

Координационная химия, 2019, T. 45, № 10, стр. 593-598
Полиядерные миртенаты Co(II) с 2,4-лутидином
Е. Н. Зорина-Тихонова 1, *, Г. Г. Александров 1, М. А. Кискин 1, Л. Л. Фролова 2, **, А. В. Кучин 2, А. А. Сидоров 1, И. Л. Еременко 1
1 Институт общей и неорганической химии им. Н.С. Курнакова РАН
Москва, Россия
2 Институт химии Коми НЦ УрО РАН
Сыктывкар, Россия
* E-mail: kamphor@mail.ru
** E-mail: frolova-ll@chemi.komisc.ru
Поступила в редакцию 26.02.2019
После доработки 15.05.2019
Принята к публикации 20.05.2019
Аннотация
Взаимодействие хлорида кобальта(II) с миртенатом калия (KМyr) с последующим добавлением 2,3-лутидина в этаноле приводит к образованию трехъядерного соединения [Co3(Мyr)6(2,4-Lut)2] (I). Центральный атом кобальта(II) связан с каждым из двух других атомов металла тремя анионами миртеновой кислоты. Взаимодействие раствора соединения I с миртенатом лития (LiMyr) приводит к формированию четырeхъядерного гетерометаллического комплекса состава [Li2Co2(Myr)6(2,4-Lut)2] (II). В соединении II можно выделить два биядерных фрагмента {LiCo(Myr)3(2,4-Lut)}, в которых атомы лития(I) связаны с атомами кобальта(II) тремя мостиковыми миртенатными анионами. Атомы лития связаны двумя атомами кислорода анионов кислоты. Соединения I, II охарактеризованы данными РСА (CIF files CCDC № 1898096 (I), 1898097 (II)).
Среди многочисленных представителей полиядерных карбоксилатов с атомами двухвалентных 3d-металлов, простейшими и наиболее распространенными являются биядерные соединения с металлофрагментом {M(μ-O2CR)4M} [1–6] и линейные трехъядерные с металлофрагментом {M(μ-O2CR)3M(μ-O2CR)3M} [7–16]. Эти металлофрагменты достаточно типичны для слоистых и каркасных полимеров с анионами дикарбоновых и поликарбоновых кислот [17–23]. Такие системы в зависимости от природы металлоцентров весьма активно изучаются различными исследователями из-за их каталитической активности, проявляемых ими магнитных и оптических характеристик или биологической активности [1, 4, 13–15, 18]. Для получения молекулярных систем используются аддукты таких простейших би- и трехъядерных фрагментов с различными нейтральными органическими донорными молекулами, причем существуют различные подходы к управлению строением металлоостова в таких структурах в зависимости от геометрических и электронных свойств апикальных органических лигандов [1–3]. Другим способом направленного влияния на строение металлокарбоксилатного остова может быть изменение природы заместителя при карбоксилатной группе, причем в случае производных Co(II), Fe(II), Mn(II) и Zn(II) [7, 12] оно примерно одинаково. Сложность точного прогнозирования строения комплексов при варьировании природы заместителя в карбоксилатном анионе и возможность получения для некоторых карбоксилатных анионов двух типов комплексов (с би- и трехъядерным строением металлокарбоксилатного остова) указывает на то, что, вероятно, термодинамическая устойчивость этих би- и трехъядерных комплексов часто бывает очень близка, а некоторые из соединений могут быть достаточно устойчивыми метастабильными продуктами, в том числе метастабильными изомерами. Это можно проиллюстрировать уникальным примером, когда в кристалле сосуществуют биядерная [Zn2(O2CPh)4(Рy)2] и трехъядерная [Zn3(O2CPh)6(Рy)2] молекулы [24]. Влияние заместителя при карбоксилатной группе можно продемонстрировать примером нафтоатных комплексов: комплекс цинка(II) с α-нафтойной кислотой (α-HNaph) с 2,3-лутидином (2,3-Lut), [Zn2(α-Naph)4(2,3-Lut)2] биядерный [7], а замена кислоты на β-нафтойную (β-HNaph) способствует формированию линейного трехъядерного комплекса [Zn3(β-Naph)6-(2,3-Lut)2] [16].
Несомненная аналогия с бензоатными комплексами наблюдается в случае комплексов карбоновых кислот с ненасыщенным заместителем, например с анионами кротоновой кислоты. Для цинка(II), как и в случае бензоатных комплексов, с хинолином получен биядерный [25, 26] и трехъядерный комплексы [27, 28].
В настоящей работе мы дополняем исследования по влиянию заместителя при карбоновой кислоте. Так, мы изучили способность формировать металлокарбоксилатный остов при использовании миртенатного аниона (Мyr–), интересного тем, что он имеет двойную связь С=С, которая может вступать в π–π-сопряжение с карбоксилатной группой, как и в случае кротонатного аниона. Однако в миртенатном анионе атомы углерода, связанные двойной связью, принадлежат объемному бициклическому фрагменту.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Анализ методом газожидкостной хроматографии (ГЖХ) проводили на хроматографе SHIMADZU GC-2010AF, колонка HP1, детектор – пламенно-ионизационный, газ-носитель – гелий. ИК-спектр миртеновой кислоты снимали в тонком слое на приборе IR Prestige 21 фирмы Shimadzu. Спектры ЯМР регистрировали на приборе Bruker AVANCE-II-300, рабочая частота 300 МГц (1Н) и 75 МГц (13С) в CDCl3 с использованием стандартных импульсных программ фирмы Bruker для одно- и двумерных экспериментов. Оптическое вращение определяли на автоматическом поляриметре P3002RS фирмы Kruss (Германия). ИК-спектры комплексов регистрировали на приборе PerkinElmerSpectrum 65 методом НПВО в интервале частот 4000–400 см–1. Элементный анализ выполняли на автоматическом C, H, N, S-анализаторе EA-3000 (EuroVector).
РCA монокристаллов комплексов I и II выполнен на дифрактометре Bruker Apex II (CCD-детектор, MoKα-излучение λ = 0.71073 Å, графитовый монохроматор) [29]. Структуры расшифрованы прямыми методами и уточнены в полноматричном анизотропном приближении для всех неводородных атомов. Атомы водорода при атомах углерода органических лигандов генерированы геометрически и уточнены в модели “наездника”. Расчеты проведены по комплексу программ SHELX-97 [30]. Кристаллографические параметры I и II приведены в табл. 1.
Таблица 1.
Кристаллографические параметры и детали уточнения структур I и II
| Параметр | Значение | |
|---|---|---|
| I | II | |
| Брутто-формула | C74H96N2O12Co3 | C78H102N4O12Co2Li2 |
| М | 1382.32 | 1419.38 |
| Т, K | 184(2) | 184(2) |
| Сингония | Триклинная | Моноклинная |
| Пр. гр. | Р1 | P21 |
| a, Å | 11.619(3) | 10.8075(14) |
| b, Å | 12.325(3) | 18.449(2) |
| c, Å | 13.635(3) | 18.927(3) |
| α, град | 111.380(3) | 90 |
| β, град | 100.092(4) | 96.460(2) |
| γ, град | 92.950(4) | 90 |
| V, Å3 | 1775.9(7) | 3749.8(8) |
| Z | 1 | 2 |
| ρ(выч.), г см3 | 1.293 | 1.257 |
| μ, мм–1 | 0.753 | 0.504 |
| θmax, град | 29.13 | 28.28 |
| Tmin/Tmax | 0.929/0.956 | 0.942/0.951 |
| Число измеренных рефлексов | 19 954 | 28 286 |
| Число независимых рефлексов | 9493 | 15 479 |
| Число рефлексов с I > 2σ(I) | 5756 | 11 664 |
| Rint | 0.0568 | 0.058 |
| Число уточняемых параметров | 648 | 883 |
| GOOF | 1.39 | 1.07 |
| R1 (I > 2σ(I)) | 0.124 | 0.057 |
| wR2 (I > 2σ(I)) | 0.378 | 0.129 |
| Остаточная электронная плотность (min/max), e/Å3 | –3.833/1.003 | –0.436/0.682 |
Полный набор рентгеноструктурных данных депонирован в Кембриджском банке структурных данных (№ 1898096 (I), 1898097 (II); deposit@ccdc.cam.ac.uk или http://www.ccdc.cam.ac.uk/ data_request/cif).
Для колоночной хроматографии использовали силикагель 60 (70–230 меш) фирмы Alfa-Aesar. Тонкослойную хроматографию (ТСХ) выполняли на пластинах “Сорбфил”, элюенты: гексан (“х. ч.”) и диэтиловый эфир (“ч. д. а.”), проявитель 10%‑ный водный раствор KMnO4. Для синтеза миртеновой кислоты использовали 1(R)(–)‑миртенол фирмы Aldriсh [α]D –47.1 (c 1.0 EtOH), пиридин (“х. ч.”), Na2CO3 (“х. ч.”), MgSO4 (“х. ч.”). Диоксид хлора (ClO2) в виде водного раствора с концентрацией 6–7 г/л предоставлен Монди Сыктывкарским лесопромышленным комплексом.
Синтез HМmyr. Через раствор (–)-миртенола (5 г, 0.033 моль) в 50 мл пиридина при перемешивании и температуре 35–40°С пропускали газообразный диоксид хлора в течение 8–10 ч. Реакцию контролировали по ТСХ и ГЖХ. Реакционную смесь охлаждали, удаляли растворитель при пониженном давлении, остаток обрабатывали 100 мл 5%-ного водного раствора Na2CO3, смесь экстрагировали Et2O для отделения нейтральных соединений. Водный слой подкисляли 10%-ным водным раствором H2SO4 (“х. ч.”) и тщательно экстрагировали Et2O. Эфирный экстракт промывали насыщенным раствором NaCl и сушили над безводным MgSO4. После удаления растворителя выход миртеновой кислоты в виде густого масла темного цвета составлял 2.0–2.2 г (40–44% на исходный миртенол). После колоночной хроматографии на SiO2 (гексан–диэтиловый эфир (10 : 1)) получили бесцветное масло, постепенно кристаллизующееся. Rf 0.56 (гексан–диэтиловый эфир (3 : 2)), [α]D –42.4 (c 0.4 EtOH). ИК-спектр (ν, см–1): 3100–2500 (уш., ν, OH), 1690 (C=O), 1630 (C=C), 1430, 1280 (ν, C–O), 1080, 960 (OH), 790, 760, 720. ЯМР 13С (δ, м.д.): 171.8 (C10), 139.8 (C2), 139.3 (C3), 40.8 (C1), 40.2 (C5), 37.6 (C6), 32.3 (C4), 31.2 (C7), 25.8 (C8), 20.9 (C9). ЯМР 1H (δ, м.д.): 0.80 (c., 3H, C8), 1.12 (д., 1H, J = 9.1, C7β), 1.34 (c., 3H, C9), 2.13 (д. д. д., 1H, С5, J1 = 5.7, J2 = 2.9, J3 = 1.5), 2.45 (д.д., 1H, C4β, J1 = 13.5, J2 = 2.9), 2.46 (д., 1Н, C4α, J = 13.5) 2.48 (д., 1Н, J = 9.1, С7α), 2.79 (т.д., 1H, J1 = 1.5, J2 = 5.7, С1), 6.99 (м., 1H, J = 1.5, С3), 12.10 (уш., 1Н, СООH).
Синтез комплексов I и II проводили на воздухе с использованием этилового спирта (96%), ацетонитрила (99%) и коммерчески доступных реагентов: CoCl2 ∙ 6H2O (“ч. д. а.”), 2,4‑лутидин (>99%), KOH (“ч. д. а.”), LiOH (“ч. д. а.”). Для получения координационных соединений использовали миртеновую кислоту с разной оптической чистотой ((–37°)–(–42.4°). Выше приведен синтез кислоты по оптимальной методике с использованием оптически чистого исходного миртенола.
Синтез [Co3(Мyr)6(2,4-Lut)2] (I). Навески KOH (0.047 г, 8.4 ммоль) и HМyr (0.1395 г, 8.4 ммоль) растворяли в этаноле (30 мл), затем добавляли CoCl2 ∙ 6H2O (0.1 г, 4.2 ммоль) и 2,4‑лутидина (0.064 мл, 6.3 ммоль). Полученную реакционную смесь фиолетового цвета перемешивали в течение 30 мин, фильтровали от выпавшего KCl и выдерживали при комнатной температуре. Образовавшиеся через 24 ч фиолетовые кристаллы, пригодные для РСА, отделяли от маточного раствора декантацией, промывали холодным EtOH и сушили на воздухе. Выход соединения I 0.110 г (57% на исходное количество кобальта(II)).
ИК-спектр (НПВО; ν, см–1): 2982 ср, 2915 ср, 2881 ср, 2822 сл, 1680 сл. ш, 1631 ср, 1622 ср, 1578 с, 1516 ср, 1494 ср, 1400 с, 1387 с, 1365 ср, 1330 с, 1321 с, 1300 ср, 1266 ср, 1238 ср, 1216 ср, 1202 ср, 1177 ср, 1150 ср, 1123 сл, 1096 ср, 1079 ср, 1055 ср, 1037 ср, 1027 ср, 966 ср, 949 ср, 926 ср, 889 ср, 847 ср, 833 ср, 807 ср, 761 с, 734 ср, 726 ср, 633 ср, 594 ср, 539 ср.
Синтез [Li2Co2(Мyr)6(2,4-Lut)2] (II). К реакционной смеси, полученной при синтезе I после фильтрации KCl приливали раствор смеси LiOH (0.010 г, 4.2 ммоль) и HMyr (0.070 г, 4.2 ммоль) в этаноле (15 мл). Полученный фиолетовый раствор концентрировали при температуре 80°С до 20 мл, после охлаждали до комнатной температуры и выдерживали 24 ч. Образовавшиеся кристаллы фиолетового цвета, пригодные для РСА, промывали холодным ацетонитрилом (–5°С) и высушивали на воздухе. Выход соединения II 0.128 г (43% на исходное количество кобальта(II)).
ИК-спектр (НПВО; ν, см–1): 2982 ср, 2935 ср, 2910 ср, 2883 ср, 2823 сл, 1634 ср, 1622 ср, 1590 с, 1554 с, 1491 ср, 1443 ср, 1395 с, 1366 ср, 1333 ср, 1315 с, 1297 ср, 1265 ср, 1235 сл, 1220 ср, 1200 ср, 1175 ср, 1149 сл, 1102 сл, 1091 ср, 1079 ср, 1057 ср, 1026 ср, 967 ср, 958 ср, 949 сл, 923 сл, 890 ср, 858 сл, 830 ср, 823 ср, 809 ср, 766 с, 728 ср, 632 ср, 604 ср, 592 ср, 540 ср.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Миртеновая кислота – производное широко распространенных в природе оптически активных бициклических монотерпенов α- и β-пиненов. В данной работе она получена окислением коммерчески доступного миртенола диоксидом хлора (промышленный продукт, используемый для отбелки целлюлозы и очистки сточных вод) в среде пиридина. Выход кислоты после выделения через водорастворимую натриевую соль и очистки колоночной хроматографией на силикагеле составлял 35–37%.
Мы установили, что реакция хлорида кобальта(II) с миртенатом калия (получен in situ взаимодействием миртеновой кислоты и гидроксида калия) с последующим добавлением 2,3-лутидина (избыток не влияет на выход продукта) в этаноле приводила к образованию ярко-фиолетового раствора, из которого впоследствии были выделены фиолетовые кристаллы соединения I. При этом реакция раствора соединения I с миртенатом лития в EtOH приводила к образованию ярко-фиолетового раствора, из которого при концентрировании выпадали фиолетовые кристаллы гетерометаллического комплекса II.
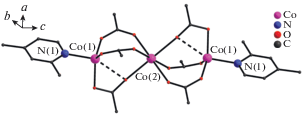
Согласно данным РСА комплекс I кристаллизуется в триклинной пр. гр. P$\bar {1}$, центр инверсии совпадает с координатами центрального атома Co(2) (рис. 1). Между собой пары атомов металлов (Co···Co 3.8192(11) Å) трехъядерной молекулы связаны тремя карбоксильными группами. Центральный атом Co(2) находится в октаэдрическом окружении шести атомов O мостиковых миртенат-анионов (Co–O 2.295(5)–2.322(5) Å). Окружение атома Co(1) представляет собой тетраэдр, состоящий из трех атомов миртенат-анионов (Co–O 1.927(5)–1.997(5) Å) и одного атома N молекул 2,4-лутидина (Co–N 2.085(5) Å). Также наблюдается формирование контакта атома Co(1) со вторым атомом O одной из мостиковых карбоксильных групп (Co(1)···O(6) 2.560(5) Å).
Рис. 1.
Молекулярное строение комплекса I (атомы водорода и циклические фрагменты аниона миртеновой кислоты не показаны).
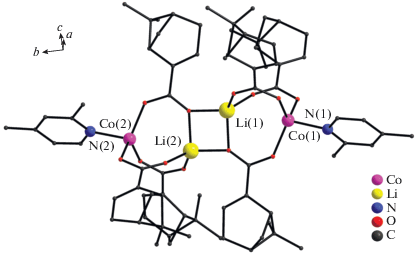
Комплекс II кристаллизуется в моноклинной пространственной группе P21. В молекуле комплекса (рис. 2) можно выделить два биядерных фрагмента {LiCo(Мyr)3(2,4-Lut)} (Co···Li 3.103(8), 3.134(8) Å), в которых атомы Li (Li···Li 2.722(8) Å) связаны между собой через два атомами O мостиковых карбоксилатных групп, формируя зигзагообразный Co–Li–Li–Co металлоостов (углы CoLiLi 122.0(3)°, 123.3(3)°). Геометрия центрального фрагмента Li2O2 близка к квадрату (Li–O 1.933(8)–1.976(8) Å, углы LiOLi 91.5(3)°, 92.9(3)°, OLiO 87.2(3)°, 88.3(3)°). Атомы кобальта и лития связаны друг с другом мостиковыми карбоксильными группами (Co–O 1.931(3)–1.963(3) Å; Li–O 1.868(8)–1.976(8) Å). Окружение каждого атома кобальта(II) достраивается до тетраэдра координацией атома N молекулы 2,4-лутидина (Co–N 2.067(4), 2.078(4) Å).
Комплекс I имеет строение, аналогичное ранее полученным комплексам с ароматическими кислотами и 2,3-лутидином [7, 8]. Можно было бы предположить, что существенную роль при формировании трехъядерной структуры играет π–π-сопряжение между O–C=O и C=C фрагментом заместителя при карбоксильной группе. Анализ структурных данных трехъядерных комплексов переходных металлов показал, что в случае анионов 2-нафтойной кислоты ориентация этих фрагментов может варьироваться от практически планарной до развернутой (торсионный угол OCC=C меняется от 5° до 73°) [7, 16]. В случае комплексов с анионами 9-антраценовой кислоты π–π-сопряжение не наблюдается (торсионный угол OCC=C меняется от 44° до 90°), что определяется взаимными стерическими напряжениями. Для сравнения также проанализированы торсионные углы в би- и трехъядрных комплексах с анионами стерически незатрудненных кротоновой и бензойной кислот, они лежат в интервалах 0°–15° [31–39] и 1°–24° [8, 40–43] соответственно. Значения торсионных углов OCC=C для вновь полученных соединений составляют 11.1(13)°–22.4(8)° для I и 2.61(4)°–17.57(3)° для II, что указывает на их близость с углами в биядерных комплексах с анионами кротоновой кислоты. Это показывает, что свободное вращение объемного заместителя в миртенатном анионе не затруднено.
Образование трехъядерного комплекса I даже на фоне избытка α-замещенного пиридина показывает, что в данном случае равновесие реакции комплексообразования смещено в сторону формирования линейного трехъядерного металлокарбосилатного остова. Подобная ситуация наблюдается при переходе от бензоатных комплексов к 2-нафтоатным: в случае бензоатов Co(II) с пиридином и его монодентатными производными можно получить оба типа комплексов [8, 44, 45], а в случае 2-нафтоатов при избытке 2,3-лутидина был выделен только линейный трехъядерный комплекс [7].
Таким образом, было показано, что анион миртеновой кислоты с кобальтом(II) в присутствии 2,3-лутидина формирует трехъядерный комплекс I и в присутствии ионов лития(I) – тетраядерный Li−Co гетерометаллический комплекс II. Анализ торсионных углов между O–C=O и C=C фрагментом заместителя при карбоксильной группе показал близость значений для вновь полученных соединений и би- и трехъядрных комплексов с анионами стерически незатрудненных кротоновой и бензойной кислот, что позволяет предположить отсутствие стерических препятствий для свободного вращения объемного заместителя в миртенатном анионе. Это не должно препятствовать образованию как описанного трехъядерного комплекса I, так и биядерного соединения, которое выделить в виде монокристаллов не удалось. Образование Li−Co комплекса II, аналога ранее полученных Li−M (M = Cu, Co, Ni, Zn) комплексов с анионами пивалиновой, трет-бутилуксусной и β-нафтойной кислот [46], подтверждает слабое влияние строения и природы заместителя при карбоксилатной группе на строение металлоостова в гетерометаллических соединения 3d-металлов.
Авторы заявляют, что у них нет конфликта интересов.
Список литературы
Фомина И.Г., Доброхотова Ж.В., Кискин М.А. и др. // Изв. АН. Сер. хим. 2007. № 9. С. 1650 (Fomina I.G., Dobrokhotova Zh.V., Kiskin M.A. et al. // Russ. Chem. Bull. 2007. V. 56. № 9. P. 1712).
Фомина И.Г., Доброхотова Ж.В., Александров Г.Г. и др. // Изв. АН. Сер. хим. 2007. № 9. С. 1660 (Fomina I.G., Dobrokhotova Zh.V., Aleksandrov G.G. et al. // Russ. Chem. Bull. 2007. V. 56. № 9. P. 1722).
Fomina I., Dobrokhotova Zh., Aleksandrov G. // Polyhedron. 2010. V. 29. P. 1734.
Сидоров А.А., Фомина И.Г., Александров Г.Г. и др. // Изв. АН. Сер. хим. 2004. № 2. С. 446 (Sidorov A.A., Fomina I.G., Aleksandrov G.G. et al. // Russ. Chem. Bull. 2004. V. 53. № 2. P. 483).
Uvarova M.A., Sinelshchikova A.A., Golubnichaya M.A. et al. // Cryst. Growth Des. 2014. V. 14. P. 5976.
Пахмутова Е.В., Малков А.Е., Михайлова Т.Б. и др. // Изв. АН. Сер. хим. 2003. № 10. С. 2006 (Pakhmutova E.V., Malkov A.E., Mikhailova T.B. et al. // Russ. Chem. Bull. 2003. V. 52. № 10. P. 2117).
Гольдберг А.Е., Кискин М.А., Николаевский С.А. и др. // Коoрд. химия. 2015. T. 41. № 3. С. 163 (Goldberg A.E., Kiskin M.A., Nikolaevskii S.A. et al. // Russ. J. Coord. Chem. 2015. V. 41. № 3. P. 182).https://doi.org/10.1134/S1070328415030021
Gavrilenko K.S., Punin S.V., Cador O. et al. // J. Am. Chem. Soc. 2005. V. 127. P. 12246.
Егоров Е.Н., Кискин М.А., Сидоров А.А., Еременко И.Л. // Изв. АН. Сер. хим. 2013. № 8. С. 1924 (Egorov E.N., Kiskin M.A., Sidorov A.A., Eremenko I.L. // Russ. Chem. Bull. 2013. V. 62. № 8. P. 1924).
Нефедов С.Е., Денисова Т.О., Долгушин Ф.М. // Изв. АН. Сер. хим. 2002. № 12. P. 2144 (Nefedov S.E., Denisova T.O., Dolgushin F.M. // Russ. Chem. Bull. 2002. V. 51. № 12. P. 2310).
Гольдберг А.Е., Николаевский С.А., Кискин М.А. и др. // Коорд. химия. 2015. Т. 41. № 12. С. 707 (Goldberg A.E., Nikolaevskii S.A., Kiskin M.A. et al. // Russ. J. Coord. Chem. 2015. V. 41. № 12. P. 777).https://doi.org/10.1134/S1070328415120015
Bu X.-H., Tong M.-L., Xie Y.-B. et al. // Inorg. Chem. 2005. V. 44. P. 9837.
Lehleh A., Beghidja A., Beghidja C. et al. // C. R. Chimie. 2015. V. 18. P. 530.
Bu X.-H., Tong M.-L., Xie Y.-B et al. // Inorg. Chem. 2005. V. 44. P. 9837.
Smolková R., Zeleňák V., Gyepes R. et al. // Polyhedron. 2018. V. 141. P. 230.
Гольдберг А.Е., Кискин М.А., Сидоров А.А., Еременко И.Л. // Изв. АН. Сер. хим. 2011. № 5. С. 829 (Goldberg A.E., Kiskin M.A., Sidorov A.A., Eremenko I.L. // Russ. Chem. Bull. 2011. V. 60. № 5. P. 849).
Xu M.-L., Zhou R., Wanga G.-Y., Ng S.W. // Acta Crystallogr. E. 2008. V. 64. P. m710.
Wang J.-J., Zhang R.-C., Cao Y.-L. et al. // New J. Chem. 2018. V. 42. P. 3551.
H. Chun. // J. Am. Chem. Soc. 2008. V. 130. P. 800.
Li Q., Yu P., Luo J., Qi C. et al. // Z. Anorg. Allg. Chem. 2017. V. 643. P. 166.
Yao Q., Sun J., Li K., Su J. et al. // Dalton Trans. 2012. V. 41. P. 3953.
Zhang Z., Nguyen H.T.H., Miller S.A et al. // J. Am. Chem. Soc. 2016. V. 138. № 3. P. 920.
Chun H., Bak W., Hong K., Moon D. // Cryst. Growth Des. 2014. V. 14. № 4. P. 1998.
Karmakar A., Sarma R.J., Baruah J.B. // Inorg. Chem. Commun. 2006. V. 9. P. 1169.
Clegg W., Little I.R., Straughan B.P. // Dalton Trans. 1986. P. 1283.
Clegg W., Hunt P.A., Straughan B.P. // Acta Crystallogr. C. 1995. V. 51. P. 613.
Clegg W., Little I.R., Straughan B.P. // Chem. Commun. 1985. P. 73.
Clegg W., Little I.R., Straughan B.P. // Inorg. Chem. 1988. V. 27. P. 1916.
SMART (control) and SAINT (integration) Software. Version 5.0. Madison (WI, USA): Bruker AXS Inc., 1997.
Sheldrick G.M. // Acta Crystallogr. A. 2008. V. 64. P. 112.
Wang Y.-Y., Shi Q., Shi Q.-Z. et al. // Polyhedron. 2000. V. 19. P. 891.
Demir S., Yolcu Z., Andac O. et al. // J. Mol. Model. 2010. V. 16. P. 1509.
Zhang X.-Y. // Acta Crystallogr. E. 2009. V. 65. P. m526.
Li X., Zhou B., Zhang J. et al. // Eur. J. Org. Chem. 2012. P. 1626.
Hamza F., Kickelbick G. // Macromolecules. 2009. V. 42. P. 7762.
Zhou C., Wang Y., Li D. et al. // Eur. J. Inorg. Chem. 2006. P. 2437.
Jana S., Cormack P.A.G., Kennedy A.R., Sherrington D.C. // Mater. Chem J. 2009. V. 19. P. 3427.
Wang Y., Liu P., Shi Q et al. // Chin. Sci. Bull. 2001. V. 46. P. 987.
Liu P., Wang Y.-Y., Li D.-S et al. // Chin. J. Chem. 2005. V. 23. P. 204.
Kwak H., Lee S.H., Kim S.H. et al. // Polyhedron. 2009. V. 28. № 3. P. 553.
Kwak H., Lee S.H., Kim S.H. et al. // Polyhedron. 2008. V. 27. № 17. P. 3484.
Hu D.-X., Chen P.-K., Luo F. et al. // Inorg. Chim. Acta. 2007. V. 360. № 15. P. 4077.
Gavrilenko K.S., Gal Y.L., Cador O. et al. // Chem. Commun. 2007. P. 280.
Catterick J., Hursthouse M.B., Thornton P., Welch A.J. // Dalton Trans. 1977. P. 223.
Catterick J., Hursthouse M.B., New D.B., Thornton P. // Chem. Commun.1974. P. 843.
Dobrokhotova Zh., Emelina A., Sidorov A. et al. // Polyhedron. 2011. V. 30. 132.
Дополнительные материалы отсутствуют.
Инструменты
Координационная химия