных весов Ленгмюра (KSV Minitrough, KSV Instrument Ltd.). Очистку барьеров из полиацеталя осуществляли последовательной промывкой этиловым спиртом, дистиллированной и деионизованной водой. Тефлоновую ванну Ленгмюра очищали путем последовательной промывки ацетоном, хлороформом, дистиллированной и деионизованной водой. Платиновую пластинку Вильгельми обрабатывали хлороформом и прокаливали на газовой горелке. Монослои выдерживали 20 мин до полного испарения растворителя и сжимали со скоростью 5 мм/мин при температуре 23 ± 2°C. Монослои переносили на кварцевые подложки при поверхностном давлении 18 мН/м со скоростью 3 мм/мин.
Для получения ПА из наночастиц золота на поверхность гидрозоля золота, налитого в стеклянный стакан, наносили 26 мкл смешанного раствора СК и ОДА в хлороформе (соотношение 1 : 1) с концентрацией 1 × 10–3 M. Нанесенный объем смеси содержал 1.66 × 1016 молекул и соответствовал расчетному количеству вещества, необходимому для формирования на поверхности субфазы в стакане диаметром 6.5 см плотноупакованного монослоя ПАВ (с площадью на молекулу 20 Å2). Формирование структуры плазмонной сетки отслеживали с помощью оптоволоконной спектроскопии поглощения/отражения. Систему оставляли на 36 или 60 мин до достижения необходимых спектральных характеристик и переносили на кварцевую или слюдяную подложку, извлекаемую вертикально из субфазы со скоростью 2 мм/мин.
Для стабилизации ПА с максимумом экстинкции при длине волны <550 нм, состоящего из отдельных наночастиц Au, температуру системы снижали до 12 ± 2°C. Для этого стакан помещали в ледяную баню, в результате чего органический слой переходил в твердое состояние и фиксировал положение наночастиц золота.
Формирование ПА на поверхности гидрозоля золота отслеживали с помощью оптоволоконного UV-vis спектрометра AvaSpec-2048 (Нидерланды), регистрируя изменения во времени интенсивности полос поглощения в спектре смешанного монослоя ПАВ. Спектры записывали каждые 2 мин, начиная с момента нанесения раствора ПАВ на поверхность субфазы.
Электронные спектры поглощения раствора EP-PDI (в кювете с длиной оптического пути 10 мм) и кварцевых подложек со сформированными монослоем EP-PDI и гибридными системами EP-PDI/ПА различного строения регистрировали с помощью спектрофотометра Shimadzu UV 2450 PC в диапазоне длин волн 190−800 нм.
Морфологию монослоев EP-PDI и гибридных систем EP-PDI/ПА исследовали в атомно-силовом микроскопе (АСМ) Nanoscope V (Veeco Instruments). Изображения были получены на воздухе в полуконтактном режиме с использованием высокоразрешающих кремниевых кантилеверов NSG01 (Tipsnano) с радиусом кривизны острия <6 нм Обработку изображений проводили с помощью программного обеспечения Gwyddion.
РУЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Для получения покрытий на основе EP-PDI на твердой поверхности раствор хромофора в хлороформе предварительно наносили на водную субфазу для формирования монослоев Ленгмюра, которые сжимали до образования максимально плотного слоя (рис. 1б).
Плотность упаковки молекул в монослое EP-PDI определяли по изотерме сжатия (рис. 2а). Оптические свойства монослоя на поверхности воды регистрировали с помощью оптоволоконной спектроскопии поглощения (рис. 2б). Сравнение спектров поглощения, записанных в процессе сжатия монослоя EP-PDI, со спектром поглощения его раствора в хлороформе (рис. 2в) показывает, что EP-PDI образует агрегаты сразу после нанесения раствора на поверхность воды. Это связано с высокой гидрофобностью хромофора и его склонностью к ароматическому стекингу. В спектрах поглощения монослоя EP-PDI присутствуют две полосы с максимумами вблизи 500 и 548 нм, что соответствуют спектру J-агрегатов EP-PDI (рис. 2б) [39].
Рис. 2.
(а) Изотерма сжатия монослоя EP-PDI на поверхности воды (скорость сжатия 5 Å2/(молекула мин). (б) Электронные спектры поглощения монослоя EP-PDI, записанные в процессе сжатия в диапазоне значений поверхностного давления Π = = 0‒18 мН/м с интервалом 3 мН/м, (в) Электронный спектр поглощения раствора EP-PDI в хлороформе с концентрацией 1 × 10–4 M

После сжатия монослой, состоящий из агрегатов EP-PDI, переносили на кварцевую или слюдяную подложку (рис. 1в). Спектр поглощения полученного покрытия совпадает со спектрами поглощения монослоя на поверхности воды. Это свидетельствует об отсутствии структурных изменений, вызванных межмолекулярными взаимодействиями, в процессе переноса монослоя с межфазной границы на твердую подложку. По данным АСМ монослои EP-PDI состоят из агрегатов с латеральным размером около 1 мкм и высотой от 10 до 20 нм, образующих разреженную упаковку (рис. 3).
Рис. 3.
АСМ-изображение и соответствующий профиль поверхности слоя EP-PDI, перенесенного с водной субфазы при Π = 18 мН/м на поверхность слюды.

Чтобы интегрировать слой EP-PDI с ПА из наночастиц золота размером 18 ± 1 нм в единую систему, ПА с заданными спектральными свойствами переносили с межфазной границы воздух/вода на твердую подложку, предварительно покрытую монослоем агрегатов EP-PDI (рис. 1г). Для сравнительного анализа были сформированы два типа таких гибридных систем, различающихся упаковкой наночастиц Au в ПА. В системе первого типа ПА представляет собой монослой, в основном, одиночных (неагрегированных) наночастиц с небольшим количеством димеров. В системе второго типа наночастицы в ПА образуют протяженные цепочки и разветвленные агрегаты, состоящие, в основном, из 10 и более частиц. На рис. 4 представлены спектры экстинкции ПА на поверхности гидрозоля золота, записанные с момента нанесения ПАВ до переноса на подложку.
Рис. 4.
Зависимость спектра экстинкции ПА на поверхности гидрозоля золота от времени; спектры записывали с интервалом 2 мин, общее время формирования ансамбля 36 (а) и 60 мин (б).
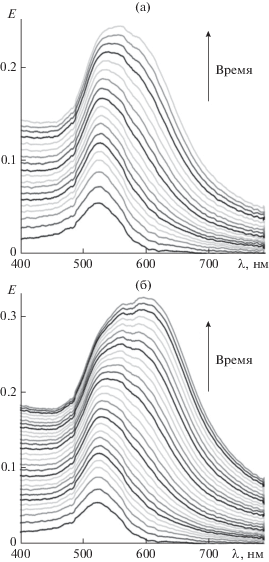
Спектры на рис. 4а соответствуют ПА, состоящему из отдельных наночастиц золота, равномерно распределенных по поверхности (см. АСМ-изображение на рис. 5а). Такая структура ПА определяет положение максимума экстинкции, точно совпадающее с положением характеристической полосы 548 нм для агрегатов EP-PDI (рис. 5в, кривые 1 и 2). Второй системе соответствуют спектры на рис. 4б. Постепенная агрегация наночастиц в цепочки (рис. 5б) приводит к сдвигу максимума экстинкции плазмонной структуры на 40 нм в длинноволновую область (до 590 нм) относительно максимума полосы поглощения слоя EP-PDI (рис. 5г, кривые 1 и 2).
Рис. 5.
(а) и (б) АСМ-изображения гибридных систем EP-PDI/ПА с максимумами экстинкции при 550 и 590 нм соответственно; (в) и (г) электронные спектры поглощения: 1 – монослоя EP-PDI, 2 – ПА из наночастиц золота с максимумом экстинкции при 550 (в) и 590 нм (г), 3 – гибридной структуры EP-PDI/ПА, 4 – сумма спектров 1 и 2.
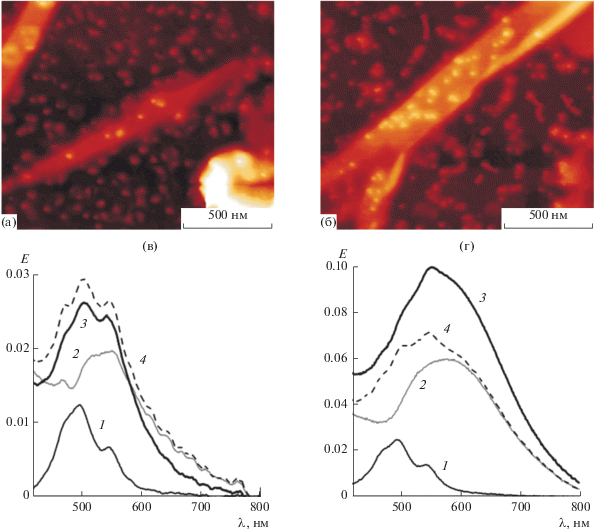
В данной работе для стабилизации ПА из отдельных наночастиц Au, равномерно распределенных в органическом слое ПАВ, с максимумом экстинкции при длине волны <550 нм был применен метод охлаждения вместо использованного ранее метода введения в гидрозоль золота наночастиц оксида церия размером 3 нм, которые выполняли функцию матрицы, стабилизирующей ПА [33]. При достижении необходимых спектральных характеристик ПА температуру системы понижали до 12 ± 2°C. Этот метод позволяет избежать влияния каких-либо дополнительных факторов, помимо упаковки наночастиц в ПА, на распространение поля плазмона в органическом слое.
Как следует из рис. 5в, система, в которой спектры органического слоя и ПА совпадают, демонстрирует аддитивное оптическое поведение компонентов, поскольку во всем исследуемом диапазоне длин волн не происходит усиления поглощения. Согласно данным АСМ, наночастицы золота в такой системе иммобилизованы как на поверхности кристаллов EP-PDI, так и на свободной от агрегатов хромофора поверхности подложки. Тем не менее, в такой системе прямой контакт между наночастицами золота и органическим материалом не приводит к возникновению резонансного усиления поглощения. Этот результат подтверждает сделанный нами ранее вывод о том, что поле плазмона одиночной металлической наночастицы, находящейся на межфазной границе воздух/органический слой, не проникает в агрегированную органическую фазу, если частица не погружена в органический слой [35].
В то же время, в случае гибридной системы, в которой максимумы экстинкции компонентов находятся далеко друг от друга, спектральная картина кардинально отличается от наблюдаемой для системы с одиночными наночастицами (рис. 5г). Система на основе агрегатов EP-PDI и ПА из цепочек наночастиц Au демонстрирует усиление экстинкции в диапазоне от 520 до 650 нм. Коэффициент усиления, рассчитанный как интегральное отношение экспериментального спектра (кривая 3) к модельному (кривая 4), составил 1.35. АСМ-изображение структуры ПА, перенесенного на поверхность слоя EP-PDI, представлена на рис. 5б. Видно, что наночастицы золота, находящиеся в контакте с хромофором, упакованы значительно плотнее, чем в описанной выше системе с совпадающим положением максимумов экстинкции компонентов. В то же время, увеличением числа наночастиц, контактирующих с хромофором, нельзя объяснить неаддитивное усиление поглощения в системе с несовпадающими максимумами поглощения компонентов. Наблюдаемое усиление оптического поглощения в такой системе подтверждает предположение о зависимости эффекта усиления оптического поглощения от взаимодействия поля плазмона с органическим веществом на границе фаз и указывает на то, что агрегированная органическая фаза является проницаемой для электрического поля коллективного плазмона, генерируемого непрерывной протяженной двумерной сеткой наночастиц золота.
ВЫВОДЫ
В работе на примере наночастиц золота и производного перилена экспериментально продемонстрированы условия возникновения эффекта усиления поглощения видимого света в ультратонких гибридных системах на основе металлических наночастиц и органического красителя, образующего J-агрегаты за счет ароматического стекинга. Установлено, что для усиления поглощения необходимо не только перекрывание спектров поглощения компонентов, но и создание такой упаковки наночастиц в ПА, которая обеспечивает проникновение поля плазмона в слой агрегированных органических молекул. Полученные результаты позволяют сделать вывод о том, что в планарной системе, состоящей из двумерного ПА, иммобилизованного на поверхности слоя агрегатов производных перилена, проникновение поля коллективного плазмона в слой возможно только в случае образования наночастицами протяженных агрегатов. Понимание закономерностей возникновения усиленных полем плазмона эффектов в гибридных системах на основе органических красителей и металлических наночастиц открывает возможность для управления оптическими свойствами таких ультратонких гибридных систем путем регулирования упаковки ПА.
Список литературы
Halas N.J., Lal S., Chang W.S., Link S., Nordlander P. // Chem. Rev. 2011. V. 111. P. 3913.
Klinkova A., Choueiri R.M., Kumacheva E. // Chem. Soc. Rev. 2014. V. 43. P. 3976.
Amendola V., Pilot R., Frasconi M., Maragò O.M., Iatì M.A. // J. Phys. Condens. Matter. 2017. V. 29. 203002.
Bosnick K., Maillard M., Brus L. // J. Phys. Chem. B. 2003. V. 107. P. 9964.
Futamata M. // Faraday Discuss. 2006. V. 132. P. 45.
Le Ru E.C., Etchegoin P.G., Meyer M. // J. Chem. Phys. 2006. V. 125. P. 204701.
Osawa M. // Bull. Chem. Soc. Jpn. 1997. V. 70. P. 2861.
Errano S., Adrid M., Pain S. Surface-Enhanced Vibrational Spectroscopy. Chichester: Wiley, 2007.
Chen Y., Munechika K., Ginger D.S. // Nano Lett. 2007. V. 7. P. 690.
El-Bashir S.M., Barakat F.M., AlSalhi M.S. // J. Lumin. 2013. V. 143. P. 43.
Lieberman I., Shemer G., Fried T., Kosower E.M., Markovich G. // Angew. Chemie - Int. Ed. 2008. V. 47. P. 4855.
Schaadt D.M., Feng B., Yu E.T. // Appl. Phys. Lett. 2005. V. 86. 063106.
Zasedatelev A.V., Dubinina T.V., Krichevsky D.M., Krasovskii V.I., Gak V.Y., Pushkarev V.E., Tomilova L.G., Chistyakov A.A. // J. Phys. Chem. C. 2016. V. 120. P. 1816.
Mandal P., Sharma S. // Renew. Sustain. Energy Rev. 2016. V. 65. P. 537.
Jang Y.H., Jang Y.J., Kim S., Quan L.N., Chung K., Kim D.H. // Chem. Rev. 2016. V. 116. P. 14982.
Lim E.L., Yap C.C., Teridi M.A.M., Teh C.H., Yusoff M.A.R., Jumali M.H.H. // Org. Electron. 2016. V. 36. № September. P. 12.
Hou W., Cronin S.B. // Adv. Funct. Mater. 2013. V. 23. P. 1612.
Chen X., Yang X., Fu W., Xu M., Chen H. // Mater. Sci. Eng. B. 2013. V. 178. P. 53.
Standridge S.D., Schatz G.C., Hupp J.T. // J. Am. Chem. Soc. 2009. V. 131. P. 8407.
Xu Q., Liu F., Liu Y., Cui K., Feng X., Zhang W., Huang Y. // Sci. Rep. 2013. V. 3. P. 2112.
Wang C.C.D., Choy W.C.H., Duan C., Fung D.D.S., Sha W.E.I., Xie F.-X., Huang F., Cao Y. // J. Mater. Chem. 2012. V. 22. P. 1206.
Xue M., Li L., Tremolet De Villers B.J., Shen H., Zhu J., Yu Z., Stieg A.Z., Pei Q., Schwartz B.J., Wang K.L. // Appl. Phys. Lett. 2011. V. 98. 253302.
Yakimov A., Forrest S.R. // Appl. Phys. Lett. 2002. V. 80. P. 1667.
Xu Q., Liu F., Meng W., Huang Y. // SPIE Micro + Nano Materials, Devices, and Applications. 2013. V. 8923. 892334.
Rai V.N., Srivastava A.K., Mukherjee C., Deb S.K. // Appl. Opt. 2012. V. 51. P. 2606.
Atwater H.A., Polman A. // Nat. Mater. 2010. V. 9. P. 865.
Daniel M., Astruc D. // Chem. Rev. 2004. V. 104. P. 293.
Lee K.S., El-Sayed M.A. // J. Phys. Chem. B. 2005. V. 109. P. 20331.
Huang X., El-Sayed I.H., Qian W., El-Sayed M.A. // J. Am. Chem. Soc. 2006. V. 128. P. 2115.
Link S., El-Sayed M.A. // J. Phys. Chem. B. 1999. V. 103. P. 4212.
Niu W., Chua Y.A.A., Zhang W., Huang H., Lu X. // J. Am. Chem. Soc. 2015. V. 137. P. 10460.
Kelly K.L., Coronado E., Zhao L.L., Schatz G.C. // J. Phys. Chem. B. 2003. V. 107. P. 668.
Zvyagina A.I., Ezhov A.A., Ivanov V.K., Arslanov V.V., Kalinina M.A. // J. Mater. Chem. C. 2015. V. 3. P. 11801.
Babenko D.I., Ezhov A.A., Turygin D.S., Ivanov V.K., Arslanov V. V., Kalinina M.A. // Langmuir. 2012. V. 28. P. 125.
Zvyagina A.I., Ezhov A.A., Meshkov I.N., Ivanov V.K., Birin K.P., König B., Gorbunova Y.G., Tsivadze A.Y., Arslanov V. V., Kalinina M.A. // J. Mater. Chem. C. 2018. V. 6. P. 1413.
Pal S.K., Chatterjee H., Ghosh S.K. // RSC Adv. 2019. V. 9. P. 42145.
Satya Bharati M.S., Lakshmi P., Byram C., Rao S.V. // 2019 Work. Recent Adv. Photonics (WRAP). 2019. P. 1.
Turkevich J. Cooper P.H.J. // Discuss. Faraday Soc. 1951. V. 55. P. 55.
Würthner F., Thalacker C., Diele S., Tschierske C. // Chemistry. 2001. V. 7. P. 2245.
Дополнительные материалы отсутствуют.
Инструменты
Коллоидный журнал