

Координационная химия, 2020, T. 46, № 12, стр. 723-732
3,6-ди-трет-бутил-2-гидрокси-4-пиридинилфенолят и комплексы олова(IV) на его основе: особенности синтеза и строения, сольватохромный эффект
А. В. Пискунов 1, *, К. И. Пашанова 1, К. А. Мартьянов 1, К. В. Арсеньева 1, А. В. Черкасов 1
1 Институт металлоорганической химии им. Г.А. Разуваева РАН
Нижний Новгород, Россия
* E-mail: pial@iomc.ras.ru
Поступила в редакцию 22.05.2020
После доработки 17.07.2020
Принята к публикации 20.07.2020
Аннотация
Разработано два синтетических подхода для получения 3,6-ди-трет-бутил-2-гидрокси-4-пиридинилфенолята (LH) – нового цвиттер-ионного редокс-активного лиганда диолатного типа. На его основе получены два гетеролигандных производных пентакоординированного олова(IV): 3,6-ди-трет-бутил-2-окси-4-пиридинилфенолято-трифенилолово(IV) (I) и 3,6-ди-трет-бутил-2-окси-4-пиридинилфенолято-дифенилхлороолово(IV) (II). Молекулярное строение лиганда LH ∙ 0.5Py и комплекса I ∙ CH3CN установлено методом РСА (CIF files CCDC № 1974166 (LH), 1974165 (I)). Показано, что лиганд LH и соединения олова(IV) на его основе демонстрируют явление сольватохромии, заключающееся в значительном гипсохромном сдвиге при увеличении полярности растворителя.
Соединения о-бензохинонового типа представляют значительный интерес для элементоорганических и координационных соединений [1], выступая в качестве редокс-активных лигандов, способных находиться в координационной сфере металлоцентра в трех окислительных состояниях: дианионном (катехолатном), анион-радикальном (семихиноновом) и нейтральном. Особый интерес органические лиганды такого типа представляют для соединений непереходных металлов, поскольку позволяют существенно расширить окислительно-восстановительные свойства соединений элементов главных подгрупп [2, 3].
Так, металлокомплексы, содержащие в своем составе парамагнитную форму лиганда указанного типа, представляют несомненный интерес с точки зрения проявляемых магнитных обменных взаимодействий как внутри-, так и межмолекулярного характера [4]. Кроме этого, уникальная способность бензохиноновых производных как редокс-активных соединений к обратимому переходу между окислительными состояниями, будучи в координационном окружении металлоцентра, позволяет наблюдать феномен валентной таутомерии [5], а также осуществлять дизайн электронной структуры металлокомплексов посредством окислительно-восстановительных превращений [6]. Вовлечение редокс-активных лигандов, связанных с комплексообразователем, в окислительно-восстановительные превращения находит применение в целом ряде стехиометрических и каталитических реакций, протекающих без изменения степени окисления комплексообразователя [7–11]. Примечательно, что большинство известных координационных соединений с подобного типа лигандными системами сформировано одно- и двухэлектронно-восстановленными редокс-формами, в то время как ряд металлокомплексов на основе нейтральных лигандов [12] немногочисленен. Весьма ограничен также список бензохиноновых производных металлов, в которых реализуется смешанный режим координации, проявляющийся в связывании редокс-активного лиганда с ионом комплексообразователя как посредством ковалентного, так и донорно-акцепторного взаимодействий [13, 14].
Настоящее исследование представляет получение 3,6-ди-трет-бутил-2-гидрокси-4-пиридинилфенолята (LH) – нового редокс-активного лиганда диолатного типа, обладающего цвиттер-ионной природой, которая сохраняется при образовании гетеролигандных комплексов олова(IV). При этом для лиганда характерен указанный выше смешанный тип связывания с координационным центром. Особый интерес представляет выраженный сольватохромный эффект исследуемых соединений, причины которого будут рассмотрены в статье.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Использованные в настоящей работе коммерческие реактивы (Aldrich) применяли без дополнительной очистки. Оловоорганические соединения [15], 3,6-ди-трет-бутил-о-бензохинон [16], 3,6-ди-трет-бутилпирокатехин [17] и 3,6-ди-трет-бутил-4-хлор-пирокатехин [18] синтезировали согласно известным методикам. Растворители, необходимые для проведения экспериментов, очищали и обезвоживали согласно стандартным процедурам [19].
Синтез 3,6-ди-трет-бутил-2-гидрокси-4-пиридинилфенолята (LH). Метод 1. В плоскодонной колбе растворяли 3,6-ди-трет-бутил-о-бензохинон (0.5 г, 2.28 ммоль) и 3,6-ди-трет-бутилпирокатехин (0.1 г, 0.45 ммоль) в избыточном количестве пиридина (25 мл, 0.31 моль). Полученную реакционную смесь кипятили с обратным холодильником при интенсивном перемешивании на магнитной мешалке в течение 2 сут. Образовавшиеся темно-красные кристаллы отделяли фильтрованием на стеклянном фильтре Шотта, промывали гептаном и сушили на воздухе. К фильтрату добавляли гептан (10 мл) и полученный раствор выдерживали сутки при комнатной температуре, после чего сформировавшийся порошкообразный ярко-красный продукт собрали на стеклянном фильтре Шотта, промывали гептаном и сушили. Суммарный выход продукта составил 0.32 г (42%).
ИК-спектр (ν, см–1): 487 сл, 538 ср, 604 ср, 630 сл, 641 сл, 681 ср, 710 с, 720 ср, 751 ср, 776 ср, 789 ср, 824 с, 849 ср, 867 сл, 877 сл, 926 сл, 953 с, 978 ср, 989 сл, 1000 сл, 1029 ср, 1060 сл, 1070 сл, 1100 сл, 1120 с, 1147 сл, 1165 ср, 1203 ср, 1262 с, 1287 с, 1308 с, 1365 с, 1432 с, 1486 ср, 1523 ср, 1580 ср, 1595 сл, 1623 с, 1725 сл, 1843 сл, 1882 сл, 2032 сл, 2998 ср, 3027 сл, 3066 сл, 3078 ср, 3091 ср, 3102 сл, 3148 сл, 3193 сл. Спектр ЯМР 1H (400 МГц; CD3OD; δ, м.д.): 1.20 (с., 9H, t-Bu); 1.36 (c., 9H, t-Bu); 6.19 (с., 1H, Hаром); 8.04–8.11 (м., 2H, H(Py)β); 8.61–8.69 (м., 1H, H(Py)γ); 9.02–9.04 (м., 2H, H(Py)α).
Пригодные для РСА кристаллы LH · 0.5Py получали кристаллизацией 3,6-ди-трет-бутил-2-гидрокси-4-пиридинилфенолята из горячего пиридина.
Синтез хлорида 1-(2,5-ди-трет-бутил-3,4-дигидроксифенил)пиридиния (LH2Cl). К раствору 4‑хлор-3,6-ди-трет-бутилпирокатехина (1 г, 3.9 ммоль) в изопропиловом спирте (20 мл) прибавляли избыточное количество пиридина (1 мл, 12.4 ммоль), после чего полученную реакционную смесь кипятили в течение 2 ч в атмосфере аргона. По окончании реакции растворители удаляли при пониженном давлении, а образовавшийся сухой остаток перекристаллизовали из смеси ацетонитрил–толуол (1 : 1). Выход бесцветных кристаллов 0.72 г (55%).
ИК-спектр (ν, см–1): 528 ср, 615 с, 645 с, 675 с, 694 сл, 708 ср, 733 ср, 769 сл, 789 с, 810 сл, 843 ср, 902 ср, 945 с, 965 с, 1028 ср, 1059 сл, 1127 с, 1162 сл, 1191 ср, 1206 ср, 1223 сл, 1248 сл, 1270 с, 1401 с, 1482 с, 1547 сл, 1591 ср, 1600 ср, 1628 с, 1797 сл, 3100 c. Спектр ЯМР 1H (400 МГц; CD3OD; δ, м.д.): 1.22 (с., 9H, t-Bu), 1.39 (с., 9H, t-Bu), 6.70 (с., 1H), 8.15–8.22 (м., 2H), 8.72–8.79 (т.т., J = 7.9, 1.3 Гц, 1H), 9.11–9.17 (м., 2H). Спектр ЯМР 13C (400 МГц; CD3OD; δ, м.д.): 28.35, 28.98, 34.15, 36.47, 117.90, 127.21, 130.14, 133.95, 137.15, 146.68, 147.03, 147.76, 148.06.
Синтез 3,6-ди-трет-бутил-2-гидрокси-4-пиридинилфенолята (LH). Метод 2. К раствору 0.5 г (1.5 ммоль) хлорида 1-(2,5-ди-трет-бутил-3,4-дигидроксифенил)пиридиния в 10 мл изопропилового спирта при интенсивном перемешивании добавляли 0.18 г (1.6 ммоль) трет-бутилата калия. Реакционная смесь принимала ярко-оранжевую окраску. По окончании реакции результирующий раствор охлаждали до 5°С. Выпавший продукт отфильтровывали, промывали водой (10 мл) и охлажденным изопропиловым спиртом (10 мл). Высушенный осадок перекристаллизовывали из метанола, после чего выделяли 0.29 г (0.97 ммоль, 65.1%) ярко-оранжевого порошкообразного продукта. Спектральные характеристики полученного соединения аналогичны таковым для 3,6-ди-трет-бутил-2-гидрокси-4-пиридинилфенолята, синтезированного по методу 1 (см. выше).
Синтез 3,6-ди-трет-бутил-2-окси-4-пиридинилфенолято-трифенилолова(IV) (I). К ярко-красному раствору LH (0.098 г, 0.295 ммоль) в метаноле (10 мл) добавляли Ph3SnOH (0.11 г, 0.295 ммоль), при этом практически мгновенно произошло выпадение розового осадка. После добавления Et3N (0.5 мл) реакционную смесь перемешивали без нагрева в течение 0.5 ч. Бордовый осадок отфильтровывали на стеклянном фильтре Шотта, промывали метанолом и высушивали. Выход 0.18 г (92.3%).
ИК-спектр (ν, см–1): 505 сл, 538 сл, 551 сл, 575 сл, 616 сл, 631 сл, 654 ср, 684 ср, 702 с, 729 с, 779 сл, 788 ср, 815 сл, 852 ср, 964 ср, 985 ср, 995 сл, 1024 сл, 1070 ср, 1096 сл, 1127 ср, 1157 сл, 1189 сл, 1201 ср 1274 с, 1315 сл, 1353 сл, 1386 сл, 1407 с, 1438 с, 1507 сл, 1570 сл, 1621 ср, 1828 сл, 1882 сл, 1933 сл, 1957 сл, 3036 сл, 3049 сл, 3062 ср, 3106 сл. Спектр ЯМР 1H (400 МГц; (CD3)2SO; δ, м.д.): 1.16 (c., 9H, t-Bu), 1.36 (c., 9H, t-Bu), 5.99 (c., 1H, –С=СН–), 7.23–7.30 (м., 9Н, –С=СН–), 7.67–7.71 (м., 6Н, ‒С=СН–), 8.02–8.09 (т., 2Н, –С=СН–), 8.58–8.65 (т., 1Н, –С=СН–), 9.18 (д., 2Н, J = 5.78 Гц, ‒С=СН–).
Красные монокристаллы состава I · CH3CN, пригодные для РСА, выращивали перекристаллизацией вещества из ацетонитрила при охлаждении насыщенного раствора в условиях отсутствия кислорода и влаги воздуха.
Синтез 3,6-ди-трет-бутил-2-окси-4-пиридинилфенолято-дифенилхлороолова(IV) (II). К ярко-красному раствору LH (0.098 г, 0.295 ммоль) в метаноле (10 мл) добавляли Ph2SnCl2 (0.10 г, 0.295 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 1 ч, после чего раствор помутнел и приобрел ярко-желтую окраску. Добавление Et3N (0.5 мл) способствовало изменению цвета раствора на красно-оранжевый. Выпавший ярко-оранжевый осадок отфильтровывали на стеклянном фильтре Шотта, промывали метанолом и высушивали. Выход 0.12 г (69.5%).
ИК-спектр (ν, см–1): 499 сл, 509 ср, 525 сл, 541 сл, 553 сл, 588 ср, 622 ср, 633(с), 655 ср, 686 ср, 703 с, 739 с, 780 сл, 788 ср, 815 ср, 851 ср, 923 сл, 961 ср, 984 с, 997 сл, 1025 сл, 1046 ср, 1058 с, 1065 с, 1097 сл, 1128 с, 1159 ср, 1185 сл, 1202 ср, 1262 с, 1318 ср, 1358 сл, 1403 с, 1431 ср, 1443 с, 1514 сл, 1578 ср, 1624 ср, 1734 сл, 1774 сл, 1831 сл, 1922 сл, 3041 сл, 3067 ср, 3075 сл, 3107 ср, 3169 ср.
Спектр ЯМР 1H (400 МГц; (CD3)2SO; δ, м.д.): 1.18 (c., 9H, t-Bu), 1.41 (c., 9H, t-Bu), 6.07 (c., 1H, ‒С=СН–), 7.24–7.36 (м., 6Н, –С=СН–), 7.68–7.70 (м., 1Н, –С=СН–), 7.80–7.85 (м., 3Н, –С=СН–), 8.05–8.11 (т., 2Н, –С=СН–), 8.60–8.68 (т., 1Н, ‒С=СН–), 9.24 (д., 2Н, J = 5.42 Гц, –С=СН).
ИК-спектры регистрировали в вазелиновом масле (Nujol) с применением кювет из KBr на ИК-Фурье-спектрометрах ФСМ-1201 (диапазон 4000–450 см–1). Cпектры ЯМР регистрировали на приборе Bruker Avance III (400 МГц) с внутренним стандартом Me4Si). Химические сдвиги соединений соотносили с тетраметилсиланом, использованным в качестве внутреннего эталона. Элементный анализ (С, Н) выполняли на анализаторе Euro EA 3000. Электронные спектры поглощения (ЭСП) получали с применением кварцевых кювет (l = 1 см) на спектрофотометре PerkinElmer Lambda-25 (диапазон 200–1100 нм). Спектры ЭПР фиксировали на Bruker EMX спектрометре (рабочая частота ~9.7 ГГц). Дифенилпикрилгидразил применяли как стандартное соединение при определении величин g-фактора (g = 2.0037).
РСА монокристаллов LH · 0.5Py и I · CH3CN проведен на дифрактометре Bruker D8 Quest (ω‑сканирование, МоКα-излучение, λ = 0.71073 Å, T = 100 K). Измерение и интегрирование экспериментальных наборов интенсивностей, учет поглощения и уточнение структур проведены с использованием программных пакетов APEX2 [20], SADABS [21] и SHELX [22]. Структуры решены прямым методом и уточнены полноматричным МНК по $F_{{hkl}}^{2}$ в анизотропном приближении для неводородных атомов. Все атомы водорода, кроме H(2A) в LH, помещены в геометрически рассчитанные положения и уточнены изотропно. Aтом H(2A) в LH найден из разностного синтеза Фурье электронной плотности и уточнен в изотропном приближении. Кристаллографические данные, параметры рентгеноструктурных экспериментов и уточнения структур приведены в табл. 1.
Таблица 1.
Кристаллографические данные и параметры уточнения структур LH ∙ 0.5Py и I ∙ CH3CN
| Параметр | Значение | |
|---|---|---|
| LH ∙ 0.5Py | I ∙ CH3CN | |
| Формула | C21.50H27.50N1.50O2 | C39H42N2O2Sn |
| М | 338.95 | 689.43 |
| Сингония | Моноклинная | Моноклинная |
| Пр. гр. | P21/n | P21/c |
| a, Å | 10.8766(5) | 12.8581(13) |
| b, Å | 15.6499(8) | 19.774(2) |
| c, Å | 12.1013(6) | 26.724(3) |
| β, град | 116.0320(10) | 91.615(2) |
| V, Å3 | 1850.88(16) | 6792.0(12) |
| Размеры кристалла, мм | 0.16 × 0.11 × 0.10 | 0.31 × 0.10 × 0.09 |
| θмакс, град | 28.81 | 28.50 |
| Z | 4 | 8 |
| ρ(выч.), г/см3 | 1.216 | 1.348 |
| µ, мм–1 | 0.078 | 0.788 |
| F(000) | 732 | 2848 |
| Измерено отражений | 26 131 | 79 852 |
| Число независимых отражений (Rint) | 4802 (0.0376) | 17 147 (0.0571) |
| Число уточняемых параметров | 251 | 838 |
| R1, wR2 (I > 2σ(I)) | 0.0452, 0.1121 | 0.0379, 0.0753 |
| R1, wR2 (по всем данным) | 0.0613, 0.1211 | 0.0620, 0.0825 |
| S(F 2) | 1.038 | 1.036 |
| Остаточная электронная плотность (max/min), e/Å3 | 0.379/–0.295 | 1.197/–0.828 |
Структуры зарегистрированы в Кембриджском банке структурных данных (CCDC № 1974166 (LH), 1974165 (I); ccdc.cam.ac.uk/structures).
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Пространственно-затрудненный 3,6-ди-трет-бутил-о-бензохинон является важнейшим стартовым реагентом, позволяющим получать производные о-бензохинонового типа с разнообразными заместителями. Так, посредством нуклеофильного присоединения спиртов были синтезированы соответствующие алкокси-замещенные о-бензохиноновые лиганды [23]. Аналогичным образом, реакции первичных аминов с 3,6-ди-трет-бутил-о-бензохиноном позволяют получать новые лигандные системы как орто-, так и пара-бензохинонового типа [24, 25]. Нуклеофильное присоединение пиридина к 3,6-ди-трет-бутил-о-бензохинону, проводимое в избыточном количестве пиридина при кипячении в присутствии 3,6-ди-трет-бутилпирокатехина, приводит к образованию 3,6-ди-трет-бутил-2-гидрокси-4-пиридинилфенолята (LH) по методу 1 (схема 1 ). Данное соединение представляет собой новый лиганд диолатного типа, обладающий цвиттер-ионной природой.
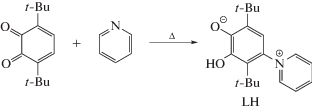
Схема 1 .
Взаимодействие пиридина с галогенсодержащими производными диоксоленовых лигандов можно рассматривать как альтернативный метод введения пиридинильного фрагмента в их структуру. Ранее описанные в литературе примеры демонстрируют, что замещение брома на пиридиний может проходить при использовании 3,4,5,6-тетрабромпирокатехина [26] как свободного, так и находящегося в анион-радикальной форме непосредственно в координационной сфере кобальта [27] и марганца [28]. В то же время, в реакциях с 3,4,5,6-тетрахлорпирокатехином пиридин выступает в качестве основания: электроноакцепторный эффект атомов хлора увеличивает способность групп ОН быть депротонированными, в связи с чем преимущественно проходит отрыв протона пиридином. Для получения продукта нуклеофильного замещения авторами предложена методика, включающая использование о-хлоранила, пиридина и NH2OH · HCl [26, 29].
В настоящей работе с целью получения хлорида 1-(2,5-ди-трет-бутил-3,4-дигидроксифенил)пиридиния в качестве стартовых компонентов были использованы пиридин и 4-хлор-3,6-ди-трет-бутилпирокатехин (cхема 2). Реализация в указанной системе нуклеофильного замещения хлорид-иона пиридином становится возможной благодаря наличию в исходном пирокатехине обладающих выраженными электронодонорными свойствами трет-бутильных групп, которые компенсируют акцепторное влияние атома хлора.
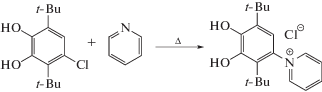
Схема 2 .
Аналогично пирокатехинам, аннелированным солями дитиокарбаматов [30], хлорид 1-(2,5-ди-трет-бутил-3,4-дигидроксифенил)пиридиния способен к взаимодействию с основаниями с образованием LH по методу 2 (схема 3 ). Обобщая вышесказанное, лиганд LH может быть синтезирован в две стадии с использованием 4-хлор-3,6-ди-трет-бутилпирокатехина в качестве стартового реагента.
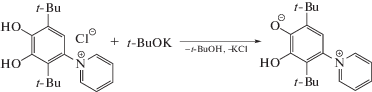
Схема 3 .
Лиганд LH способен вступать в окислительно-восстановительные реакции с такими окислителями, как оксид свинца(IV) или катион феррициния. Так, при взаимодействии LH с (Cp2Fe)PF6 в абсолютном метаноле наблюдается возникновение сигнала в спектре ЭПР, отвечающего образованию соответствующего феноксильного радикала (схема 4 ).
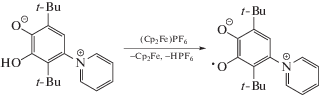
Схема 4 .
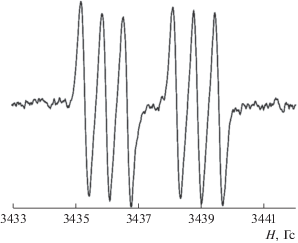
Сверхтонкая структура спектра ЭПР феноксила (дублет 1 : 1 триплетов 1 : 1 : 1) отвечает взаимодействию неспаренного электрона с протоном феноксильного кольца и магнитным ядром азота пиридинильного фрагмента (рис. 1). Параметры спектра ЭПР: gi = 2.0045, ai(1H) = 0.291 мТл, ai(14N) = 0.067 мТл.
При взаимодействии эквимольных количеств оловоорганических соединений (Ph3SnOH или Ph2SnCl2) c LH в присутствии основания в среде метанола конечными продуктами являются гетеролигандные комплексы пентакоординированного олова(IV) I и II (схема 5 ). Соответствующие реакции протекают в аэробных условиях с количественными выходами.
Схема 5 .
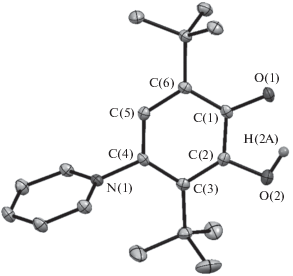
Соединение LH представляет собой цвиттер-ион (схемы 1 и 3 , рис. 2), в котором атом азота N(1) заряжен положительно, а отрицательный заряд сконцентрирован на атоме кислорода O(1). Центральный хиноидный фрагмент в LH практически плоский (среднее отклонение атомов от плоскости составляет 0.03 Å). Искажение шестичленного углеродного кольца значительно меньше, чем в исходном 3,6-ди-трет-бутил-о-бензохиноне [31]: длины связей C–C в LH составляют 1.387(2)–1.432(2) Å и сопоставимы с аналогичными значениями в молекуле бензола (1.40 Å). Расстояние C(1)–O(1) равно 1.308(2) Å и значительно короче длины связи C–O для протонированного атома кислорода (C(2)–O(2) = 1.372(2), O(2)–H(2A) = = 0.91(2) Å); по-видимому, вследствие сопряжения неподеленной электронной пары O(1) с π-системой ароматического кольца. Двугранный угол между плоскостями шестичленных циклов равен 78.11(3)°. Геометрические характеристики пиринидильного заместителя (C–N = 1.348(2), 1.350(2) Å; C–C = 1.375(2)–1.384(2) Å) сопоставимы с аналогичными в молекуле пиридина. Длина связи N(1)–C(4) составляет 1.461(2) Å и отражает ее донорно-акцепторную природу. Несмотря на неэквивалентность заместителей в 4,5-положениях углеродного кольца, трет-бутильные группы в LH находятся практически в заслоненной конформации.
Рис. 2.
Молекулярная структура соединения LH. Тепловые эллипсоиды атомов приведены с 50%-ной вероятностью. Атомы водорода (кроме H(2A)) не показаны для ясности.
Комплекс I кристаллизуется в моноклинной пространственной группе P21/с. В независимой области кристаллической ячейки содержится две кристаллографически независимых молекулы IА и IВ, геометрические характеристики которых близки между собой (молекулярная структура I показана на рис. 3). В каждой из них металлоцентр связан с двумя атомами кислорода лиганда LH и тремя атомами углерода фенильных заместителей. Таким образом, формально координационное число ионa Sn(IV) в I равно пяти. Стоит отметить, что взаимное расположение лигандов в IA и IB несколько отличается друг от друга. Так, если координационный полиэдр Sn(IV) в IB можно описать как сильно искаженную тригональную бипирамиду (τ5 = 0.64 [32]), в апикальных позициях которой находится атомы C(20B) и О(1В), то геометрия координационного окружения Sn(IV) в IА (рис. 4) является переходной между тетрагональной пирамидой и тригональной бипирамидой (τ5 = 0.53 [32]). Кроме того, в молекулах IA и IB положениe фенильных заместителей С(26) несколько отличается друг от друга. Так, в молекуле IA торсионный угол O(2)Sn(1)C(26)C(27) равен 171.0(2)° и значительно больше аналогичного угла в IB (113.3(2))°.
Рис. 3.
Молекулярная структура соединения I (кристаллографически независимые молекулы А и В). Тепловые эллипсоиды приведены с 50%-ной вероятностью. Атомы водорода и трет-бутильные группы не показаны для ясности.
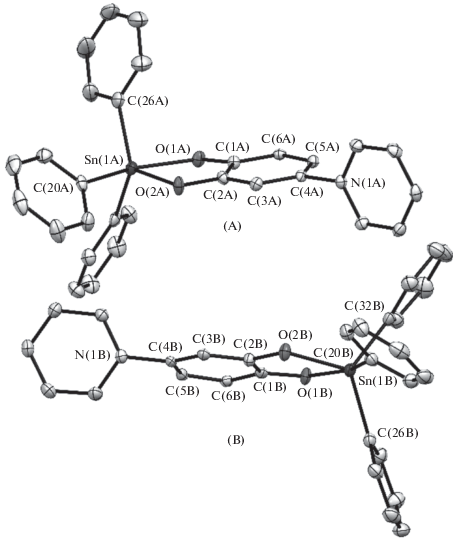
Рис. 4.
Молекулярная структура соединения IА (а) и координационный полиэдр иона Sn(1A) (б). Тепловые эллипсоиды приведены с 50%-ной вероятностью. Атомы водорода (а, б) и трет-бутильные группы (б) не показаны для ясности.
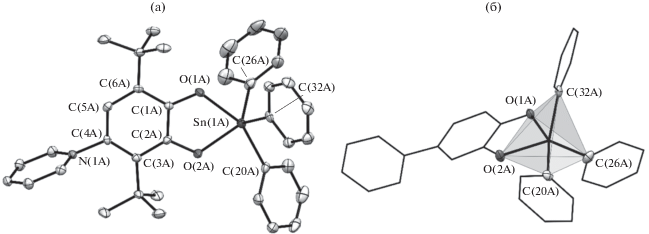
Лиганды LH координированы ионами Sn(IV) несимметрично. Длины связей Sn(1)–O(2) (2.071(2), 2.059(2) Å) меньше суммы ковалентных радиусов 2.09 Å для данных элементов [33] и позволяют охарактеризовать эти взаимодействия как ковалентные. Расстояния Sn(1)–O(1) значительно длиннее (2.170(2), 2.178(2) Å), но не превышают суммы ван-дер-ваальсовых радиусов атомов олова и кислорода (3.70 Å) [33] и свидетельствуют о донорно-акцепторной природе этих взаимодействий. Ранее было продемонстрировано, что такая особенность координации лигандов встречается в гидрокси-парахиноновых производных Sn(IV), однако во всех известных случаях лиганд не обладал цвиттер-ионной природой [13, 14]. В отличие от соединения LH, где расстояния C–O значительно отличаются друг от друга (1.308(2), 1.372(2) Å), в комплексе I эти длины связей близки между собой и лежат в узком интервале значений 1.316(3)–1.339(3) Å. При этом длины связей N–C и C–C в центральном шестичленном кольце лиганда и его пиридинильном заместителе в I сопоставимы с аналогичными характеристиками для молекулы LH (табл. 2). В целом, окислительное состояние лиганда в I соответствует катехолатной редокс-форме соединений о-бензохинонового типа [34]. Пиринидильный заместитель ориентирован практически перпендикулярно хиноидному фрагменту лиганда: двугранный угол между плоскостями шестичленных циклов несколько больше, чем в LH (78.11(3)°), и равен 88.28(8)° (IA) и 83.76(8)° (IB). Как и в LH, трет-бутильные заместители в I находятся в заслоненной конформации.
Таблица 2.
Избранные длины связей (Å) для соединений LH и I
| Связь | I | Связь | LH | I | Связь | LH | I |
|---|---|---|---|---|---|---|---|
| Sn(1)–O(1) | 2.170(2) | O(2)–H(2A) | 0.91(2) | N(1)–C(4) | 1.461(2) | 1.464(3) | |
| 2.178(2) | O(1)–C(1) | 1.308(2) | 1.316(3) | 1.461(3) | |||
| Sn(1)–O(2) | 2.071(2) | 1.319(3) | N(1)–C(15) | 1.348(2) | 1.341(3) | ||
| 2.059(2) | O(2)–C(2) | 1.372(2) | 1.324(3) | 1.337(3) | |||
| Sn(1)–C(20) | 2.180(2) | 1.339(3) | N(1)–C(19) | 1.350(2) | 1.341(3) | ||
| 2.190(2) | C(1)–C(2) | 1.432(2) | 1.429(3) | 1.338(2) | |||
| Sn(1)–C(26) | 2.128(3) | 1.414(3) | С(15)–C(16) | 1.376(2) | 1.371(3) | ||
| 2.137(3) | C(2)–C(3) | 1.392(2) | 1.402(3) | 1.367(4) | |||
| Sn(1)–C(32) | 2.140(3) | 1.403(3) | С(16)–C(17) | 1.384(2) | 1.380(4) | ||
| 2.146(3) | C(3)–C(4) | 1.409(2) | 1.393(3) | 1.375(4) | |||
| 1.396(3) | С(17)–C(18) | 1.382(2) | 1.376(4) | ||||
| C(4)–C(5) | 1.387(2) | 1.394(3) | 1.373(4) | ||||
| 1.386(3) | C(18)–C(19) | 1.375(2) | 1.371(3) | ||||
| C(5)–C(6) | 1.395(2) | 1.389(3) | 1.368(3) | ||||
| 1.386(3) | |||||||
| C(1)–C(6) | 1.410(2) | 1.398(3) | |||||
| 1.403(3) |
ЭСП LH, а также комплексов I и II на его основе зарегистрированы в аэробных условиях в диапазоне длин волн 200–1100 нм. Измерения проведены для широкого ряда растворителей (табл. 3), в которых исследуемые соединения продемонстрировали растворимость, достаточную для проведения эксперимента.
Таблица 3.
Положения максимумов (λ, нм) наиболее длинноволновых полос поглощения в спектрах LH, I и II
| Растворитель | LH | Растворитель | I | II |
|---|---|---|---|---|
| Уксусная кислота | 317 | Диметилсульфоксид | 481 | 458 |
| Метанол | 451 | Диметилформамид | 487 | 464 |
| Этанол | 465 | Ацетонитрил | 491 | 463 |
| Изопропанол | 497 | Ацетон | 509 | 482 |
| Диметилформамид | 530 | Тетрагидрофуран | 541 | 510 |
| Диметилсульфоксид | 531 | Дихлорметан | 561 | 535 |
Установлено, что сам лиганд и синтезированные на его основе металлокомплексы демонстрируют ярко выраженный сольватохромный эффект в видимой и ближней ультрафиолетовой областях электронного спектра. Для выяснения причины возникновения сольватохромии были рассмотрены различные свойства растворителей: εr, μ, $n_{{\text{D}}}^{{{\text{2}}0}},$ $E_{{\text{T}}}^{{\text{N}}},$ DNN и AN. При этом выявлено, что превалирующей характеристикой среды, вызывающей изменение электронного спектра, является полярность растворителя. Так, наблюдаемый гипсохромный сдвиг спектров лиганда LH (рис. 5, 2–6), имеющих схожий вид, отвечает возрастанию полярности растворителя. Положение максимума наиболее длинноволновой полосы при этом хорошо коррелирует с нормализованным эмпирическим параметром полярности $E_{{\text{T}}}^{{\text{N}}}$ (рис. 6а).
Рис. 5.
ЭСП LH, зарегистрированные в уксусной кислоте (1), метаноле (2), этаноле (3), изопропаноле (4), диметилформамиде (5), диметилсульфоксиде (6).
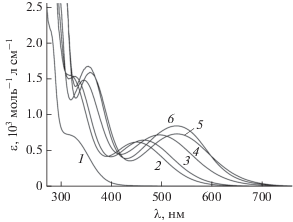
Рис. 6.
Зависимость $E_{{\text{T}}}^{{\text{N}}}$/λ (а) для LH. Растворители – уксусная кислота (1), метанол (2), этанол (3), изопропанол (4), диметилформамид (5), диметилсульфоксид (6). Зависимость Р/λ (б) для комплексов I (ромбы) и II (круги). Растворители – диметилсульфоксид (1), диметилформамид (2), ацетонитрил (3), ацетон (4), тетрагидрофуран (5), дихлорметан (6).
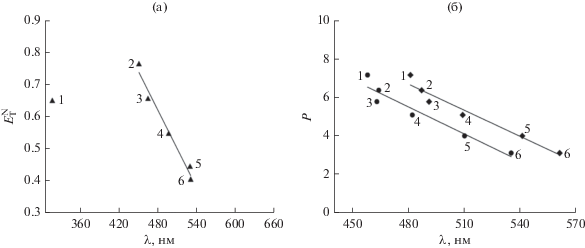
Данная характеристика предложена Димротом и Райхардтом [35] и описывает энергию электронного перехода (при Т = 25°С), соответствующего длинноволновому поглощению стандартного N-феноксипиридиниевого бетаинового красителя, схожего по своему строению с соединением LH. Другим фактором, обуславливающим сольватохромию LH, выступает степень акцепторных свойств (электрофильности) растворителя. Возрастание акцепторного числа AN растворителя [35], позволяющего оценить его Льюисову кислотность, приводит к сдвигу ЭСП в коротковолновую область. С учетом цвиттер-ионного характера изученного лиганда в данном случае определяющим фактором, очевидно, выступает способность растворителя эффективно сольватировать отрицательно заряженный атома кислорода лиганда LH. Примечательно, что вид ЭСП LH, записанного в уксусной кислоте (рис. 5, 1), существенно отличается от рассмотренных выше (рис. 5, 2–6), и положение максимума не соответствует тенденции (рис. 6а). Очевидно, что в растворе уксусной кислоты происходит протонирование отрицательно заряженного кислородного атома с образованием пиридиниевой соли, аналогичной соединению LH2Cl.
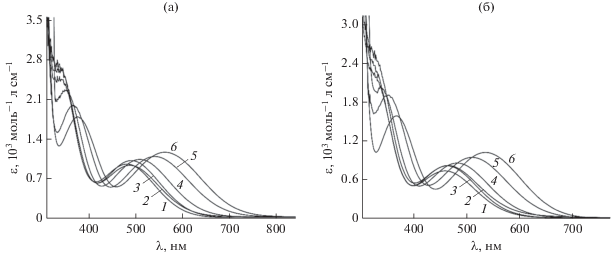
Полярность растворителя выступает доминирующим фактором наблюдаемой сольватохромии и в случае комплексных соединений олова(IV) I и II (рис. 7). В данном случае гипсохромный сдвиг наиболее длинноволновых полос поглощения наилучшим образом коррелирует (рис. 6б) с возрастанием индекса полярности растворителей Р ', предложенного Снайдером [36] и учитывающего совокупную способность среды к протонодонорным, протоноакцепторным и диполь-дипольным взаимодействиям с растворенным веществом. Примечательно, что металлокомплексы I и II демонстрируют схожий вид ЭСП и характер сольватохромного эффекта. При этом замена одной фенильной группы у атома металла на галоген вызывает смещение полос поглощения в коротковолновую область в общем случае более чем на 20 нм (табл. 3), что делает лиганд LH удобной тестовой платформой для оценки электронных свойств заместителей в координационных и элементоорганических соединениях.
Рис. 7.
ЭСП комплексов олова(IV) I (а) и II (б), зарегистрированные в диметилсульфоксиде (1), диметилформамиде (2), ацетонитриле (3), ацетоне (4), тетрагидрофуране (5), дихлорметане (6).
В результате проведенных исследований разработано два синтетических подхода для получения нового цвиттер-ионного редокс-активного лиганда диолатного типа. Его комплексообразующие способности апробированы на примере органических производных олова(IV). Обнаружено, что сольватохромный эффект, присущий исходному лиганду, сохраняется и в построенных на его основе комплексах. Установлено, что электронные спектры поглощения полученных соединений обладают высокой чувствительностью к распределению заряда в молекулах.
Авторы заявляют об отсутствии конфликта интересов.
Список литературы
Abakumov G.A., Piskunov A.V., Cherkasov V.K. et al. // Russ. Chem. Rev. 2018. V. 87. № 5. P. 393.
Чегерев М.Г., Пискунов А.В. // Коорд. химия. 2018. Т. 44. № 2. С. 109 (Chegerev M.G., Piskunov A.V. // Russ. J. Coord. Chem. 2018. V. 44. P. 258). https://doi.org/10.1134/S1070328418040036
Ершова И.В., Пискунов А.В. // Коорд. химия. 2020. Т. 46. № 3. С. 132 (Ershova I.V., Piskunov A.V. // Russ. J. Coord. Chem. 2020. V. 46. P. 154). https://doi.org/10.1134/S1070328420030021
Poddel’sky A.I., Cherkasov V.K., Abakumov G.A. // Coord. Chem. Rev. 2009. V. 253. P. 291.
Tezgerevska T., Alley K.G., Boskovic C. // Coord. Chem. Rev. 2014. V. 268. P. 23.
Miller J.S., Min K.S. // Angew. Chem. Int. Ed. 2009. № 2. V. 48. P. 262.
Broere D.L.J., Plessius R., van der Vlugt J.I. // Chem. Soc. Rev. 2015. V. 44. P. 6886.
Dzik W.I., van der Vlugt J.I., Reek J.N.H., de Bruin B. // Angew. Chem. Int. Ed. 2011. V. 50. P. 3356.
Luca O.R., Crabtree R.H. // Chem. Soc. Rev. 2013. V. 42. P. 1440.
Lyaskovskyy V., de Bruin B. // ACS Catal. 2012. V. 2. P. 270.
Fedushkin I.L., Nikipelov A.S., Morozov A.G. et al. // Chem. Eur. J. 2012. V. 18. P. 255.
Abakumov G.A., Cherkasov V.K., Piskunov A.V. et al. // Dokl. Chem. 2009. № 1. V. 427. P. 168.
Piskunov A.V., Meshcheryakova I.N., Fukin G.K. et al. // Russ. J. Coord. Chem. 2014. V. 40. № 4. P. 205. https://doi.org/10.1134/S1070328414040083
Piskunov A.V., Meshcheryakova I.N., Fukin G.K. et al. // Russ. J. Coord. Chem. 2017. V. 43. № 12. P. 816. https://doi.org/10.1134/S1070328417120077
Кошечков К.А., Землянский Н.Н., Шевердина Н.И., Панов Е.М. Методы элементоорганической химии. Германий, олово, свинец. М.: Наука, 1968. 704 с.
Morris A.M., Pierpont C.G., Finke R.G. // Inorg. Chem. 2009. V. 48. P. 3496.
Мещерякова И.Н., Шавырин А.С., Черкасов А.В., Пискунов А.В. // Изв. АН. Сер. хим. 2019. № 7. С. 1414.
Garnov V.A., Nevodchikov V.I., Abakumov G.A. et al. // Russ. Chem. Bull. 1985. P. 2589.
Гордон А., Форд Р. Спутник химика. М.: Мир, 1976. 543 с. (Gordon A.J., Ford R.A. The Chemist’s Companion. N.Y.: Wiley Intersci. Publ., 1972. 537 p).
Smart APEX2. Madison (WI, USA): Bruker AXS Inc., 2014.
Krause L., Herbst-Irmer R., Sheldrick G.M., Stalke D. // J. Appl. Cryst. 2015. V. 48. P. 3.
Sheldrick G.M. // Acta Crystfllogr. C. 2015. V. 71. P. 3.
Шурыгина М.П., Дружков Н.О., Арсеньев М.В. и др. // Журн. орган. химии. 2011. Т. 47. С. 490.
Абакумов Г.А., Черкасов В.К., Кочерова Т.Н. и др. // Изв. АН. Сер. хим. 2006. С. 1151.
Дружков Н.О., Мещерякова И.Н., Черкасов А.В., Пискунов А.В. // Изв. АН. Сер. хим. 2020. № 1. С. 49.
Bakewell N., Thavarajah R., Motevalli M., Sheriff T.S. // New J. Chem. 2017. V. 41. P. 15411.
Panja A., Frontera A. // Eur. J. Inorg. Chem. 2018. V. 7. P. 924.
Panja A., Jana N.C., Patra M. et al // J. Mol. Cat. A. 2016. V. 412. P. 56.
Sheriff T.S., Watkinson M., Motevallia M., Lesin J.F. // Dalton Trans. 2010. V. 39. P. 53.
Kuropatov V.A., Cherkasov V.K., Fukin G.K., Abakumov G.A. // Russ. Chem. Bull. 2011. V. 60. P. 2291.
Fukin G.K., Cherkasov A.V., Shurygina M.P. et al. // Struct. Chem. 2010. V. 21. P. 607.
Addison A.W., Rao T.N., Reedijk J. et al. // Dalton Trans. 1984. № 7. P. 1349.
Бацанов С.С. // Журн. неорган. химии. 1991. V. 36. P. 3015.
Brown S.N. // Inorg. Chem. 2012. V. 51. P. 1251.
Райхардт К. Растворители и эффекты среды в органической химии. М.: Мир, 1991. С. 763.
Snyder L.R. // J. Chromatogr. 1974. V. 92. № 2. P. 223.
Дополнительные материалы отсутствуют.
Инструменты
Координационная химия