

Коллоидный журнал, 2021, T. 83, № 6, стр. 702-714
Термические превращения в механохимически активированных системах MeOx/C (Me = Mo, Mn, Bi, V)
А. Н. Стрелецкий 1, *, Г. А. Воробьева 1, И. В. Колбанев 1, А. Б. Борунова 1, А. В. Леонов 2
1 Федеральный исследовательский центр химической физики им. Н.Н. Семенова РАН
119991 Москва, ул. Косыгина, 4, Россия
2 Московский государственный университет им. М.В. Ломоносова, химический факультет
119992 Москва, Ленинские горы, 1, стр. 3, Россия
* E-mail: str1945@yandex.ru
Поступила в редакцию 06.08.2021
После доработки 10.08.2021
Принята к публикации 13.08.2021
Аннотация
Методами термогравиметрии и калориметрии в сочетании с масс-спектроскопией, а также с помощью рентгеновской дифракции исследованы термические превращения в механически активированных системах MoO3/C, MnO2/C, Bi2O3/C и V2O5/C, перспективных в качестве материалов для электродов суперконденсаторов и ионных батарей. Установлено, что кристаллическая структура активированных высокодисперсных нанокомпозитов устойчива до 250–350°C в зависимости от природы оксида. Проанализирована реакционная способность образующегося “слабосвязанного” кислорода в различных оксидах при МА при восстановлении углеродом. В случае МоО3 и MnO2 существенное снижение температуры начала восстановления оксидов углеродом обусловлено более низкими температурами разложения оксидов с выделением кислорода в результате активации. Сопоставлено взаимодействие механически активируемых оксидов с углеродом и с алюминием и CO. Оказалось, что присутствие углерода понижает температуру восстановления MnO2, практически не влияет на скорость реакции в случае МоO3 и Bi2O3 и повышает температуру превращения V2O5.
1. ВВЕДЕНИЕ
Хорошо известно, что механическая активация (МА) двухкомпонентной смеси сопровождается возникновением самых разнообразных дефектов в компонентах, в том числе наносостояний и наночастиц, появлением реакционноспособных центров, а также перемешиванием компонентов и гомогенизацией смеси (см., например, [1–3]). Одним из следствий МА является существенное ускорение химических превращений при последующем прогреве активированных композиций.
Настоящая статья посвящена анализу термических превращений в механически активированных системах MeOx/C (Me = Mo, Mn, Bi, V). Интерес к МА и термическим превращениям в этих системах после активации обусловлен различными причинами. Значительное внимание уделено возможностям механохимического приготовления тугоплавких карбидов металлов. Наиболее широко используемый способ – это МА смесей оксидов металлов с углеродом и последующий их прогрев [1–3]. Таким способом, в частности, приготовлены наноразмерные карбиды молибдена [4–7] и ванадия [8, 9]. Применение МА позволяет существенно снизить температуру синтеза и является экономически целесообразной. Другой вариант этого метода заключается в добавлении к смеси MeOx/C Al [10], Mg [11, 12], Ca [13] или Zn [14], окисление которых кислородом из MeOx термодинамически очень выгодно, и совместный помол трехкомпонентной смеси. В результате температура синтеза карбидов MeCx резко понижается, и достаточно часто удается механохимически синтезировать карбиды при формально комнатной температуре. Авторы объясняют этот эффект развитием при помоле самораспространяющегося высокотемпературного синтеза, инициируемого окислением введенных дополнительно металлов.
Для выяснения физических причин ускорения реакций оксидов с углеродом при МА целесообразно выяснить природу и реакционную способность дефектов, которые возникают при МА индивидуальных оксидов, а также сопоставить взаимодействие активированных оксидов с другими реагентами. Дефектная структура MoO3, V2O5 и Bi2O3, подвергнутых МА, детально исследована [15–17]. Проанализированы закономерности восстановления как исходных оксидов, так и активированных в газовой среде на примере систем МоО3/Н2 [18–21] и V2O5/СО [22, 23]. В [24] показано, что МА приводит к приобретению MnOx высокой каталитической активности в процессах гидрирования CO2. Проведен большой цикл работ (см., например, обзоры [25–27] и работы [28–30]) по созданию механически активированных энергонасыщенных композитов с использованием систем, состоящих из двух твердых компонентов (Al, Mg + MeOx). Оказалось, что такие композиты обладают рядом преимуществ (например, более высокой скоростью горения) по сравнению с энергонасыщенными композитами, приготовленными традиционными способами [28]. Накопленные данные позволили высказать предположение [27, 31], что важнейшими причинами ускорения химических превращений при МА являются образование в оксидах реакционноспособного “слабосвязанного” кислорода и формирование большой поверхности контакта компонентов. Одной из целей настоящей работы является проверка этого предположения для термических реакций в механически активированных системах MeOx/C (Me = Mo, Mn, Bi, V), а также сравнение реакционной способности активированных оксидов с углеродом и алюминием.
Еще одна причина интереса к системам MeOx/C обусловлена тем, что многие из них являются перспективными материалами для электродов суперконденсаторов (см. например, обзоры [32, 33] и работы, посвященные системам МоО3/C [34, 35] и V2O5/C [36]). Одним из способов приготовления этих систем является МА. Можно указать на обзор [37] и работы по использованию МА для приготовления материалов электродов из MoO3/C [38], V2O5/C [39, 40] и MnO2/C [41, 42]. Отметим, что в предыдущей статье [43] нами были исследованы возможности МА систем MeOx/C (Me = Mo, Mn, V, Bi, Sb, Tb, Cd, Pb; C = графит) для приготовления материалов электродов суперконденсаторов. В контексте этого направления предполагается проверить термическую стабильность механически активированных композитов MeOx/C.
2. ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Список исследованных систем, их состав, весовое содержание графита в смеси, продолжительность t и доза D механической активации, а также величина удельной поверхности S приведены в табл. 1. Образцы 1–3 и 6–10 из этой таблицы – те же, что были приготовлены в работе [43]. МА этих смесей проводили в вибрационной мельнице SPEX-8000 на воздухе. Результаты расчета дозы активации и данные по удельной поверхности также приведены в [43]. Исходный MnO2 (образец 4 в табл. 1) активировали на воздухе в гораздо более мощной мельнице конструкции Аронова. После активации он имел удельную поверхность 31 м2/г (образец 5 в табл. 1).
Таблица 1.
Список и стехиометрия исследованных систем. [C] – массовое содержание графита в смеси, t – продолжительность МА, D – доза МА, S – удельная поверхность композита, T(ДСК) – значения максимальной температуры прогрева в ДСК-эксперименте
| № | Система | [C], мас. % | t, мин | D, кДж/г | S, м2/г | T(ДСК), °C |
|---|---|---|---|---|---|---|
| 1 | MoO3/5С | 28 | 0 | 0 | 800 | |
| 2 | 30 | 1.0 | 31.9 | 350, 500, 600, 800 | ||
| 3 | 180 | 6.0 | 51.6 | 350, 500, 600, 800, 850 | ||
| 4 | MnO2 | 0 | 0 | 0 | 0.5 | 1000 |
| 5 | MnO2 | 0 | 40 | 7.2 | 31 | 300, 600, 1000 |
| 6 | 5MnO2/C | 2.6 | 180 | 6.3 | 12.2 | 550 |
| 7 | MnO2/5С | 41 | 180 | 6.3 | 97 | 550 |
| 8 | Bi2O3/5С | 11 | 180 | 4.8 | 29.3 | 260, 450, 700, 700 |
| 9 | V2O5/С | 6 | 90 | 2.8 | 10.9 | 300, 520, 650 |
| 10 | V2O5/5С | 24 | 90 | 2.8 | 23.3 | 300, 520, 650 |
Для всех образцов, представленных в табл. 1, проведены измерения методами дифференциальной сканирующей калориметрии (ДСК) и термогравиметрии (ТГ) с масс-спектральным анализом их состава. Термогравиметрические исследования проводились на приборе синхронного термического анализа STA 449C (NETZSCH, Германия), сопряженного с масс-спектрометром Aeolos-32 в среде He высокой чистоты марки “55” (99.9995%) при различных значениях температуры вплоть до 1000°С. В последнем столбце табл. 1 для каждого из образцов приведены значения максимальной температуры для каждого прогрева в ДСК-ячейке.
После каждого прогрева проводили дифракционные измерения. Фазовый состав образцов определяли с помощью рентгенофазового анализа на дифрактометре Дрон-3М. Количественный фазовый состав устанавливали с помощью программы Phan% [44], величины областей когерентного рассеяния и микроискажений рассчитывали (с учетом геометрической ширины) по программе Profile [44].
3. ЭКСПЕРИМЕНТАЛЬНЫЕ ДАННЫЕ
3.1. Система MoO3/5C
Для системы MoO3/C были исследованы три образца состава MoO3/5C (28 мас. % C) с разной продолжительностью МА (№№ 1–3 в табл. 1). Результаты ДСК, ТГ и масс-спектрального анализа этих образцов суммированы на рис. 1 и 3, а дифракционные измерений до и после их прогрева – на рис. 2 и 4 и в табл. 2.
Рис. 1.
Уменьшение массы (1a, 2a, 3a) и выделение CO2 (1b, 2b, 3b) при прогреве исходной (1a, 1b) и активированных 30 (2a, 2b) и 180 мин (3a, 3b) смесей MoO3/5C в He до 800°C.

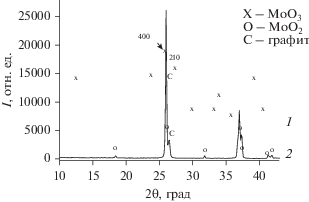
Рис. 3.
(а) Уменьшение массы (1), тепловые эффекты (2) и ионные токи, соответствующие значениям m/z = 28, 44, 18 и 32, при прогреве активированной 180 мин смеси MoO3/5C в He до 850°C. (б) Фрагмент рис. 3а в температурном диапазоне 740–850°C. Обозначения те же.

Рис. 4.
(а) Дифрактограммы смеси МоО3/5С после 30 мин МА (1) и прогрева до 350 (2), 500 (3), 600 (4) и 800°C (5) в ДСК-ячейке. (б) Дифрактограммы такой же смеси после 180 мин МА (1) и прогрева до 350 (2), 500 (3), 800 (4) и 850°C (5).

Рис. 5.
(а) Изменение массы (1),тепловые эффекты (2) и ионные токи, соответствующие значениям m/z = 32 и 44 (выделению O2 и CO2), при прогреве в He образца активированного 40 мин MnO2. (б) Дифрактограммы MnO2 после 40 мин МА (1) и прогрева до 300 (2), 600 (3) и 1000°C (4).

Таблица 2.
Количественный фазовый состав и изменение массы ΔM/M системы МоО3/5С до и после ее помола и прогрева до разных значений температуры T
| Образец | T, °C | Фазовый состав, мас. % | ΔM/M, % | ||||
|---|---|---|---|---|---|---|---|
| MoO3 | C | Mo4O11 | MoO2 | Mo2C | |||
| Исходный (№ 1) | 25 | 72 | 28 | ||||
| 800 | 33.4 | 66.6 | –11.8 | ||||
| После 30 мин МА (№ 2) |
25 | 63 | 37 | ||||
| 350 | 63 | 37 | –0.93 | ||||
| 500 | 42.7 | 39.6 | 12.0 | 5.7 | –2.18 | ||
| 600 | + | 57.1 | 42.9 | –6.2 | |||
| 800 | 21.4 | 78.6 | –12.15 | ||||
| После 180 мин МА (№ 3) |
25 | + | Следы | ||||
| 350 | + | + | –1.38 | ||||
| 500 | 55.9 | 44.1 | –5.11 | ||||
| 800 | 57.4 | 42.6 | –12.84 | ||||
| 850 | 17.9 | 3.6 | 78.5 | –38.59 | |||
Рис. 6.
(а) Тепловые эффекты (1), изменение массы (2) и ионные токи, соответствующие значениям m/z = 32 и 44 (выделению O2 и CO2), при прогреве в He образца 5MnO2/C, активированного в течение 180 мин. (б) Дифрактограммы активированных образцов 5MnO2/C (1) и MnO2/5C (2), прогретых до 550°C.

На рис. 1 приведены данные об изменении массы и кинетике выделения CO2 при прогреве в ДСК-ячейке всех трех образцов МоО3/5С. Прогрев неактивированной смеси сопровождается уменьшение массы (кривая 1а) с выделением CO2 (кривая 1б) в температурном диапазоне 450–760°C. Образование CO2 при восстановлении оксида сопровождается слабо выраженным экзотермическим эффектом (кривая не приведена).
На рис. 2 сопоставлены дифрактограммы этой смеси до и после прогрева в инертной среде. Для исходного образца до прогрева зафиксированы обе фазы, МоО3 и C (рис. 2, кривая 1 и табл. 2), причем исходный образец МоО3 текстурирован. Для изотропного образца МоО3 соотношение интенсивностей линий 400 и 210 составляет I(400)/I(210) = 0.45. В случае исходного МоО3 интенсивность линии 400 аномально высока и отношение I(400)/I(210) = 1.2. После прогрева до 800°C обнаружены только фазы MoO2 и графита. Таким образом, при прогреве происходит брутто-реакция
(1)
$\begin{gathered} {\text{Mo}}{{{\text{O}}}_{3}} + {\text{C}} \to {\text{Mo}}{{{\text{O}}}_{2}} + {\text{C}}{{{\text{O}}}_{2}} \\ (\Delta G_{{298}}^{^\circ } = - 123.1\,\,{{{\text{кДж}}} \mathord{\left/ {\vphantom {{{\text{кДж}}} {{\text{моль}}}}} \right. \kern-0em} {{\text{моль}}}}\,\,[7]). \\ \end{gathered} $Следовательно, можно полагать, что уменьшение массы образца обусловлено расходованием части графита и кислорода оксида на образование CO2.
Как следует из рис. 1, МА приводит к снижению температуры восстановления оксида при прогреве образцов. Кривая 2а резкого уменьшения массы после 30 мин активации смещена в низкотемпературную область на 80°C, а кривая 3а (180 мин активации) – на 170°C относительно неактивированного образца (кривая 1а). Для обоих активированных образцов МоО3 выделение CO2 начинается при 100°C. Более интенсивное выделение CO2 (кривая 2b) начинается при 420°C для образца после 30 мин активации и при 350°C для образца после 180 мин активации (кривая 3b). Выделение углекислого газа завершается при 700 и 650°C соответственно для образцов 2 и 3 табл. 1.
Для образца МоО3/5С после 180 мин активации наблюдается резкое дополнительное уменьшение массы при температуре 770°C (кривая 3а на рис. 1). Для более четкого выявления второй стадии восстановления в следующем опыте такой же образец прогрели до 850°C (рис. 3). На рис. 3а, помимо данных о потере массы и ионном токе, отвечающем CO2 (m/z = 44), приведены также калориметрические данные и величины ионных токов, соответствующих воде (m/z = 18), CO и N2 (m/z = 28) и кислороду (m/z = 32). На рис. 3б высокотемпературная часть этого опыта показана в увеличенном масштабе. Отчетливо видно, что вторая, высокотемпературная стадия потери массы сопровождается интенсивным эндоэффектом.
Анализ масс-спектральных данных подтверждает, что первая стадия восстановления оксида молибдена сопровождается выделением CO2 со следами CO, а на второй стадии основным выделяющимся газом является CO с небольшой примесью CO2. Действительно, как видно на рис. 3а, при температуре около 550°C интенсивность тока, обусловленного ионом ${\text{CO}}_{2}^{ + }$ (m/z = 44), возрастает примерно на порядок величины одновременно с уменьшением массы образца. Фоновое значение тока ионов с m/z = 28 обусловлено следами азота в остаточных газах. Небольшое увеличение этого ионного тока, имеющее место одновременно с ростом с пика, отвечающего m/z = 44, обусловлено ионом CO+ – осколком CO2. Соотношение интенсивностей ионных токов CO+ и ${\text{CO}}_{2}^{ + },$ как следует из рис. 3а, равно примерно 0.15, т.е. чуть выше табличного значения 0.11. Ионный ток, обусловленный остаточным кислородом (m/z = 32), в этом температурном диапазоне не изменяется. При высоких температурах (рис. 3б) наблюдается другая картина. Прирост ионного тока, отвечающего m/z = 28, примерно на порядок больше прироста тока ионов с m/z = 44. Это однозначно указывает на то, что основным газом, выделяющимся на второй стадии, является CO. В то же время некоторое небольшое количество CO2 также выделяется из образца. Следует отметить, что в начале второй стадии резко падает давление кислорода, т.е. можно полагать, что на начальном этапе этой стадии процесса кислород из газовой фазы также принимает участие в окислении углерода.
Для более детального исследования отдельных стадий процесса восстановления МоО3 углеродом был проведен прогрев его образцов до ряда промежуточных значений температуры, которые были выбраны исходя из данных о потере массы, и для всех образцов они приведены в табл. 1. После каждого прогрева анализировали фазовый состав образцов методом рентгеновской дифракции.
Дифрактограммы обоих активированных образцов МоО3/5С до и после их прогрева в ДСК-ячейке до разной температуры приведены на рис. 4, а результаты количественного фазового анализа сопоставлены в табл. 2. В этой таблице также суммированы данные о потере образами массы (ΔM/М) при прогреве.
После МА смеси МоО3/5С текстура МоО3 исчезает, уширяются линии оксида и графита, новые линии не появляются. После 30 мин активации видны обе фазы – МоО3 и C (кривая 1 на рис. 4а), соотношение фаз близко к исходной стехиометрии (табл. 2), I(400)/I(210) = 0.6, удельная поверхность S = 32 м2/г (табл. 1). После 180 мин активации (кривая 1 на рис. 4б) линия графита почти исчезла, остался только МоО3, соотношение интенсивностей I(400)/I(210) = 0.48, т.е. близко к значению для изотропного оксида, S = = 52 м2/г.
Как следует из рис. 4 и табл. 2, после прогрева до 350°C в обоих активированных образцах МоО3/5С присутствуют только обе исходные фазы. В образце № 2 (30 мин активации) после прогрева до 500°C наряду с MoO3 рентгеновская дифракция обнаруживает продукты восстановления оксида – промежуточную фазу Mo4O11 и фазу MoO2. Прогрев этого образца до 600°C приводит к исчезновению фазы MoO3, и в нем содержится около 60 мас. % фазы Mo4O11 и 40 мас. % MoO2. Наконец, при прогреве до 800°C промежуточная фаза Mo4O11 полностью восстанавливается до MoO2. Таким образом, из анализа дифракционных данных следует, что первая стадия восстановления протекает по реакции (1) через образование промежуточного продукта Мо4О11, при этом до 300°C фазовый состав не изменяется, а конечный продукт MoO2 начинает возникать при 500°C. В образце МоО3/5С, подвергнутом активации в течение 30 мин, восстановление оксида молибдена до MoO2 завершается к 800°C.
Сопоставим дифракционные данные с результатами ТГ-анализа. В расчете на 100 г в смеси имеются 72 г MoO3 + 28 г C, т.е. 48 г Mo + 24 г O + + 28 г C. В реакции (1) CO2 в смеси образуется из кислорода оксида и углерода. На это расходуется 1/3 кислорода (24 г × 1/3 = 8 г) и столько углерода, сколько надо для образования CO2 (8 × 12)/32 = 3 г. Таким образом, общие потери массы кислорода и углерода должны составлять 11 г на 100 г смеси, т.е. 11%. Эта цифра достаточно близка к экспериментальным данным о потерях массы, приведенным в табл. 2.
Для образца № 3 с большей дозой активации (180 мин) первая стадия протекает несколько быстрее, чем для образца № 2. В результате не удалось обнаружить промежуточный продукт Mo4O11, а при прогреве до 800°C зарегистрировано появление Mo2C. При 850°C фаза Mo2C становится доминирующей.
Таким образом, для образцов с большой дозой активации при 780–810°C начинается вторая стадия восстановления по брутто-реакции
(2)
${\text{2Mo}}{{{\text{O}}}_{2}} + 5{\kern 1pt} {\text{C}} \to {\text{M}}{{{\text{o}}}_{{\text{2}}}}{\text{C}} + 4{\text{CO}}{\kern 1pt} .$(2а)
$\begin{gathered} {\text{Mo}}{{{\text{O}}}_{2}} + 2{\text{С}} \to {\text{Mo}} + 2{\text{CO}} \\ (\Delta G_{{298}}^{^\circ } = 275.9\,\,{{{\text{кДж}}} \mathord{\left/ {\vphantom {{{\text{кДж}}} {{\text{моль}}}}} \right. \kern-0em} {{\text{моль}}}}\,\,{\text{[}}7{\text{]}}), \\ \end{gathered} $(2б)
$\begin{gathered} 2{\text{Mo}} + {\text{C}} \to {\text{M}}{{{\text{o}}}_{{\text{2}}}}{\text{C}} \\ (\Delta G_{{298}}^{^\circ } = - 53.1\,\,{{{\text{кДж}}} \mathord{\left/ {\vphantom {{{\text{кДж}}} {{\text{моль}}}}} \right. \kern-0em} {{\text{моль}}}}\,\,{\text{[}}7{\text{]}}). \\ \end{gathered} $В табл. 3 суммированы основные характеристики двух стадий взаимодействия МоО3 с C.
Таблица 3.
Характеристики двух стадий взаимодействия оксида молибдена с графитом по реакциям (1) и (2). T0.5 и Tfin – температуры полупревращения и завершения реакции, ΔMexp и ΔMcalc – экспериментальные и расчетные значения уменьшения массы
| Образец MoO3 |
MoO3 + C → MoO2 + CO2 (1) экзотермическая реакция |
MoO2 + C → Mo2C + CO (2) эндотермическая реакция |
||||
|---|---|---|---|---|---|---|
| T0.5 | Tfin | ΔMexp, % | ΔMcalc, % | ΔT, °C | ΔMexp, % | |
| Исходный | 680 | 720 | 11.80 | 11 | ||
| После 30 мин МА | 600 | 660 | 12.15 | |||
| После 180 мин МА | 540 | 620 | <12.84 | 780–850 | 25.75 | |
Подводя итог сказанному, можно сделать следующие утверждения.
1. Первая стадия восстановления МоО3 углеродом протекает по брутто-уравнению (1) с выделением CO2 через промежуточную стадию образования фазы Mo4O11. Возникновение активных состояний при МА МоО3 снижает температуру полупревращения оксида на 1-ой стадии его восстановления до MoO2 на 100–150°C. При этом превращение начинается, по-видимому, при 100–150°C.
2. Для образца MoO3/5C с максимальной дозой МА и максимальной удельной поверхностью (примерно 52 м2/г) удалось провести полное восстановление оксида до карбида (брутто-реакции (1) и (2)) при температуре прогрева до 850°C. Вторая реакция является эндотермической и сопровождается выделением CO.
3. При прогреве до 350°C концентрация фазы МоО3 в системе остается неизменной. Потеря массы при этом составляет 1.38%, что соответствует формальной стехиометрии MoO2.88. До температуры 500°C в системе сохраняется некоторое количество кристаллической фазы МоО3 наряду с другими кристаллическими фазами. При этом потеря массы составляет 5.11%; это соответствует формальной стехиометрии MoO2.54.
Полученные результаты в целом совпадают с литературными данными и вносят в них некоторые уточнения. В [6, 7] отмечается, что реакция (1) может протекать в процессе механической обработки, однако следы продукта (MoO2) удается наблюдать только при очень длительной активации. В нашем случае была осознанно выбрана небольшая доза активации [43] (D ≤ 6 кДж/г), и следов MoO2 после помола обнаружено не было. Гипотеза о том, что сформировавшийся при МА композит имеет структуру ядро/оболочка, где оболочка состоит из углерода, подтверждается данными [4, 7]. Температурные диапазоны первой и второй стадий восстановления в нашей работе и по данным [4, 6] достаточно близки. При малых дозах активации удалось методом рентгеновской дифракции подтвердить образование промежуточной фазы Mo4O11. Эта фаза наблюдалась при восстановлении образцов неактивированного МоО3 в атмосфере H2 [20, 21] или CO [22, 23], однако для МА-образцов МоО3/C она ранее, по-видимому, не наблюдалась. В нашей работе также экспериментально подтверждены теоретические предсказания [7] того, что реакция (2) сопровождается эндотермическим эффектом и при ее протекании выделяется, прежде всего, CO. Можно отметить, что нам не удалось обнаружить промежуточную фазу Mo на второй стадии восстановления (реакции (2а), (2б)). Как следует из [7], это обусловлено достаточно высоким содержанием графита в смеси в нашем случае.
Обсудим возможные причины снижения температуры восстановления МоО3 углеродом в результате МА. Одной из них является образование наноразмерной композитной структуры, где наночастицы оксида покрыты аморфным углеродом, при этом удельная поверхность смеси составляет 52 м2/г, т.е. в результате МА происходит существенное увеличение поверхности контакта компонентов. Второй причиной является, по-видимому, образование “слабосвязанного” кислорода в МоО3. В [31] показано, что при прогреве в вакууме активированного образца МоО3 из него при температуре 200–450°C (см. рис. 7 в [31]) выделяется в газовую фазу кислород в количестве до 8 × 1019 молекула/г, при том что в отсутствие МА кислород начинает выделяться в газовую фазу только при температуре выше 500°C. В результате обеднения решетки кислородом формируется фаза Мо4О11, причем количество выделившегося кислорода примерно в два раза превышает необходимое для протекания реакции 4МоО3 → → Mo4O11 + O. В присутствии “внешнего” кислорода в газовой фазе выделение “решеточного” кислорода прекращается, но в молекулярно плотных композитах МоО3/C и МоО3/Al ничто не мешает решеточному кислороду диффундировать во второй компонент и реагировать с ним с выделением тепла.
Рис. 7.
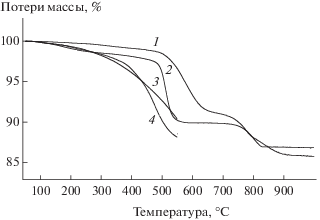
Сравнение термогравиметрических данных при разложении в He исходного (1) и активированного (2) MnO2, а также активированных смесей 5MnO2/C (3) и MnO2/5C (4). Образцы 4–7 табл. 1.
Данные рис. 1 свидетельствуют о том, что восстановление исходного МоО3 (в котором нет “слабосвязанного” кислорода), регистрируемое по выделению CO2 (кривая 1b), начинается только при 500°C, т.е. при температуре, соответствующей началу разложения с выделением кислорода обычного МоО3. Из активированных образцов МоО3 (кривые 2b и 3b) выделение CO2 начинается при 100–200°C, увеличивается с ростом температуры; при 400°С этот процесс ускоряется, и далее выделение CO2 продолжается до температуры 500°C и выше. Можно предположить, что протекание реакции восстановления МоО3 при низкой температуре непосредственно связано с образованием “слабосвязанного” кислорода при МА оксида и его выделением при прогреве. Некоторое запаздывание по температуре может быть обусловлено как диффузионными процессами, так и существенно более высокой скоростью разогрева в нашем случае по сравнению со скоростью разогрева в опытах, результаты которых приведены на рис. 7 работы [31]. При температуре выше 500°C реакция восстановления в активированных системах инициируется уже обычным путем, а не за счет разложения “слабосвязанного” кислорода.
3.2. Система MnO2/C
Известно, что MnO2 разлагается с выделением кислорода при умеренных значениях температуры, поэтому до опытов с активированными системами MnO2/C было проверено влияние МА на разложение MnO2.
В первых экспериментах проверили термическую устойчивость исходного MnO2 (образец 4 в табл. 1), а также механически активированного оксида без добавок углерода (образец 5 в табл. 1). На рис. 5а приведены кривые термического разложения MnO2 после его активации. Видно, что существуют три температурные области уменьшения массы: 1) до 400°C, 2) 400–550°C, 3) 700–820°C (кривая 1). При температуре ниже 400°C уменьшение массы обусловлено выделением воды и следов CO2. Во втором и третьем диапазонах температуры наряду с уменьшением массы наблюдается выделение из образца газообразного кислорода (кривая М32), сопровождающееся эндотермическими тепловыми эффектами (кривая 2).
Дифракционные данные для образцов MnO2, прогретых в ДСК-ячейке до температуры 300, 600 и 1000°C, приведены на рис. 5б. Видно, что при прогреве до 300°C (кривая 2) фазовый состав MnO2 не изменился. После прогрева до 600°C на дифрактограмме зарегистрированы линии только фазы Mn2O3, а после прогрева до 1000°C в образце обнаружена только фаза Mn3O4. Можно полагать, что разложение MnO2 протекает в две стадии:
(3)
$2{\text{Mn}}{{{\text{O}}}_{2}} \to {\text{M}}{{{\text{n}}}_{{\text{2}}}}{{{\text{O}}}_{3}} + {\text{O (}}400{\kern 1pt} - {\kern 1pt} 550^\circ {\text{C)}},$(4)
$3{\text{M}}{{{\text{n}}}_{{\text{2}}}}{{{\text{O}}}_{3}} \to 2{\text{M}}{{{\text{n}}}_{{\text{3}}}}{{{\text{O}}}_{4}} + {\text{O (}}700{\kern 1pt} - {\kern 1pt} 820^\circ {\text{C)}}{\text{.}}$Следует отметить, что температурные диапазоны протекания реакций (3) и (4) определены из данных о потери массы. При этом экспериментальные значения потери массы соответствуют рассчитанным по уравнениям (3) и (4).
Для неактивированных образцов наблюдаются те же процессы (3) и (4), но температуры разложения MnO2 и выделения кислорода сдвинуты в высокотемпературную область на 50–100°C. Необходимо учитывать, что описанные выше эксперименты проведены в инертной среде. Если разложение MnO2 проводить на воздухе, то соответствующие значения температуры будут выше.
Таким образом, MnO2 термически разлагается по уравнениям (3) и (4), а МА приводит к снижению температур разложения.
Для системы MnO2/C исследованы два образца с малым (5MnO2/C, 2.6% C) и большим (MnO2/5C, 41% C) содержанием графита (образцы 6 и 7 в табл. 1).
На рис. 6а приведены данные о тепловом эффекте (кривая 1), изменении массы (кривая 2), выделении CO2 и уровне остаточного кислорода при прогреве активированного образца 5MnO2/C до 550°C. Сравнение рис. 6а и 5а, показывает, что введение даже небольшого количества углерода в систему приводит к полному прекращению выделения кислорода в газовую фазу, но сопровождается интенсивным выделением CO2. Для системы MnO2/5С результаты ТГ и масс-спектрометрии примерно такие же, однако потеря массы ΔМ/М к 550°C в этом случае немного больше (табл. 4).
Таблица 4.
Фазовый состав, относительная убыль кислорода ΔM(O)/MnO2 и потеря массы ΔM/M при прогреве двух образцов смеси MnO2/C до 550°C в атмосфере He
| Образец | Фазовый состав, мас. % | ΔM(O)/MnO2 | ΔM/M, % | |||
|---|---|---|---|---|---|---|
| MnO2 | Mn2O3 | Mn3O4 | С | |||
| 5MnO2/C | 100 | – | 0.25 | 9.61 | ||
| MnO2/5С | 6.8 | 19.4 | 31.4 | 42.4 | 0.27 | 11.05 |
Дифракционные данные для обеих систем после их прогрева до 550°C приведены на рис. 6б, а результаты фазового анализа суммированы в табл. 4. После прогрева активированного образца с малым содержанием графита обнаружена только фаза Mn2O3, т.е. реакция (3) в этом случае прошла так же, как и в образце без графита. Однако выделения кислорода в газовую фазу не зафиксировано. Кислород реагирует с графитом, и в газовую фазу выделяется CO2. Для образца с большим содержанием графита после прогрева до 550°C зарегистрировано значительное количество фазы Mn3O4 (табл. 4), которая для образца без углерода формируется при температуре на 200°C выше. В то же время в образце сохранилось небольшое количество исходного MnO2. В целом “глубина” восстановления MnO2 оказалась несколько больше. Параметр ΔM(O)/MnO2 отражает относительную убыль кислорода в реакциях восстановления оксида. Для реакции MnO2 → MnO3/2 этот параметр равен (2 – 3/2)/2 = 0.25. Рассчитанный из данных количественного фазового анализа этот параметр для системы MnO2/5С оказался немного больше и равен 0.27.
На рис. 7 сопоставлены данные по уменьшению массы при прогреве в гелии исходного (кривая 1) и активированного (кривая 2) образцов MnO2, а также активированных смесей 5MnO2/C (кривая 3) и MnO2/5C (кривая 4). Первая стадия разложения исходного образца по реакции (3) протекает при 500–650°C. МА снижает температуру его разложения на 50–100°C (кривая 2). В случае активации систем MnO2/C (кривые 3 и 4 на рис. 7, сравнить с кривой 2) дополнительное уменьшение массы заметно, начиная с 300°C, а выделение CO2 наблюдается, начиная с 100°C (рис. 6а). Таким образом, даже незначительная добавка углерода (2.6 мас. % в системе 5MnO2/C) приводит к существенному ускорению восстановления MnO2. В системе MnO2/5С c высоким содержанием углерода появляется фаза, которая в отсутствие углерода возникает только выше 750°C, т.е. при температуре на 200°C большей.
Итак, в присутствии графита восстановление MnO2 существенно ускоряется. Известно, что разложение MnO2 с выделением кислорода является равновесной реакцией, зависящей от давления кислорода в газовой среде. Выше мы упоминали о замедлении разложения MnO2 при прогреве на воздухе. Можно полагать, что активированный графит является “химическим” перехватчиком выделяющегося атомарного кислорода и сдвигает равновесие в сторону разложения MnO2.
3.3. Система Bi2O3/5C
На дифрактограммах механически активированной системы Bi2O3/5C наблюдаются только уширенные линии моноклинной фазы Bi2O3(mncl), а линии графита выделить не удается [43]. Данные ДСК, ТГ и масс-спектрометрии, полученные при прогреве этой системы, приведены на рис. 8а, а дифрактограммы после ее прогрева до 260, 450 и 700°C – на рис. 8б.
Рис. 8.
(а) Уменьшение массы (1), тепловые эффекты (2) и ионные токи, соответствующие значениям m/z = 28, 44, 18, 32, при прогреве активированной в течение 180 мин смеси Bi2O3/5C в He. (б) Дифрактограммы смеси Bi2O3/5C после МА и прогрева в ДСК-ячейке до 260 (1), 450 (2) и 700°C (3).

Рис. 9.
Сопоставление процессов восстановления Bi2O3 в атмосфере CO [17] и графитом. Уменьшение массы (кривые 1, 2, 3) и выделение CO2 (1а, 2а, 3а) при восстановлении Bi2O3 в СО (1, 2) и прогреве смеси Bi2O3/5C (3); 1 – неактивированный образец.

Рис. 10.
(а) Уменьшение массы (1, 2) и ионные токи CO2 (1а и 2а) при прогреве активированных в течение 90 мин смесей V2O5/C (1, 1а) и V2O5/5C (2, 2а) в He. (б) Дифрактограммы смеси V2O5/5C, активированной в течение 90 мин и прогретой до разной температуры в He.

Результаты количественного фазового анализа образцов Bi2O3/5C, прогретых до этих значений температуры, а также данные об уменьшении их массы суммированы в табл. 5. Как видно из их сопоставления, прогрев до 260°C сопровождается небольшим уменьшением массы образца, обусловленным выделением воды. При этом фазовый состав системы не изменяется. В диапазоне 300–600°C происходит значительное уменьшение массы образца, сопровождающееся выделением CO2. Исходя из соотношения интенсивностей пиков, отвечающих значениям m/z = 28 и 44, можно утверждать, что содержание CO в выделяющихся газах незначительно. При температуре выше 600°C масса образца не изменяется, а на дифрактограмме зарегистрирована фаза Bi с небольшой примесью Bi2O3, но в другой, тетрагональной модификации. Таким образом, в температурном диапазоне 260–600°C протекает реакция
(5)
${\text{B}}{{{\text{i}}}_{{\text{2}}}}{{{\text{O}}}_{3}} + 5{\kern 1pt} {\text{C}} \to 2{\text{Bi}} + 1.5{\kern 1pt} {\text{C}}{{{\text{O}}}_{2}} + 3.5{\kern 1pt} {\text{C}}.$Таблица 5.
Уменьшение массы ΔM/M и фазовый состав активированной системы Bi2O3/5C после ее прогрева в гелии до разных значений температуры T
| T, °C | 20 | 260 | 450 | 700 |
| ΔM/M, % | 0 | –0.62 | –5.29 | –14.19 |
| Фазовый состав, мас. % | 100 Bi2O3(mncl) | 100 Bi2O3(mncl) | 74 Bi2O3(mncl), 26 Bi | 91 Bi, 9 Bi2O3(tetr) |
Таблица 6.
Фазовый состав и потери массы ΔM/M при прогреве двух смесей V2O5/C до 550°C в атмосфере гелия
| T, °C | V2O5/С | V2O5/5С | ||
|---|---|---|---|---|
| фазовый состав, мас. % | ΔM/M, % | фазовый состав, мас. % | ΔM/M, % | |
| 20 | 100 V2O5 | 0 | 100 V2O5 | 0 |
| 300 | 100 V2O5 | –0.96 | 100 V2O5 | –1.00 |
| 520 | 93 V6O13, 7 V2O5 | –7.94 | 79 V6O13, 21 V2O5 (С) | –6.93 |
| 650 | 100 VO2 | –12.49 | 100 VO2 (С) | –10.92 |
Согласно оценке, уменьшение массы образца в результате реакции (5) должно составлять 12.5%, что близко к экспериментальному значению (см. табл. 5): 14.19% – 0.62% = 13.57%.
Так как при прогреве механически активированного Bi2O3 в условиях ДСК-эксперимента выделения кислорода в газовую фазу не происходит, то закономерности восстановления оксида висмута углеродом были сопоставлены с реакциями Bi2O3 + СО [17] и Bi2O3 + Al [29]. На рис. 9 приведены данные об убыли массы и выделении CO2 при прогреве в CO неактивированного (кривые 1, 1а) и активированного (кривые 2, 2а) Bi2O3 [17]. На этот же график нанесены кривые 3, 3а убыли массы и выделения CO2 при восстановлении Bi2O3 углеродом, полученные в настоящем разделе. Для неактивированного образца взаимодействие с CO начинается только при 350°C, и к 600°C количество продукта (Bi) составляет примерно 10 мас. % [17]. Для активированного образца восстановление в CO начинается при температуре 200–250°C, и к 600°C количество Bi достигает примерно 60 мас. % [17]. Как видно на рис. 9, в присутствии графита выделение CO2 начинается тоже при 250°C, резкое ускорение процесса происходит при 350°C, и к 600°C реакция почти завершается (табл. 5).
Как следует из рис. 7 работы [29], восстановление висмута алюминием происходит в интервале температуры 400–700°C. Таким образом, добавка углерода не приводит к существенному ускорению восстановления активированного оксида висмута, и этот процесс протекает примерно в том же температурном диапазоне, что и реакция Bi2O3 с Al или CO.
3.4. Система V2O5/C
На рис. 10а приведены кривые уменьшения массы и выделения CO2 при прогреве двух активированных образцов: V2O5/C и V2O5/5C (№№ 9 и 10 в табл. 1). Пример дифрактограмм образцов V2O5/5C после их прогрева до разной температуры приведен на рис. 10б, а результаты фазового анализа и данные о потери массы суммированы в табл. 6. Несмотря на то, что по содержанию углерода системы V2O5/C и V2O5/5C различаются в 4 раза (табл. 1), результаты для них оказались близкими.
На ТГ-кривых можно выделить три участка. При этом до температуры 300°C уменьшение массы обусловлено, в основном, выделением воды без изменения фазового состава образцов. Характерной особенностью этой системы является наличие двух четко выраженных температурных максимумов выделения CO2. Анализ фазового состава систем до и после этих максимумов позволил выяснить, что первый максимум обусловлен восстановлением V2O5 до V6O13, а второй – восстановлением V6O13 до VO2 по реакциям (6) и (7) соответственно:
(6)
${{{\text{V}}}_{{\text{2}}}}{{{\text{O}}}_{5}} + {\text{C}} \to {{{\text{V}}}_{{\text{6}}}}{{{\text{O}}}_{{13}}} + {\text{C}}{{{\text{O}}}_{2}}(300{\kern 1pt} - {\kern 1pt} 520(540)^\circ {\text{С}}),$(7)
${{{\text{V}}}_{{\text{6}}}}{{{\text{O}}}_{{13}}} + {\text{C}} \to {\text{V}}{{{\text{O}}}_{2}} + {\text{C}}{{{\text{O}}}_{2}}(520(540){\kern 1pt} - {\kern 1pt} 650^\circ {\text{С}}).$Расчетное уменьшение массы для системы V2O5/C к 520°C составляет 7.1%, а к 650°C – 11.3%, что близко к экспериментальным значениям (7.94% – 0.96% = 6.98% и 12.49% – 0.96% = 11.53% соответственно).
Сопоставляя эти результаты с данными по взаимодействию механически активированного V2O5 c Al (пока не опубликованными), можно отметить, что взаимодействие с Al начинается при более низкой температуре, чем с углеродом. При 300°C в смеси с графитом (табл. 6) восстановление еще не началось, а в смеси с алюминием уже происходит.
4. ЗАКЛЮЧЕНИЕ
В табл. 7 суммированы данные о термической стабильности исследованных композитов. В качестве основного критерия было выбрано сохранение кристаллической структуры оксидов. Видно, что до 250–350°C кристаллическая структура оксидов сохраняется. Следует отметить, что это ориентировочные данные, так как с одной стороны шаг по температуре при измерениях был очень большим, а с другой стороны, например, в системе МоО3/5С при прогреве до 350°C кристаллическая структура, судя по дифракционным данным, не изменилась, но потеря массы составила 1.38%, что соответствует формальной конечной стехиометрии MoO2.88. Отметим, что стехиометрия фазы Мо4О11 равна MoO2.75, т.е. достаточно близка.
Таблица 7.
Максимальные значения температуры, при которых фазовый состав композитов еще не изменился
| Композит | MnO2/5С | MoO3/5С | Bi2O3/5С | V2O5/С |
|---|---|---|---|---|
| Температура | 250°C | 350°C | 260°C | 300°C |
С позиций возникновения при МА “слабосвязанного” кислорода, рассматриваемые оксиды могут быть разделены на три группы. К первой группе относится MnO2, который легко разлагается с выделением кислорода по реакции (3) при 500–600°C в случае прогрева в инертной атмосфере со следами кислорода. При прогреве на воздухе, в присутствии 150 тор O2, температура выделения кислорода повышается на сотни градусов, что указывает на существование в этом процессе некоторого равновесия. МА снижает температуру разложения MnO2 на 50–100°C (рис. 7). Возможно, это связано с образованием “слабосвязанного” решеточного кислорода при МА, но следует проверить в будущем возможную роль уменьшения размера частиц при помоле. Присутствие в активированной системе даже небольшого количества углерода (2.6 мас. %) приводит к существенному увеличению скорости восстановления оксида при значениях температуры 300–450°C. Возможно, углерод функционирует как “химический насос”, связывая выделяющийся кислород и тем самым сдвигая равновесие в сторону разложения.
Ко второй группе оксидов относится МоО3, МА которого снижает температуру начала разложения с 500°C до 200°C за счет образования слабосвязанного кислорода. В [31] удалось зафиксировать выделение кислорода в условиях его низкого равновесного давления в температурном диапазоне 200–400°C. Возникновение дефектных состояний в виде слабосвязанного кислорода приводит к резкому снижению температуры начала реакции восстановления МоО3 углеродом по реакции (1). В этом же диапазоне температуры начинается термическая реакция между MoO3 и Al. Взаимодействие МоО3 с углеродом после МА протекает в две стадии, по реакциям (1) и (2): восстановление до МоО2 и затем образование МоС2. Экспериментально подтверждена эндотермичность второй стадии и то, что основным выделяющимся газом является CO.
Наконец, к третьей группе оксидов относятся Bi2O3 и V2O5, для которых выделение кислорода из МА-образцов непосредственно не наблюдали, а их активность следует из химического взаимодействия, например, с алюминием или CO. Оказалось, что восстановление активированного Bi2O3 углеродом протекает в том же температурном диапазоне, что и восстановление этого оксида в атмосфере CO. Особенность восстановления V2O5 углеродом по сравнению с его взаимодействием с Al заключается в четком разделении последовательных реакций (6) и (7), однако они протекают при более высоких температурах, чем в активированных смесях V2O5 с Al.
Подводя итог, можно констатировать, что наличие углерода по-разному влияет на реакционную способность активированных оксидов. В случае MnO2 присутствие углерода сопровождается повышением химической активности, температурный диапазон взаимодействия МоО3 и Bi2O5 с C и Al практически одинаков, а реакция V2O5 с C протекает немного медленнее, чем с Al.
Список литературы
Suryanarayana C. // Prog. Mater. Sci. 2001. V. 46. P. 1.
Baláž P., Achimovičová M., Baláž M., Billik P., Cherkezova-Zheleva Z., Criado J.M., Delogu F., Dutková E., Gaffet E., José F., Martinéz G., Kumar R., Mitov I., Rojac T., Senna M., Streletskii A., Wieczorek-Ciurowa K. // Chem. Soc. Rev. 2013. V. 42. P. 7571.
El-Eskandarany M.S. Mechanical Alloying: For Fabrication of Advanced Engineering Materials. Jackson, MS, USA: University Press of Mississippi, 2013.
Bokhonov B., Borisova Yu., Korchagin M. // Carbon. 2004. V. 42. P. 2067.
Díaz Barriga Arceo L.,Orozco E., Mendoza-León H., Palacios González E., Leyte Guerrero F., Garibay Febles V. // J. Alloys Compd. 2007.V. 434–435. P. 799.
Saghafi M., Ataie A., Heshmati-Manesh S. // Int. J. Refract. Hard Met. 2011. V. 29. P. 419.
Khabbaz S., Honarbakhsh-Raouf A., Ataie A., Saghafi M. // Int. J. Refract. Hard Met. 2013. V. 41. P. 402.
Kurlov A.S., Gusev A.I. // Inorg. Mater. 2013. V. 49. P. 347.
Zaki Z.I., El-Sadek M.H., Ali H.H., Ahmed H. // Materials. 2020. V. 13. P. 4408.
Xia Z.P., Shen Y.Q., Shen J.J., Li Z.Q. // J. Alloys Compd. 2008.V. 453. P. 185.
Torabi O., Golabgir M.H., Tajizadegan H., Torabi H. // Int. J. Refract. Hard Met. 2014. V. 47. P. 18.
Hossein-Zadeh M., Razavi M., Safa M., Abdollahi A., Mirzaee O. // J. King Saud Univ. Eng. Sci. 2016.V. 28. P. 207.
Hossein-Zadeh M., Mirzaee O. // Adv. Powder Technol. 2014. V. 25. P. 978.
Hoseinpur A., Khaki J.V., Marashi M.S. // Mater. Res. Bull. 2013. V. 48. P. 399.
Сивак М.В., Стрелецкий А.Н., Колбанев И.В., Леонов А.В., Дегтярев Е.Н., Перменов Д.Г. // Коллоид. журн. 2015. Т. 77. С. 355.
Kokorin A.I., Streletskii A.N., Kolbanev I.V., Borunova A.B., Degtyarev E.N., Leonov A.V., Permenov D.G., Konstantinova E.A. // J. Phys. Chem. C. V. 122. P. 2019.
Стрелецкий А.Н., Колбанев И.В., Воробьева Г.А., Леонов А.В., Борунова А.Б., Дубинский А.А. // Коллоид. журн. 2019. Т. 81. С. 617.
Saghafi M., Heshmati-Manesh S., Ataie A., Khodadadi A.A. // Int. J. Refract. Hard Met. 2012. V. 30. P. 128.
Chaudhury S., Mukerjee S.K., Vaidya V.N., Venugopal V. // J. Alloys Compd. 1997. V. 261. P. 105.
Dang J., Zhang G.-H., Chou K.C., Reddy R.G., He Y., Sun Y. // Int. J. Refract. Hard Met. 2013. V. 41. P. 216.
Lalik E. // Catal. Today. 2011. V. 169. P. 85.
Samsuri A., Saharuddin T.S.T., Salleh F., Othaman R., Hisham M.W.M., Yarmo M.A. // Mater. Sci. Forum. 2016. V. 840. P. 299.
Samsuri A., Saharuddin T.S.T., Salleh F., Othaman R., Hisham M.W.M., Yarmo M.A. // Malays. J. Anal. Sci. 2016. V. 20. P. 382.
Ochirkhuyag A., Sapi A., Szamosvolgyi A., Kozma G., Kukovecz A., Konya Z. // Phys. Chem. Chem. Phys. 2020. V. 22. P. 13999.
Dreizin E.L. // Prog. Energy Combust. Sci. 2009. V. 35. P. 141.
Dreizin E.L., Schoenitz M. // J. Mater. Sci. 2017. V. 52. P. 11789.
Streletskii A.N., Sivak M.V., Dolgoborodov A.Yu. // J. Mater. Sci. 2017. V. 52. P. 11810.
Dolgoborodov A.Yu. // Combust. Explos. Shock Waves. 2015. V. 51. P. 86.
Стрелецкий А.Н., Воробьева Г.А., Колбанев И.В., Леонов А.В., Кириленко Г.В., Гришин Л.И., Долгобородов А.Ю. // Коллоид. журн. 2019. Т. 81. С. 625.
Streletskii A.N., Kolbanev I.V., Vorobieva G.A., Dolgoborodov A.Yu., Kirilenko V.G., Yankovskii B.D. // J. Mater. Sci. 2018. V. 53. P. 13550.
Сивак М.В., Стрелецкий А.Н., Колбанев И.В., Леонов А.В., Дегтярев Е.Н. // Коллоид. журн. 2016. Т. 78. С. 618.
Zhi M., Xiang C., Li J., Li M., Wu N. // Nanoscale . 2013. V. 5. P. 72.
Borenstein A., Hanna O., Attias R., Luski S., Brousse T., Aurbach D. // J. Mater. Chem. A. 2017. V. 5. P. 12653.
Liu X., Wu Y., Wang H., Wang Y., Huang C., Liu L., Wang Z. // RSC Adv. 2020. V. 10. P. 17497.
Jiang Y., Yan X., Cheng Y., Zhang Y., Xiao W., Gan L., Tang H. // RSC Adv. 2019. V. 9. P. 13207.
Haris M., Atiq S., Abbas K., Naseem S. // Mater. Sci. Eng. B. 2019. V. 243. P. 19.
Schlem R., Burmeister C.F., Michalowski P., Ohno S., Dewald G.F., Kwade A., Zeier W.G. // Adv. Energy Mater. 2021. 2101022.
Tao T., Glushenkov A.M., Zhang C., Zhang H., Zhou D., Guo Z., Liu H.K., Chen Q., Hu H., Chen Y. // J. Mater. Chem. 2011. V. 21. P. 9350. https://doi.org/10.1039/c1jm10220f
Wang L., Wang Y., Zhu X., Zhao Y. // J. Alloys Compd. 2019. V. 792. P. 418.
Srikanth M., Rahman M. M., Li L.H., Cai Q., Chen Y. // RSC Adv. 2016. V. 6. P. 35287.
Moazzen E., Kucuk K., Aryal S., Timofeeva E.V., Segre C.U. // J. Power Sources. 2020. V. 448. 227374.
Cao J., Zhang D., Zhang X., Wang S., Han J., Zhao Y., Huang Y., Qin J. // Appl. Surf. Sci. 2020. V. 534. 147630.
Стрелецкий А.Н., Колбанев И.В., Борунова А.Б., Леонов А.В., Нищак О.Ю., Перменов Д.Г., Иванова О.П. // Коллоид. журн. 2021. Т. 83. С. 690.
Shelekhov E.V., Sviridova T.A. // Met. Sci. Heat Treat. 2000. V. 42. № 8. P. 309.
Дополнительные материалы отсутствуют.
Инструменты
Коллоидный журнал