

Координационная химия, 2020, T. 46, № 1, стр. 31-39
Синтез серосодержащих производных цимантрена с потенциальными фото- и электрохимическими свойствами
Е. С. Келбышева 1, *, Ю. А. Гордей 2, М. Г. Езерницкая 1, А. Ф. Смольяков 1, 3, Л. Н. Телегина 1
1 Институт элементоорганических соединений им. А.Н. Несмеянова РАН
Москва, Россия
2 Российский химико-технологический университет им. Д. И. Менделеева
Москва, Россия
3 Российский экономический университет им. Г.В. Плеханова
Москва, Россия
* E-mail: kellena80@mail.ru
Поступила в редакцию 03.07.2019
После доработки 05.08.2019
Принята к публикации 15.08.2019
Аннотация
С целью получения серосодержащих производных цимантрена (циклопентадиенилмарганецтрикарбонил (Cym = C5H4Mn(CO)3)) изучено направление гидролиза изотиоурониевых солей алкилцимантренов в различных условиях, а также последующие химические превращения продуктов гидролиза этих солей. Установлено, что эффективным способом получения алкилцимантренилтиолов является гидролиз под действием NaOH изотиоурониевых солей в смеси EtOH–ацетон (4 : 1) в течение 2 ч, либо восстановление LiAlH4 соответствующих дисульфидов, образующихся с хорошими выходами при гидролизе изотиоурониевых солей в течение 6 ч. Изучены электрохимические свойства CymCH(CH3)SSCH(CH3)Cym до и после облучения в растворе светом Hg-лампы при λ = 365 нм. Методом РСА установлено строение комплексов CymCH(CH3)SSCH(CH3)Cym (I) и CymCH2SCH2Cym (II) (CIF file CCDC № 1921952 и 1921953 соответственно).
Изучение свойств различных металлоорганических соединений, в частности на основе циклопентадиенилмарганецтрикарбонила (цимантрена Cym = C5H4Mn(CO)3), показало их эффективность при использовании в каталитических [1], фотохимических [2], фотохромных [3], биологических [4], хемосенсорных [5] и других системах [6]. Таким образом, разработка методов введения в молекулу цимантрена разного рода функциональных групп является актуальной задачей. Тиолы и сульфиды – активные группы, легко подвергающиеся реакциям алкилирования [7] и трансалкилирования [8], окисления и восстановления [9], участвуют в фотореакциях [10], а также важны для поддержания окислительно-восстановительного баланса в белках [11], используются при создании флуоресцентных биозондов [12, 13], улавливателей ионов тяжелых металлов [14].
В настоящей работе с целью получения S-содержащих производных цимантрена, а именно тиолов, дисульфидов и сульфидов – CymCH(CH3)SSCH(CH3)Cym (I) и CymCH2SCH2Cym (II) – был изучен в различных условиях гидролиз соответствующих изотиоурониевых солей алкилцимантренов, являющийся ключевой стадией в получении тиопроизводных цимантрена. Также были проведены химические превращения продуктов гидролиза и их производных.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
В работе использовали безводный тетрагидрофуран (TГФ), полученный перегонкой над бензофенонкетилнатрием или натрием в атмосфере аргона. Применяемые в синтетических процедурах коммерчески доступные изоамилбромид (Aldrich) и гидразингидрат (Aldrich) использовались без дополнительной очистки. Синтез бромметилцимантрена (CymCH2Br) (III) и бромэтилцимантрена (CymCH(CH3)Br) (IV) проводили согласно [15].
Спектры ЯМР 1H и 13C (внутренний стандарт – растворитель (δ от Me4Si): ацетон – 2.05, хлороформ – 7.26, ДМСО – 2.50 м.д.) регистрировали на спектрометре Bruker Avance 400 (400.13 и 100.61 МГц соответственно). Сигналы в ЯМР 1H и 13C спектрах определяли при помощи экспериментов JMODECHO и 2D корреляционного анализа, в том числе COSY, NOESY. ИК-спектры регистрировали на Bruker IR Fourier спектрометре с разрешением 2 cм–1 в KBr ячейках. EI масс спектры регистрировали на Kratos MS 890 и Finnigan POLARIS Q спектрометрах при 70 эВ. Ход реакций и чистоту продуктов контролировали методом аналитической ТСХ на пластинах Silufol UV-245 (Kavalier). Для колоночной хроматографии использовали силикагель 60 (Merck, размер зерен 0.040–0.063 мм).
Синтез 2-цимантренилметил-2-тиопсевдомочевины гидробромида (V · HBr). К раствору III (3.1 г, 10.4 ммоль) в 25 мл ацетона при перемешивании в атмосфере аргона добавляли тиомочевину (0.9 г, 11.1 ммоль). Реакционную смесь перемешивали при 40°C в течение 3 сут. Растворитель удаляли в вакууме и остаток промывали EtOAc. Выход 3.7 г (96%).
| Найдено, %: | C 32.21; | H 3.00; | N 7.89; | S 8.78; | Br 21.41; | Mn 14.4. |
| Для C10H10N2O3SBrMn | ||||||
| вычислено, %: | C 32.19; | H 2.70; | N 7.52; | S 8.59; | Br 21.41; | Mn 14.7. |
ЯМР 1H (25°С, ДMСO-d6, δ, м.д. (J, Гц)): 4.16 (с., 2H, CH2), 5.00 (м., 2H, H–Cp), 5.20 (м., 2H, H–Cp), 9.05 (с., 1.5H, NH), 9.21 (с., 1.5H, NH).
Синтез 2-цимантренил-1-этил-2-тиопсевдомочевины гидробромида (VI · HBr). К раствору IV (5.0 г, 16.2 ммоль) в 25 мл ацетона при перемешивании в атмосфере аргона добавляли тиомочевину (1.5 г, 20.0 ммоль). Реакционную смесь перемешивали при 40°C в течение 3 сут. Растворитель удаляли в вакууме и остаток промывали EtOAc. Выход 5.9 г (95%).
| Найдено, %: | C 33.97; | H 3.03; | N 7.19; | S 8.04; | Br 21.54; | Mn 14.0. |
| Для C11H12N2O3SBrMn | ||||||
| вычислено, %: | C 34.13; | H 3.12; | N 7.24; | S 8.24; | Br 20.64; | Mn 14.2. |
ЯМР 1H (25°С, ацетон-d6, δ, м.д. (J, Гц)): 1.76 (д., 3H, CH3, J = 6.6), 4.89 (м., 1H, H-Cp), 5.03 (м., 1H, H–Cp), 5.12 (кв., 1H, CH, J = 6.6), 5.35 (м., 1H, H–Cp), 5.40 (м., 1H, H–Cp), 8.99 (с., 1.5H, NH), 10.02 (с., 1.5H, NH).
Общая методика гидролиза изотиомочевин (A). К раствору соответствующей изотиомочевины V · HBr rили VI · HBr (1 экв.), растворенной в 40 мл соответствующего растворителя (смесь EtOH–ацетон в разных соотношениях), добавляли раствор NaOH (2 экв.) в 10 мл Н2О. Реакционную смесь перемешивали при кипячении в течение 2–6 ч. Затем добавляли 100 мл Н2О и водный раствор экстрагировали EtOAc (3 × 100 мл). Органические слои промывали водой и насыщенным раствором NaCl и сушили над Na2SO4.
Cинтез цимантренилметилтиола (VII) выполняли по общей методике гидролиза изотиомочевин (A) исходя из V · HBr (3.0 г, 8.0 ммоль) и NaOH (0.6 г, 15.7 ммоль) в смеси растворителей EtOH–ацетон (4 : 1) и кипятили в течение 2 ч. Выход 1.2 г (60%).
ЯМР 1H (25°С, ацетон-d6, δ, м.д. (J, Гц)): 3.61 (с., 2H, CH2), 4.94 (м., 2H, H–Cp), 5.13 (м., 2H, H–Cp).
Cинтез цимантренил-1-этилтиола (VIII) выполняли по общей методике гидролиза изотиомочевин (A) исходя из VI · HBr (6.0 г, 15.7 ммоль) и NaOH (1.2 г, 31.4 ммоль) в смеси растворителей EtOH–ацетон (4 : 1) и кипятили в течение 2 ч. Выход 0.8 г (20%).
ЯМР 1H (25°С, ацетон-d6, δ, м.д. (J, Гц)): 3.61 (с., 2H, CH2), 4.94 (м., 2H, H–Cp), 5.13 (м., 2H, H–Cp).
Cинтез ди(цимантренилметил)дисульфида (IX) выполняли по общей методике гидролиза изотиомочевин (A) исходя из V · HBr (1.0 г, 2.7 ммоль) и NaOH (0.2 г, 5.3 ммоль) в смеси растворителей EtOH–ацетон (1 : 1) и кипятили в течение 6 ч. Выход 0.5 г (36%); Тпл = 78–79°C (гексан–EtOAc).
| Найдено, %: | C 43.40; | H 2.31; | S 12.93; | Mn 21.9. |
| Для C18H12O6S2Mn2 | ||||
| вычислено, %: | C 43.37; | H 2.41; | S 12.85; | Mn 22.1. |
ЯМР 1H (25°С, CDCl3, δ, м.д. (J, Гц)): 3.42 (с., 2H, CH2 4.75 (м., 2H, H–Cp), 4.85 (м., 2H, H–Cp). ЯМР 13C (25°С, CDCl3, δ, м.д. (J, Гц)): 30.17 (CH2), 82.14 (2С–Cp), 83.28 (2С–Cp), 101.29 (Ci–Cp), 224.83 (3CO).
Cинтез ди(цимантренил-1-этил)дисульфида (I) выполняли по общей методике гидролиза изотиомочевин (A) исходя из VI · HBr (2.0 г, 8.4 ммоль) и NaOH (0.4 г, 15.7 ммоль) в смеси растворителей EtOH–ацетон (1 : 1) и кипятили в течение 6 ч.
Изомер 1: выход 1.6 г (38%); Тпл = 67–68°C (гексан–EtOAc).
| Найдено, %: | C 45.58; | H 3.21; | S 11.98; | Mn 20.8. |
| Для C20H16O6S2Mn2 | ||||
| вычислено, %: | C 45.64; | H 3.06; | S 12.18; | Mn 20.9. |
ЯМР 1H (25°С; CDCl3; δ, м.д. (J, Гц)): 1.53 (д., 6H, CH3, J = 7.0), 3.46 (кв., 2H, CH, J = 7.0), 4.63 (м., 2H, H–Cp), 4.80 (м., 2H, H–Cp), 4.86 (м., 2H, H–Cp), 4.93 (м., 2H, H–Cp). ЯМР 13C (25°С; CDCl3; δ, м.д. (J, Гц)): 20.07 (2C–CH3), 43.06 (2СН), 79.47 (C–Cp), 83.13 (C–Cp), 83.45 (C–Cp), 83.52 (C–Cp), 105.12 (Ci–Cp), 224.58 (3C–CO).
Изомер 2: выход 1.5 г (35%); Тпл = 82–83°C (гексан–EtOAc).
| Найдено, %: | C 45.41; | H 3.01; | S 11.80; | Mn 20.6. |
| Для C20H16O6S2Mn2 | ||||
| вычислено, %: | C 45.64; | H 3.06; | S 12.18; | Mn 20.9. |
ЯМР 1H (25°С; CDCl3; δ, м.д. (J, Гц)): 1.52 (д., 6H, CH3, J = 7.0), 3.45 (кв., 2H, CH, J = 7.0), 4.62 (м., 2H, H–Cp), 4.81 (м., 2H, H–Cp), 4.89 (м., 2H, H–Cp), 4.92 (м., 2H, H–Cp). ЯМР 13C (25°С; CDCl3; δ, м.д. (J, Гц)): 20.10 (2C–CH3), 43.02 (2C–СН), 79.43 (C–Cp), 83.14 (C–Cp), 83.43 (2C–Cp), 105.25 (Ci–Cp), 224.59 (3C–CO).
Cинтез (дицимантренилметил)сульфида (II) выполняли по общей методике гидролиза изотиомочевин (A) исходя из V (1.0 г, 2.7 ммоль) и NaOH (0.2 г, 5.3 ммоль) в смеси растворителей EtOH–ацетон (1 : 1) и кипятили в течение 6 ч. Выход 0.25 г (20%); Тпл = 81–82°C (гексан–EtOAc).
| Найдено, %: | C 46.36; | H 2.51; | S 6.84; | Mn 23.4. |
| Для C18H12O6SMn2 | ||||
| вычислено, %: | C 46.37; | H 2.59; | S 6.88; | Mn 23.6. |
ЯМР 1H (25°С; CDCl3; δ, м.д. (J, Гц)): 3.38 (с., 2H, CH2), 4.69 (м., 2H, H–Cp), 4.77 (м., 2H, H–Cp). ЯМР 13C (25°С; CDCl3; δ, м.д. (J, Гц)): 36.65 (С–CH2), 82.39 (2C–Cp), 83.81 (2C–Cp), 99.79 (Ci–Cp), 224.52 (3С–CO).
Синтез цимантренилметилизоамилсульфида (Х). К суспензии K2CO3 (5.5 г, 40 ммоль) в 20 мл ДМФА при перемешивании в атмосфере аргона добавляли VII (2.0 г, 8.0 ммоль) в 10 мл ДМФА и выдерживали 30 мин. Затем к реакционной смеси добавляли по каплям изоамилбромид (1.5 мл, 12 ммоль) и грели при 60°С в течение 6 ч. Полученную реакционную смесь выливали в ледяную воду (100 мл) и продукт экстрагировали EtOAc (3 × 75 мл), органический слой сушили над MgSO4. Растворитель удаляли в вакууме и Х выделяли с помощью колоночной хроматографии (элюент: гексан–EtOAc (4 : 1)). Выход 1.5 г (60%).
| Найдено, %: | C 52.38; | H 5.52; | S 9.86; | Mn 17.0. |
| Для C14H17O3SMn | ||||
| вычислено, %: | C 52.50; | H 5.35; | S 10.01; | Mn 17.2. |
ЯМР 1H (25°С; ацетон-d6, δ, м.д. (J, Гц)): 0.80 (д., 6H, CH3, J = 6.5), 1.48 (кв., 2H, CH2, J = 7.8), 1.68 (м., 1H, CH), 2.61 (т., 2H, CH2, J = 7.6), 3.41 (с., 2H, CH2–S), 4.88 (м., 2H, H–Cp), 5.01 (м., 2H, H–Cp). ЯМР 13C (25°С; CDCl3; δ, м.д. (J, Гц)): 22.29 (2С–СН3), 27.47 (С–СН), 28.50 (С–CH2), 30.29 (С–CH2S), 38.19 (С–CH2S), 82.06 (2C–Cp), 83.01 (2C–Cp), 102.67 (Ci–Cp), 224.25 (3С–CO).
Синтез цимантренилэтилизоамилсульфида (ХI) выполняли аналогично Х исходя из VIII (2.0 г, 8.0 ммоль), K2CO3 (5.5 г, 40 ммоль) и изоамилбромидa (1.5 мл, 12 ммоль). Выход 1.2 г (44%).
| Найдено, %: | C 53.78; | H 5.48; | S 9.09; | Mn 16.5. |
| Для C15H19O3SMn | ||||
| вычислено, %: | C 53.89; | H 5.73; | S 9.59; | Mn 16.4. |
ЯМР 1H (25°С; CDCl3; δ, м.д. (J, Гц)): 0.92 (д., 6H, CH3, J = 6.5), 1.45 (м., 2H, CH2, J = 7.8), 1.52 (д., 3Н, СН3), 1.68 (м., 1H, CH), 2.55 (м., 2H, CH2), 3.41 (м., 1H, CH–S), 4.59 (м., 1H, H–Cp), 4.75 (м., 1H, H–Cp), 4.79 (м., 1H, H–Cp), 4.90 (м., 1H, H–Cp). ЯМР 13C (25°С; CDCl3; δ, м.д. (J, Гц)): 21.65 (C–CH3), 22.28 (2С–СН3), 27.57 (С–СН), 29.17 (С–CH2), 36.81 (С–CH), 38.34 (С–CH2S), 79.27 (C–Cp), 82.78 (C–Cp), 82.83 (C–Cp), 82.99 (C–Cp), 107.91 (Ci–Cp), 225.14 (3С–CO).
Общая методика восстановления сульфидов и дисульфидов LiAlH4 (B). К суспензии LiAlH4 (1.0 экв.) в 100 мл ТГФ в атмосфере аргона добавляли по каплям соответствующий сульфид или дисульфид (1.0 экв.) в 40 мл ТГФ при температуре реакционной смеси 30°С. Перемешивали в течение 3 ч при комнатной температуре. В реакционную смесь добавляли 300 мл эфира, охлажденного до 0°С, нейтрализовали насыщенным раствором NH4Cl (25 мл), подкисляли 15%-ным раствором HCl до рН 5.5. Осадок отделяли декантацией, растворяли в 15%-ной HCl и раствор экстрагировали EtOAc (3 × 200 мл). Органические слои объединяли и промывали H2O (2 × 200 мл). Сушили над Na2SO4, упаривали. Остаток очищали методом колоночной хроматографии (элюент: гексан–EtOAc (2 : 1)).
Общая методика получения несимметричных сульфидов из изотиоурониевых солей в присутствии KOH и гидразингидрата (C). К 1 экв. изотиурониевой соли прибавляли 1 экв. соответствующего бромида и при перемешивании медленно добавляли раствор КОН (2 экв.) в 6 мл гидразингидрата. Смесь перемешивали 11 ч при 20–25°С, экстрагировали дихлорметаном. Экстракт сушили над MgSO4. Продукт очищали с помощью колоночной хроматографии (элюент: гексан–EtOAc (3 : 1)). Спектральные характеристики и данные элементного анализа полностью идентичны описанным выше несимметричным сульфидам.
Общая методика получения симметричных сульфидов из цимантренилалкилбромидов в присутствии этаноламина и серы (D). К смеси 1 экв. моноэтаноламина и 10 экв. гидразингидрата прибавляли 3 экв. порошкообразной серы. Смесь перемешивали 2 ч при 80–85°С, охлаждали до комнатной температуры и добавляли порциями при перемешивании 3 экв. соответствующего цимантренилалкилбромида. Перемешивали реакционную смесь в течение 6 ч при комнатной температуре. Продукты экстрагировали CH2Cl2, сушили над MgSO4 и очищали с помощью колоночной хроматографии (элюент: гексан–EtOAc (3 : 1)) с последующей раскристаллизацией.
РСА. Экспериментальные наборы данных получены на дифрактометре SMART APEX II [16] (графитовый монохроматор, λ(MoKα) = 0.71073 Å, ω-сканирование). Структуры расшифрованы прямым методом и уточнены в полноматричном анизотропном приближении по F 2 для неводородных атомов. Позиции атомов водорода рассчитаны исходя из геометрических соображений и уточнены изотропно наложением ограничений на длины связей C–H и величины их эквивалентных тепловых параметров (Uэкв(H)). Для метильных групп расстояния С–Н зафиксированы на 0.96 Å, величины Uэкв(H) равны 1.5 Uэкв(С), где Uэкв(С) – атом углерода метильной группы. В случае остальных атомов водорода эти величины составляют 0.92 Å и 1.2 Uэкв(С). Все расчеты проведены с использованием комплекса программ SHELTL PLUS 5.1 и SHELX-2014 [17, 18]. Основные кристаллографические данные и параметры рентгеноструктурного эксперимента для соединений I и II приведены в табл. 1.
Таблица 1.
Кристаллографические данные и параметры уточнения для соединений I и II
| Параметр | Значение | |
|---|---|---|
| I | II | |
| М | 526.33 | 466.22 |
| T, K | 120 | 120 |
| Cингония | Pомбическая | Mоноклинная |
| Пр. гр. | P212121 | P21/c |
| Z | 4 | 4 |
| a, Å | 6.9630(4) | 7.1315(6) |
| b, Å | 13.6145(7) | 10.7852(8) |
| c, Å | 22.8148(12) | 23.2587(18) |
| β, град | 90 | 96.5250(10) |
| V, Å3 | 2162.8(2) | 1777.3(2) |
| ρ(выч.), г см–3 | 1.616 | 1.742 |
| μ, см–1 | 13.94 | 15.71 |
| F(000) | 1064 | 936 |
| 2θmax, град | 50 | 50 |
| Число измеренных отражений | 26 421 | 22 972 |
| Число независимых отражений | 5758 | 5515 |
| Число отражений с I > 2σ(I) | 5363 | 4903 |
| Количество уточняемых параметров | 274 | 244 |
| R1, wR2 (I > 2σ(I)) | 0.0361, 0.0826 | 0.0324, 0.0799 |
| R1, wR2 (по всем отражениям) | 0.0397, 0.0849 | 0.0378, 0.0837 |
| GOОF | 0.999 | 1.032 |
| Остаточная электронная плотность (max/min), e Å–3 | 1.428/–0.732 | 0.628/–0.658 |
Координаты атомов и другие параметры структуры I и II депонированы в Кембриджском банке структурных данных (№ 1921952 и 1921953 соответственно); deposit@ccdc.cam.ac.uk или http:// www.ccdc.cam.ac.uk/data_request/cif).
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Синтез цимантренилалкилтиолов осуществляли стандартным методом, используемым для получения бензилтиолов [19], гидролиза соответствующих изотиоурониевых солей алкилцимантренов. Изотиоурониевые соли CymCH2SC(NH)NH2. HBr (V ∙ HBr) и CymCH(CH3)SC(NH)NH2. HBr (VI ∙ HBr) получали с количественным выходом из III и IV и тиомочевины (cхема 1a). Проведение их последующего гидролиза в присутствии NaOH при кипячении осуществлялось, в отличие от описанной в [19] методики, в органических растворителях в связи с абсолютной нерастворимостью цимантренильных солей в воде. Гидролиз соединений V ∙ HBr и VI ∙ HBr в смеси EtOH–ацетон (4 : 1) в течение 2 ч приводит к получению цимантренилалкилтиолов CymCH2SH (VII) и CymCH(CH3)SH (VIII) с выходами 60 и 20% соответственно (cхема 1a). Однако кроме тиолов VII и VIII наблюдалось образование побочных продуктов.
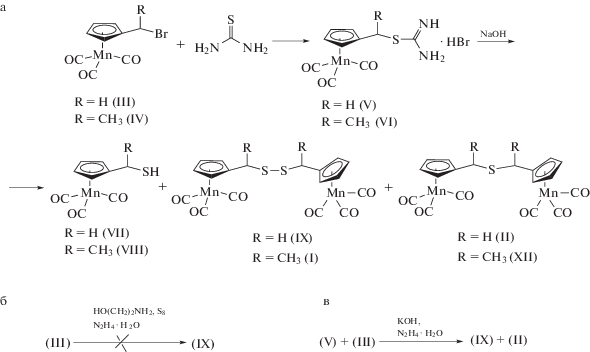
Схема 1 .
Изменение условий гидролиза (время реакции и соотношение растворителей) позволяло смещать направление реакции к тем или иным продуктам (табл. 2). Например, при проведении гидролиза V ∙ HBr в смеси растворителей EtOH–ацетон (1 : 1) в течение 2 ч происходит преимущественное образование тиола VII (выход 54%), а выход побочных продуктов CymCH2SSCH2Cym (IX) и II оказывается малым (12% для соединения IX и 7% для II), а их соотношении составляет 2 : 1 (табл. 2). Увеличение времени протекания реакции до 6 часов приводит к образованию исключительно продуктов IX и II (выход 36% для соединения IX и 20% для II) (табл. 2). В случае гидролиза рацемической изотиоурониевой соли VI ∙ HBr наблюдается образование только тиола VIII и одного побочного продукта реакции I, который нам удалось разделить на две фракции переосаждением из 96%-ного EtOH, представляющие собой мезо-форму и смесь энантиомеров. Изменение времени протекания реакции и соотношения растворителей не приводят к появлению второго продукта XII, но изменяет соотношение соединений VIII : I с 1 : 1 при кипячении в течение 2 ч на 0 : 1 при проведении реакции в течение 6 ч. Таким образом, увеличение времени гидролиза до 6 ч в обоих случаях приводят к получению только побочных продуктов.
Таблица 2.
Соотношение продуктов IX к II в реакции щелочного гидролиза изотиуроновой соли V
| Время реакции, ч | Соотношение EtOH : ацетон | ||
|---|---|---|---|
| 4 : 1 | 1 : 1 | 1 : 4 | |
| 2 | 5 : 1 | 2 : 1 | 3 : 1 |
| 4 | 6 : 1 | 2 : 1 | 4 : 1 |
| 6 | 6 : 1 | 2 : 1 | 4 : 1 |
Строение всех полученных соединений было доказано методами ЯМР 1Н и 13С, масс-, ИК-спектрометрией, элементным анализом, а также в случае соединений I и II методом РСА. Анализ спектров ЯМР 1Н соединений IX и II показал, что сигналы от протонов SСН2-группы и Ср-кольца практически совпадают (табл. 3). При этом существенная разница в положении сигналов от углерода SСН2-группы наблюдается в спектрах ЯМР 13С для IX и II и сигналы находятся при 30.17 и 36.65 м.д. соответственно. Из литературных данных [20, 21] известно, что наличие в молекуле двух атомов серы сдвигает сигналы от углерода СН2-группы в слабое поле по сравнению с моносульфидом. В нашем случае наблюдается аналогичная картина (табл. 3). Спектры 1Н и 13С для двух энантиомерных форм I практически идентичны с максимальной разницей в химических сдвигах 0.02 м.д. для всех протонов в спектрах ЯМР 1Н и 0.13 м.д. для химического сдвига для Ci–Ср углерода.
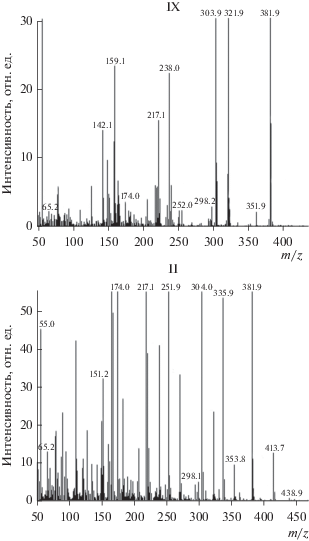
Анализ масс-спектров моно- и дисульфидов показал близкую картину. Сульфиды обычно характеризуются интенсивным пиком молекулярного иона. Однако в случае производных циклопентадиенилмарганецтрикарбонила редко удается зафиксировать молекулярный ион при ионизации методом электронного удара, что связано с моментальной потерей групп СО (одной или нескольких) [22, 23]. Для соединения II были зафиксированы интенсивный пик с m/z 382 [M–3СО] и пики низкой интенсивности m/z 438 [M–СО], 354 [M–4СО] и 298 [M–6СО] (pис. 1). Наблюдаются интенсивные молекулярные пики m/z 217 [M–SCH2Cym] и 251 [M–CH2C5H4 + 2]. Известно, что дисульфиды в случае ионизации электронным ударом выбрасывают один или два атома серы [24]. В масс-спектре IX имеется интенсивный пик иона m/z 382. Таким образом, в масс-спектре IX мы наблюдаем пики ионов аналогичные и для II (pис. 1). Пик с m/z 322 соответствует сдваиванию двух цимантренилметильных фрагментов с потерей четырех СО. Описанная фрагментация, а также наличие большого количества других интенсивных пиков характерна для поливариативных процессов, протекающих при фрагментации органических соединений серы (сульфидов и дисульфидов) под электронным ударом [24]. Масс-спектры двух мезо-форм I совпадают по фрагментации. В данном случае первоначально происходит разрыв связи S–S, что приводит к образованию ионов: [SC2H5C5H4Mn(CO)3 + 2], [SC2H5C5H4Mn] и [C2H5C5H4Mn] с m/z 265, 180, 162.
Структура комплексов I и II подтверждена методом РСА (pис. 2). В обоих комплексах основные длины связей и валентные углы лежат в узком интервале и близки к комплексам цимантренильного типа [25]. Проведeнное структурное исследование установило, что соединение I представляет собой рацемический двойник. В кристалле I комплекс находится в общем положении. Отметим, что длины связей С–С в циклопентадиенильном кольце альтернированы. Наибольшая длина связи С–С 1.423(1), к которой карбонильная группа С=О находится в цис-положении (рис. 2). Данный факт характерен для циклопентадиенильных соединений марганца, что ранее было описано в работе Хофмана [26]. В кристалле I молекулы образуют зигзагообразные цепочки вдоль кристаллографической оси 21. В отличие от молекулы I, имеющей хиральные центры, соединение II хиральных центров не имеет и кристаллизуется в центросимметричной пр. гр. Молекула II так же как и I образует зигзагообразные цепочки вдоль кристаллографической оси c. Длины связей С–С в циклопентадиенильном кольце также как и в предыдущем случае альтернированы, что согласуется с результатами работы Хофмана.
Рис. 2.
Структуры соединений I и II и их основные длины связей и валентные углы. Для I: Mn(1)–С(1) 2.151, Mn(1)–C(6) 1.796, Mn(2)–C(12) 2.135, Mn(2)–C (17) 1.787, S(1)–S(2) 2.040, S(1)–C(9) 1.849, S(2)–C(11) 1.851, C(1)–C(9) 1.496, C(11)–C(12) 1.502 Å и C(1)Mn(1)C(6) 111.8°, C(12)Mn(2)C(17) 93.6°, Mn(1)C(1)C(9) 128.62°, Mn(2)C(12)C(11) 125.53°, S(2)S(1)C(9) 105.21°, S(1)S(2)C(11) 103.00°; для II: Mn(1)–С(1) 2.153 Mn(1)–C(6) 1.798, Mn(2)–C(11) 2.153, Mn(2)–C(16) 1.797, S(1)–C(9) 1.816, S(2)–C(10) 1.824, C(1)–C(9) 1.496, C(10)–C(11) 1.498 Å и C(1)Mn(1)C(6) 95.54°, C(11)Mn(2)C(16) 91.96°, Mn(1)C(1)C(9) 129.11°, Mn(2)C(11)C(10) 125.81°, C(9)S(1)C(10) 98.54°.
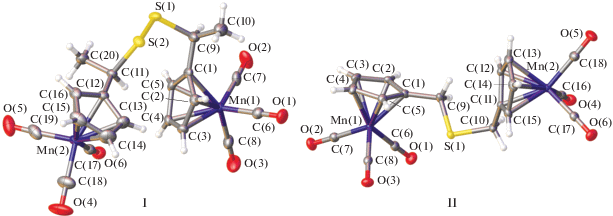
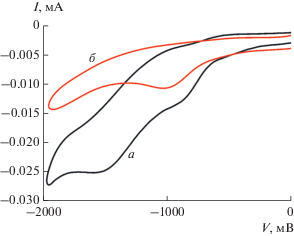
Рис. 3.
Циклическая вольтамперограмма в 0.1 М растворе тетрафторбората тетраэтиламмония: в присутствии 5.2 × 10–3 М соединения I (a), после облучения соединения I в течение 6 мин (б). Скорость сканирования 100 мВ/с.
Были проведены попытки селективного выделения моносульфида II и дисульфида IX с использованием разработанных ранее эффективных методик для органических аналогов [27]. Метод для получения дисульфидов заключался в восстановлении элементной серы до дисульфид-анионов с использованием системы гидразингидрат–моноэтаноламин [28]. Однако в нашем случае дисульфид выделен не был (схема 1 б), в связи с заменой бромид-иона на OH-группу в данных условиях и выделением с 74%-ным выходом гидроксиметилцимантрена. Селективный метод получения симметричных и несимметричных моносульфидов [27] в нашем случае также оказался неэффективен. Взаимодействие изотиоуроновой соли V ∙ HBr с III приводит к образованию смеси дисульфида IX и моносульфида II в соотношении 1 : 1 (выходы 18 и 19% соответственно) (схема 1 в). Соединения IX и II раскристаллизовывали из смеси гексан–EtOAc (5 : 1).
Мы предполагаем, что образование сульфидов и дисульфидов во всех реакциях связано с тем, что первоначально в ходе гидролиза образуется тиол, который окислительно сдваивается за счет взаимодействия атомов серы с атомом марганца. Это предположение подтверждается тем, что различные комплексы цимантрена могут выступать в качестве катализаторов в реакции фотопревращения тиолов в дисульфиды [1, 29]. Факт участия марганца в реакции сдваивания также косвенно подтверждается тем, что в наших условиях гидролиз бензилизотиоурониевой соли не приводит к образованию какого-либо количества продуктов сдваивания, а образуется исключительно бензилтиол с выходом более 94% независимо от времени гидролиза. А гидролиз бензилизотиоурониевой соли [30] NaOH в смеси растворителей EtOH–ацетон (1 : 1) в течение 6 ч в присутствии 1 мол. экв. цимантрена приводит к появлению в реакционной смеси симметричного дибензилдисульфида с выходом 24%.
Дисульфиды IX и I, в отличие от II, легко восстанавливаются литийалюминийгидридом до соответствующих тиолов VII и VIII с практически количественным выходом. Тогда как восстановление литийалюминийгидридом соединения II приводит к развалу и выделению метилцимантрена [31] с выходом 62%, что подтверждает предполагаемую структуру соединений.
Алкилирование тиолов VII и VIII проводили по стандартной методике в ДМФА в присутствии калия двууглекислого (cхема 2). Выходы изоамилцимантренилметилсульфида CymCH2SC5H11 (X) и изоамилцимантренилэтилсульфида CymCH(CH3)SC5H11 (XI) 60 и 44% соответственно.
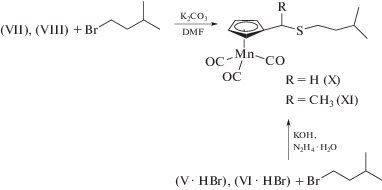
Схема 2 .
Более эффективным оказался способ получения несимметричного сульфида X из изотиурониевой соли V. Превращение V в X осуществляли в системе гидразингидрат–щелочь в соответствии с методикой, изложенной в статье [27]. Выход сульфида X по данной методике составил 65%, тогда как общий выход X по двустадийной методике, описанной выше, был в 2 раза ниже (36%) (cхема 2).
Данные, полученные на основе циклической вольтамперометрии, показывают, что восстановление соединения I проходит в две стадии с последовательным участием двух электронов (pис. 3). Значение потенциала у катодного пика на кривой (–1.49 В) оказывается близко к значению потенциала катодного пика дифенилдисульфида [32]. После облучения светом Hg лампы при λ = 365 нм в течение 6 мин этот пик полностью исчезает и остается только пик при –0.9 В (pис. 3), вид циклической вольтамперограммы остается без изменений в течение 3 ч после облучения. Изменения вольтамперограммы при фотолизе соединения I связаны с тем, что происходит отрыв лиганда СО от атома марганца. Это приводит к образованию стабильного дикарбонильного комплекса, который восстанавливается в одну стадию с переносом одного электрона.
Таким образом, были разработаны методы получения ряда серосодержащих производных цимантрена с различными функциональными группами, такими как меркапто, сульфидная и дисульфидная, изучено направление реакций гидролиза соответствующих изотиурониевых солей. Было показано, что полученные соединения могут быть легко модифицированы с целью создания фотохромных систем на их основе. Изучены электрохимические свойства соединения I до и после облучения.
Авторы заявляют об отсутствии конфликта интересов.
Список литературы
Fraser R., van Rooyen P.H., de Lange J. et al. // J. Organomet. Chem. 2017. V. 840. P. 11.
Maldonado T., Ferraudi G., Lappin A.G. et al. // ChemPhotoChem. 2018. V. 2. P. 1.
Kelbysheva E.S., Telegina L.N., Rodionov A.N. et al. // Eur. J. Inorg. Chem. 2016. V. 23. P. 3767.
Ravera M., Moreno-Viguri E., Paucar R. et al. // Eur. J. Med. Chem. 2018. V. 155. P. 459.
Dewangan S., Barik T., Mishra S. et al. // Appl. Organomet. Chem. 2018. P. 1.
Assim K., Jeschke J., Jakob A. et al. // Thin Solid Films. 2016. V. 619. P. 265.
Khurana J.M., Sahoo P.K. // Synth. Commun. 1992. V. 22. № 12. P. 1691.
Nawrot D., Kolenic M., Kunes J. et al. // Tetrahedron. 2018. V. 74. P. 594.
Zeida A., Babbush R., González Lebrero M.C. et al. // Chem. Res. Toxicol. 2012. V. 25. № 3. P. 741.
Banuls M.-J., Gonzalez-Martínez M. A., Sabek J. et al. // Anal. Chim. Acta. 2019. V. 1060. P. 103.
Rudyk O., Eaton Ph. // Redox Biology. 2014. V. 2. P. 803.
Jianjun Du, Weijun Ma, Quanyong Gu et al. // Sensors Actuators. B. 2019. V. 287. P. 118.
Deyan Gong, Jiaxi Ru, Ting Cao et al. // Sensors Actuators. B. 2018. V. 258. P. 72.
Sharma P., Mourya M., Choudhary D. et al. // Sensors Actuators. B. 2018. V. 268. P. 310.
Келбышева Е.С., Телегина Л.Н., Ершова Е.А. и др. // Изв. АН. Сер. хим. 2017. № 2. С. 327.
APEX II. Software package. Madison (WI, USA): Bruker AXS Inc., 2005.
Sheldrick G.M. // Acta Crystallogr. A. 2008. V. 64. P. 112.
Sheldrick G.M. // Acta Crystallogr. C. 2015. V. 71. P. 3.
Zhdanko A.G., Gulevich A.V., Nenajdenko V.G. // Tetrahedron. 2009. V. 65. P. 4692.
Enthaler S. // ChemCatChem. 2011. V. 3. P. 666.
Benevides P.J.C., Young M.C.M., Giesbrecht A.M. et al. // Phytochemistry. 2001. V. 57. P. 743.
Nekrasov Yu.S., Sukharev Yu.N., Tepfer E.E. et al. // Eur. J. Mass Spectr. 2002. V. 8. № 3. P. 247.
Sizoi V.F., Nekrasov Yu.S., Sukharev Yu.N. et al. // J. Organomet. Chem. 1980. V. 202. P. 83.
Лебедев А.Т. Масс-спектрометрия в органической химии. М.: БИНОМ лаборатория знаний, 2003. 493 с.
Smol’yakov A.F., Dolgushin F.M., Ginzburg A.G. et al. // J. Mol. Struct. 2012. V. 1014. P. 81.
Albright T.A., Hofmann P., Hoffmann R. // J. Am. Chem. Soc. 1977. V. 99. P. 7546.
Леванова Е.П., Вахрина В.С., Грабельных В.А. и др. // Журн. орган. химии. 2015. Т. 51. № 2. С. 175.
Дерягина Э.Н., Руссавская Н.В., Паперная Л.К. и др. // Изв. АН. Сер. хим. 2005. № 11. С. 2395.
Tan K.Y.D., Kee J.W., Fan W.Y. // Organometallics. 2010. V. 29. P. 4459.
Hickey S.M., White J.M., Pfeffer F.M. et al. // Synlett. 2015. V. 26. № 12. P. 1759.
Linehan J.C., Wallen S.L., Yonker C.R. et al. // J. Am. Chem. Soc. 1997. V. 119. P. 10170.
Borsari M., Cannio M., Gavioli G. // Electroanalysis. 2003. V. 15. P. 1192.
Дополнительные материалы отсутствуют.
Инструменты
Координационная химия