

Координационная химия, 2020, T. 46, № 4, стр. 221-231
Исследование возможности применения реакций ионного обмена для синтеза β-дикетиминат-халькогенидных комплексов неодима и самария. Неожиданное восстановление Sm(III) до Sm(II)
О. А. Миронова 1, 2, Т. С. Сухих 1, 2, С. Н. Конченко 1, 2, Н. А. Пушкаревский 1, 2, *
1 Институт неорганической химии им. А.В. Николаева СО РАН
Новосибирск, Россия
2 Новосибирский государственный университет
Новосибирск, Россия
* E-mail: nikolay@niic.nsc.ru
Поступила в редакцию 25.09.2019
После доработки 23.10.2019
Принята к публикации 31.10.2019
Аннотация
Исследованы возможности протекания реакций ионного обмена между дииодо-β-дикетиминатными комплексами неодима(III) и самария(III) [Ln(Nacnac)I2(Тhf)2] (Ln = Nd (I), Sm (II); Nacnac = HC$\left\{ {{\text{C}}\left( {{\text{Me}}} \right){\text{N}}\left( {{\text{2}},{\text{6}} - {{{\text{C}}}_{{\text{6}}}}{\text{H}}_{{\text{4}}}^{i}{\text{P}}{{{\text{r}}}_{{\text{2}}}}} \right)} \right\}_{{\text{2}}}^{-};$ Thf = тетрагидрофуран), и моно- и дихалькогенидами калия K2Qn (Q = S, Se, Te; n = 1, 2) в тетрагидрофуране. Показано, что реакции ионного обмена иодидных лигандов на дихалькогенидные в этих условиях не протекают. Взаимодействие I с K2Se приводит к образованию комплекса двухвалентного самария [Sm(Nacnac)I(Thf)2] (III). Предложена последовательность стадий, приводящая к образованию этого комплекса, включающая восстановление стерически загруженного бис(дикетиминатного) комплекса.
Катионы лантаноидов являются жесткими кислотами Льюиса и преимущественно образуют комплексы с O- и N-донорными лигандами. Комплексы лантаноидов с лигандами, координирующимися атомом тяжелого халькогена (например, моно- и полихалькогенид или халькогенолат-анионы, где халькоген Q = S, Se или Te), как правило, менее стабильны по отношению к гидролизу и окислению [1]. Внимание к таким соединениям обусловлено проявлением физико-химических свойств, интересных для практических приложений, прежде всего эффективной ИК-люминесценции [2]. Кроме того, молекулярные комплексы можно использовать как предшественники для термальных синтезов лантаноид-халькогенидных (нано-)материалов, обладающих магнитными [3–5] и фотоэлектрическими [6] свойствами. По данным Кембриджского банка структурных данных [7], среди известных комплексов лантаноидов с халькоген-донорными лигандами значительно преобладают халькогенолаты (QR–) и комплексы с различными халькогензамещенными оксоанионами. Подавляющее большинство последних содержит донорные атомы серы, например дитиокарбаматы, дитиофосфинаты [8]. Исключительно редки молекулярные комплексы, содержащие халькогенидные или полихалькогенидные фрагменты в координационной сфере одного или нескольких катионов лантаноидов. Для кинетической стабилизации таких (поли-)халькогенидных комплексов используются различные объемные полидентатные лиганды. Так, известен ряд комплексов с мостиковыми или хелатными лигандами ${\text{Q}}_{n}^{{{\text{2}}--}}$ (Q = S, Se, Te), стабилизированных замещенными циклопентадиенидными (n = 1–3 [9–12]) или формамидинатными лигандами (n = 1, 2, 4 [13–15]). Также известны единичные примеры комплексов с трис-пиразолилборатными (n = 1, 2, 4, 5 [16, 17]), β-дикетиминатными (n = 1 [18, 19]), силиламидными (n = 1, 2 [20]) или аминовыми (n = 1, 2 [21, 22]) лигандами. Моноядерные халькогенидные комплексы представлены пентахалькогенидами (Q = S, Se; n = 5), стабилизированными трис-пиразолилборатным [17] или иминохиноновым [23] лигандами, а также комплексным соединением [DyI2(Thf)5]+ [DyI2S5(Thf)2]– (Тhf = тетрагидрофуран), не содержащем объемных органических лигандов в координационной сфере диспрозия [24]. Изучено достаточно большое количество полиядерных (“кластерных”) халькогенидных комплексов, содержащих внутренние моно- и дихалькогенидные лиганды [2]. Все эти комплексы получены с использованием окислительно-восстановительных реакций, в которых комплекс лантаноида (восстановитель) взаимодействует с источником халькогена (окислителем), таким как элементарный халькоген, фосфинхалькогенид (Ph3PS, nBu3PTe), или полихалькогенидный комплекс переходного металла [25]. Использование других реакций синтеза полихалькогенидных комплексов лантаноидов, не связанных с восстановлением халькогенидного предшественника, в литературе не описано. Единственное исключение – реакция замещения хлорид-ионов в циклопентадиенилдихлоридах неодима и самария LnCpxCl2 при взаимодействии с Na2Se5, приводящие к образованию уникальных гексаядерных комплексов [${\text{Cp}}_{{\text{6}}}^{x}$Ln6(µ6-Se)(µ-Se2)6]2– (Cpx = η5-C5H5, η5-tBuC5H4; Ln = Nd, Sm), содержащих внутриполостной шестикоординированный атом селена [26, 27].
В настоящей работе мы исследовали возможность получения моно- и дихалькогенидных комплексов самария и неодима, стабилизированных объемным N,N'-бис(2,6-диизопропилфенил)-β-дикетиминатным лигандом (Nacnac–, схема 1 ), с помощью реакций ионного обмена.
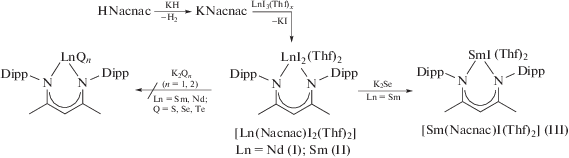
Схема 1 .
Дикетиминатные лиганды (L) дают возможность модифицировать заместители как при донорных атомах азота, так и в углеродном скелете для достижения необходимого стерического и электронного влияния. Кроме того, такие лиганды могут выполнять роль антенны для сенсибилизации люминесценции ионов лантаноидов благодаря системе сопряженных связей.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Все операции с легкоокисляющимися и гидролизующимися веществами проводили в атмосфере очищенного аргона или в вакууме с использованием стандартной аппаратуры Шленка и цельнопаяных ампул. Обработку соединений и подготовку анализов проводили в аргоновом перчаточном боксе. Растворители очищали перегонкой в атмосфере аргона над натрием или натрий-калиевым сплавом в присутствии бензофенон-кетила, хранили над сплавом Na–K до использования и переносили путем переконденсации в вакууме. Исходные комплексы [Ln(Nacnac)I2(Тhf)2] (Ln = Nd (I), Sm (II)) получали в растворе in situ без выделения при взаимодействии эквимолярных количеств иодида лантаноида (NdI3 ∙ 3.5Thf или SmI3) и K(Nacnac), а также в кристаллическом виде по методике [28]. Соединение SmI2 получали по методике [29]. Остальные исходные вещества квалификации не ниже “ч. д. а.” использовали в коммерчески доступном виде.
ИК-спектры (ν, см–1) регистрировали на спектрометре FT-801 (Simex) в таблетках KBr. Элементные анализы на C, H, N выполняли на приборе Vario Micro Cube (Elementar). Элементный анализ на Se выполняли обратным иодометрическим титрованием [30]. Анализ методом энергодисперсионной рентгеновской спектроскопии (ЭДС) проводили с помощью электронного микроскопа Hitachi TM 3000, оборудованного анализатором Bruker Nano.
Синтезы K2Qn (Q = S, Se, Te; n = 1, 2) проводили по методике [31] из простых веществ в Тhf в присутствии нафталина; растворитель и нафталин удаляли нагреванием в вакууме. Для тщательной очистки K2Se от примесей K2Se2 и металлического калия методику модифицировали. Смесь элементов (K: 0.766 г, 19.6 ммоль; порошок Se: 0.725 г, 9.18 ммоль) и нафталина (0.117 г, 0.913 ммоль) в Тhf перемешивали в вакуумированном сосуде, состоящим из двух секций, соединенных под прямым углом и снабженным вакуумным краном с тефлоновым вентилем. Избыток калия брали для заведомо полного восстановления селена; смесь перемешивали в течение 48 ч (для завершения реакции обычно достаточно 6 ч [31]). Для удаления избытка калия раствор, содержащий нафталид калия, декантировали во вторую секцию. Не открывая сосуд, растворитель и нафталин переконденсировали в первую секцию путем ее охлаждения. Суспензию перемешивали, давали осесть осадку K2Se, раствор сливали во вторую секцию. Процедуру промывки повторяли до исчезновения зеленого окрашивания промывного раствора, определяемого нафталидом калия. Осадок K2Se высушивали в вакууме при нагревании до 70°С. Выход 1.09 г (75%). Анализ показал достаточную чистоту продукта (найдено: 49.5% Se, вычислено: 50.24% Se).
Альтернативный синтез K2Se проводили из элементов в жидком аммиаке [32]. Продукт высушивали в вакууме без нагревания. Анализ указал на присутствие одной молекулы аммиака (эксп. 46.3% Se, для K2Se · NH3 расч. 45.33% Se), что согласуется с упомянутой методикой. Необходимое для удаления аммиака высушивание в вакууме при 250°С не проводили, поскольку аммиак не влияет на ход дальнейших реакций.
Реакция взаимодействия [Ln(Nacnac)I2(Тhf)2] с халькогенидами калия. К смеси SmI3 (276 мг, 0.519 ммоль) и K(Nacnac) (237 мг, 0.52 ммоль) добавляли 5 мл Тhf и перемешивали в течение 12 ч. К образовавшемуся красному раствору II с осадком KI добавляли K2Se (82 мг, 0.52 ммоль) и реакционную смесь перемешивали при нагревании (75°С) в течение 3 сут. Темный раствор отделяли от оранжевого осадка центрифугированием. Содержание тяжелых элементов в осадке по данным ЭДС K : I : Sm : Se = 31.8(6) : 16.8(10) : 5.0(4) : : 18.6(11) ат. %. Раствор концентрировали в вакууме до объема в 1 мл. Образовавшееся масло разбавляли 3 мл гексана и смесь запаивали в вакуумированной двухсекционной Г-образной ампуле. Ампулу размещали так, чтобы нижняя секция с раствором находилась при температуре 40°С, а верхняя секция со смесью продуктов реакции – при комнатной температуре. За счет градиента температур растворитель переносили в верхнюю часть и в течение нескольких дней экстрагировали растворимые соединения, которые собирались в нижней части ампулы. Полученный раствор сконцентрировали до небольшого объема, при этом образовывалась смесь серо-коричневых игольчатых кристаллов комплекса [Sm(Nacnac)I(Тhf)2] (III), красных кристаллов комплекса II и небольшого количества кристаллов SmI2(Thf)5 · 1.25Thf (IV · 1.25Thf, согласно РСА). Образовавшийся кристаллический продукт отделяли декантацией и высушивали в вакууме. В верхней части ампулы осталось небольшое количество темного мелкокристаллического осадка иодида самария(II) (отношение содержания Sm : I = 11.4(9) : 22.6(15) ат. % согласно ЭДС). Взаимодействие эквимолярных количеств заранее выделенного комплекса II с K2Se привело к таким же результатам.
Реакции с другими халькогенидами калия и с комплексами неодима проводили аналогичным образом (исходные соединения и их количество указаны в табл. 1). После нагревания в течение 3 сут изменения цвета раствора не наблюдалось. После отделения осадка центрифугированием растворы упаривали в вакууме. ИК-спектры твердых остатков оказались идентичны для каждого лантаноида и соответствовали спектрам комплексов [Ln(Nacnac)I2(Тhf)2] [28].
Таблица 1.
Исходные соединения, использованные для реакций обмена
| Реагенты | Q = S | Q = Se | Q = Te | |||
|---|---|---|---|---|---|---|
| n = 1 | n = 2 | n = 1 | n = 2 | n = 1 | n = 2 | |
| SmI3 (или [Sm(Nacnac)I2(Тhf)2]) | 191/0.360 | 105/0.198 | 276/0.519 | (354/0.366) | 101/0.190 | (114/0.118) |
| KL | 165/0.360 | 91/0.20 | 237/0.519 | 87/0.19 | ||
| K2Qn | 40/0.36 | 29/0.20 | 82/0.52 | 87/0.37 | 39/0.19 | 43/0.13 |
| NdI3 ∙ 3.5Тhf (или [Nd(Nacnac)I2(Тhf)2]) | 164/0.211 | 84/0.11 | 121/0.155 | 94/0.12 | (201/0.209) | 92/0.12 |
| KL | 97/0.21 | 50/0.11 | 71/0.16 | 56/0.12 | 54/0.12 | |
| K2Qn | 23/0.21 | 16/0.11 | 25/0.16 | 29/0.12 | 43/0.21 | 40/0.12 |
ИК-спектр (ν, см–1; комплексы Sm): 3055 сл, 2960 с 2927 ср, 2891 ср, 2868 ср, 1528 с. ушир, 1459 ср, 1429 ср, 1394 с. ушир, 1359 ср, 1338 сл, 1313 с, 1262 с, 1167 ср, 1109 ср, 1096 ср, 1033 сл. пл., 1012 с, 925 с, 875 ср, 853 ср. ушир, 838 ср, 794 сл. пл., 790 с, 757 ср, 693 сл, 667 сл, 636 сл. ИК-спектр (ν, см–1; комплексы Nd): 3063 сл, 2961 с, 2929 ср, 2868 с, 1515 с. ушир, 1462 ср, 1435 с, 1382 с. ушир, 1314 ср, 1256 ср, 1169 ср, 1105 ср, 1039 сл. пл., 1015 с, 930 с, 844 ср. ушир, 797 с, 787 сл. пл., 760 сл, 754 ср, 720 сл, 701 сл, 631 ср.
Прямой синтез комплекса [Sm(Nacnac)I(Тhf)2] (III). К смеси SmI2 (171 мг, 0.423 ммоль) и K(Nacnac) (202 мг, 0.442 ммоль) добавляли 7 мл Thf и перемешивали 2 ч при комнатной температуре. Образовавшийся осадок KI отделяли центрифугированием. Раствор медленно концентрировали в вакууме до объема в 0.5 мл, при этом продукт кристаллизуется в виде массы мелких темных игольчатых кристаллов. Растворитель удаляли, кристаллы промывали эфиром (2 порции по 1 мл) и высушивали в вакууме. Выход III 204 мг (60%).
По данным элементного анализа, соединение содержит около 5% примеси KNacnac. ЭДС: отношение Sm : I = 4.4(8) : 3.6(5) ат. %.
ИК-спектр (ν, см–1): 3058 сл, 2960 с, 2928 ср, 2869 ср, 1624 ср, 1551 с, 1489 сл, 1463 ср, 1436 ср, 1381 ср, 1363 ср, 1323 ср, 1274 ср, 1254 ср, 1174 ср, 1099 сл, 1059 сл, 1022 сл, 934 сл, 838 сл, 787 ср, 759 ср, 731 ср, 701 ср.
Реакция взаимодействия SmI3 с K2Se. Смесь SmI3 (57 мг, 0.11 моль), K2Se (25 мг, 0.16 моль) и 2 мл Тhf запаивали в вакуумированной ампуле. Ампулу выдерживали в течение 24 ч при 45°С (протекание реакции не было заметно). Затем температуру повышали до 70°С и смесь выдерживали еще 15 ч. Произошло изменение цвета раствора с желтого на бесцветный, а также образование оранжевого осадка. Более длительное выдерживание смеси при той же температуре в течение 20 сут не привело к дальнейшим изменениям.
Реакция взаимодействия SmI2 с K2Se2. Смесь SmI3 (49 мг, 0.12 моль), K2Se2 (16 мг, 0.07 моль) и 2 мл Тhf запаивали в вакуумированной ампуле. Ампулу выдерживали в течение 20 сут при 70°С. После 40 ч протекания реакции не отмечено никаких изменений. После 5 сут образовались одиночные желтые кристаллы, параметры ячейки которых идентичны таковым для SmI3(Тhf)3.5 (по данным рентгеновской дифракции на нескольких монокристаллах, сравнение со структурой TOVZAF в КБСД). В течение следующих 15 сут наблюдали увеличение количества желтых кристаллов и постепенное обесцвечивание темно-синего раствора (цвета, характеристичного для SmI2). Коричневый осадок K2Se2, сохранившийся к концу эксперимента, и не полностью обесцветившийся раствор свидетельствуют о неполном протекании реакции.
РСА соединений III (две модификации) и IV проведен по стандартной методике на автоматическом четырехкружном дифрактометре Bruker Apex DUO, оснащенном двухкоординатным CCD детектором (MoKα-излучение, λ = 0.71073 Å, графитовый монохроматор) при 150 K. Интенсивности отражений измерены методом φ- и ω-сканирования узких (0.5°) фреймов. Поглощение учтено по программе SADABS [33]. Все отобранные кристаллы IV – немероэдрические двойники, поэтому массив полученных данных для одного из них обработан с учетом двух доменов с матрицей двойникования (1 0.031 –0.123 0 –1 0 0 0 –1), BASF 0.471, поглощение учтено по программе TWINABS (Version 2012/1). Структуры III и IV расшифрованы прямым методом и уточнены полноматричным МНК в анизотропном для неводородных атомов приближении по комплексу программ SHELXT [34] и SHEXL [35] с использованием программной оболочки Olex2 [36]. На последних стадиях уточнения структуры IV использовали данные в формате HKLF 5. Сольватные молекулы Тhf в этой структуре уточнены в изотропном приближении. Атомы H во всех структурах локализованы геометрически и уточнены в приближении жесткого тела. Кристаллографические данные, детали экспериментов и уточнения приведены в табл. 2.
Таблица 2.
Кристаллографические данные и результаты уточнения структур III и IV
| Параметр | Значение | ||
|---|---|---|---|
| IIIа | IIIб | IV · 1.25Thf | |
| Брутто-формула | C37H57N2O2ISm | C37H57N2O2ISm | C25H50O6.25I2Sm |
| M | 839.09 | 839.09 | 854.80 |
| Пр. гр. | P21/n | P1 | P1 |
| a, Å | 8.8354(6) | 8.5558(7) | 13.0752(10) |
| b, Å | 39.068(2) | 18.8555(16) | 22.7268(17) |
| c, Å | 22.3398(14) | 23.909(2) | 23.9186(18) |
| α, град | 90 | 88.869(3) | 69.185(3) |
| β, град | 98.594(2) | 80.891(3) | 84.109(3) |
| γ, град | 90 | 86.627(3) | 89.264(3) |
| V, Å3 | 7624.7(8) | 3801.7(6) | 6606.5(9) |
| Z | 8 | 4 | 8 |
| ρ(выч.), г/см3 | 1.462 | 1.466 | 1.719 |
| μ, мм–1 | 2.381 | 2.388 | 3.679 |
| F(000) | 3392.0 | 1696.0 | 3344.0 |
| Размер кристалла, мм | 0.26 × 0.13 × 0.05 | 0.33 × 0.05 × 0.05 | 0.31 × 0.28 × 0.12 |
| Диапазон сбора данных по 2θ, град | 2.118–51.478 | 1.726–49.426 | 1.83–51.462 |
| Диапазоны h, k, l | –10 ≤ h ≤ 10, –37 ≤ k ≤ 47, –27 ≤ l ≤ 26 | –10 ≤ h ≤ 7, –22 ≤ k ≤ 22, –28 ≤ l ≤ 28 | –15 ≤ h ≤ 15, –27 ≤ k ≤ 27, –29 ≤ l ≤ 29 |
| Число измеренных рефлексов | 68526 | 28301 | 40 003 |
| Число независимых рефлексов (Rint, Rσ) | 14 478 (0.0578, 0.0593) | 12 767 (0.0641, 0.1119) | 40 003 (0.0523, 0.0745) |
| Число уточняемых параметров | 947 | 884 | 1086 |
| Число ограничений | 594 | 242 | 665 |
| GOOF по F 2 | 1.039 | 0.946 | 1.099 |
| R-фактор (I > 2σ(I)) | R1 = 0.0441, wR2 = 0.0775 |
R1 = 0.0464, wR2 = 0.0688 |
R1 = 0.0593, wR2 = 0.1184 |
| R-фактор (все данные) | R1 = 0.0751, wR2 = 0.0855 |
R1 = 0.0981, wR2 = 0.0822 |
R1 = 0.1076, wR2 = 0.1455 |
| Δρmax/Δρmin, e Å–3 | 1.36/–0.81 | 0.72/–0.73 | 3.15/–2.18 |
Данные РСА депонированы в в Кембриджском банке структурных данных (CCDC (№ 1951605 (II), 1951606 (IIIa), 1951607 (IIIб); http://www.ccdc.cam.ac.uk/conts/retrieving.html).
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
В реакциях обмена галогенидных лигандов на халькогенидные в координационной сфере ионов лантаноидов наилучшим уходящим галогенидом должно быть наиболее “мягкое” основание Льюиса – иодид-анион. Поэтому мы проверяли возможность реакций обмена иодид-ионов в комплексах [Nd(Nacnac)I2(Тhf)2] (I) и [Sm(Nacnac)I2(Тhf)2] (II), синтез которых описан ранее [28]. Как правило, образование нерастворимого в органическом растворителе осадка KI приводит к смещению равновесия реакции ионного обмена, что в данном случае может привести к образованию халькогенидных комплексов (уравнение (1), Solv = молекулы растворителя, например Тhf в качестве лигандов).
(1)
$\begin{gathered} {\text{LnL}}{{{\text{I}}}_{2}}{{\left( {{\text{Solv}}} \right)}_{m}} + {{{\text{K}}}_{{\text{2}}}}{{{\text{Q}}}_{n}}({\text{тв}}.) \to \\ \to {\text{LnL}}\left( {{{{\text{Q}}}_{n}}} \right){{\left( {{\text{Solv}}} \right)}_{x}} + 2{\text{KI}}{\kern 1pt} \downarrow ({\text{тв}}.). \\ \end{gathered} $Наиболее оптимальной средой для проведения реакции является апротонный растворитель средней полярности. Слабополярный или несольватирующий растворитель приведет к очень низкой скорости реакции из-за незначительной концентрации ионов в растворе. Растворитель высокой полярности (DMSO, DMF) или растворитель, способный давать хелатные комплексы (например, DME или N,N-тетраметилэтилендиамин), могут приводить к изоляции катионов лантаноидов в виде сольвато-комплексов [Ln(Solv)x]3+. Следствием этого будет полная диссоциация исходного комплекса и перераспределение стабилизирующего лиганда и халькогенидных лигандов между катионами. Протонные растворители (например, спирты) могут оказаться достаточно сильными кислотами для протонирования стабилизирующего анионного лиганда, что приведет к его уходу в виде нейтральной молекулы. Поэтому в качестве растворителя выбран Тhf, который способен в некоторой степени сольватировать ионы щелочных металлов и в то же время является достаточно лабильным лигандом, способным уходить из координационной сферы лантаноидов [28, 37, 38]. В реакциях обмена иодидных лигандов на халькогенидные возможны побочные процессы, такие как более глубокое перераспределение ионов, нежели обмен двух иодид-ионов на халькогенид. При этом может произойти образование нерастворимых халькогенидов лантаноидов, например согласно уравнению (2) (здесь и далее сольватные молекулы Тhf в уравнениях реакций опущены).
(2)
$\begin{gathered} 3{\text{LnL}}{{{\text{I}}}_{{\text{2}}}} + 3{{{\text{K}}}_{{\text{2}}}}{{{\text{Q}}}_{n}} \to {\text{L}}{{{\text{n}}}_{2}}{{\left( {{{{\text{Q}}}_{n}}} \right)}_{3}}{\kern 1pt} \downarrow + \\ + \,\,{\text{Ln}}{{{\text{L}}}_{{\text{2}}}}{\text{I}} + 5{\text{KI}}{\kern 1pt} \downarrow + \,\,{\text{KL}}. \\ \end{gathered} $В результате этого побочного процесса должны образовываться комплексные формы с бόльшим содержанием дикетиминатных лигандов, например [LnL2]+ или [LnL3]. Исключить возможность их образования можно при помощи объемных лигандов L, делающих образование бис- или трис-(дикетиминатных) комплексов невыгодным по стерическим причинам. Лиганд Nacnac является подходящим с точки зрения стерического влияния. Действительно, гомолептические комплексы [Ln(Nacnac)2] известны только для двухвалентных ионов лантаноидов (Ln = Sm2+, Eu2+, Yb2+ [38–40]), в то время как для трехвалентных ионов лантаноидов, обладающих заметно меньшими ионными радиусами (на ~0.2 Å [41]), такие комплексы малоустойчивы по стерическим причинам. Единственный известный комплекс [TmIII(Nacnac)2]+ был выделен только в присутствии некоординирующего аниона [B(C6F5)4]– [42].
Дополнительное препятствие для реакции ионного обмена – низкая растворимость исходных халькогенидов K2Qn в органических не сильно полярных растворителях, что может нивелировать движущую силу реакции ионного обмена. Тем не менее можно оценить, что образование двух молей KI (${{\Delta }_{f}}G_{{{\text{298}}}}^{^\circ }$ = –324.9 кДж/моль) из одного моля K2S (${{\Delta }_{f}}G_{{{\text{298}}}}^{^\circ }$ = –364.0 кДж/моль [43]) дает существенный термодинамический выигрыш в 285.8 кДж. Этого может быть достаточно для протекания реакции замещения при условии, что энергии Гиббса образования иодидного и халькогенидного комплексов лантаноида в растворах отличаются на меньшую величину. Для остальных (ди-)халькогенидов калия K2Qn (n = 1, 2; Q = S, Se, Te) в доступной литературе отсутствуют данные об энергиях Гиббса образования, поэтому напрямую оценить такой выигрыш нельзя, но можно ожидать что он будет еще больше из-за ожидаемо меньших энтальпий образования других K2Qn по сравнению с K2S.
Для исследования возможности протекания реакций замещения реагенты перемешивали в ТГФ при комнатной температуре, при этом весь исходный комплекс лантаноида находился в растворе с концентрацией около 0.02 М. Во всех случаях за 8 ч перемешивания не наблюдалось признаков протекания реакции. Далее температуру поднимали до 60°С и после 72 ч перемешивания, раствор отделяли и упаривали его в вакууме. ИК‑спектр растворенного вещества сравнивали с известным спектром β-дикетиминат-дииодидного комплекса I или II. Ранее показано [28], что полосы поглощения, соответствующие колебаниям сопряженной системы β-дикетиминатного лиганда, весьма чувствительны к лигандному окружению в комплексе. По аналогии с известными β-дикетиминат-халькогенидными комплексами скандия [18, 19] мы полагаем, что продукты реакции обмена иодид-ионов на (ди-)халькогениды должны быть достаточно растворимы в Тhf, и их присутствие приведет к изменению частот колебаний в ИК-спектре. Однако во всех случаях, кроме взаимодействия II с K2Se, ИК-спектры выделенных из растворов соединений не отличались от таковых для исходных комплексов I или II. Таким образом, даже продолжительный контакт реагентов при повышенной температуре не приводит к образованию молекулярных халькогенидных комплексов.
В единственном случае при взаимодействии I с K2Se в течение 24 ч (60°С) цвет смеси меняется с исходного светло-желтого на коричневый. После 72 ч из раствора выделили черные игольчатые кристаллы комплекса [SmII(Nacnac)I(Thf)2] (III). Это соединение образуется с выходом ~15%, оно кристаллизуется одновременно с непрореагировавшим исходным комплексом II. Более длительное время реакции приводит к постепенному исчезновению в растворе комплекса II. Но в этом случае кристаллизация III при концентрировании раствора сопровождается выделением нерастворимого побочного продукта неустановленного строения, от которого можно избавиться путeм перекристаллизации из смеси ТГФ–гексан (1 : 5 по объему). При этом добиться заметного повышения выхода III не удается. Образование комплекса III в массе вещества доказано путeм рентгеновского дифрактометрического исследования монокристалла и подтверждения тех же параметров ячейки для нескольких кристаллов, отобранных из массы визуально однотипных кристаллических осадков, полученных в независимых экспериментах. Соотношение количеств тяжелых атомов Sm : I = 1 : 1 подтверждено методом ЭДС. Для более полной характеризации комплекс III получали по прямой реакции дииодида самария SmI2(Тhf)2 с одним эквивалентом K(Nacnac).
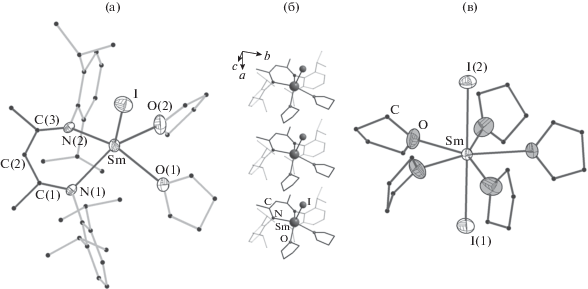
При кристаллизации комплекса III, полученного по реакции II с K2Se, а также по прямой реакции получили две полиморфные модификации IIIa и IIIб. Условия кристаллизации отличались тем, что в случае IIIa соединение закристаллизовалось при выпаривании раствора в Тhf, а случае IIIб перекристаллизацию проводили из смеси Тhf–гексан (1 : 5 по объему). Тем не менее обе модификации не включают сольватный растворитель. Каждая структура содержит по две кристаллографически независимые молекулы, и молекулярное строение комплексов в обеих модификациях практически идентично (табл. 3). Координационное окружение атома самария представляет собой тетрагональную пирамиду с основанием, образованным двумя атомами азота лиганда Nacnac– и двумя атомами кислорода молекул Тhf (рис. 1a), атом иода находится в вершине пирамиды. Координационное число (КЧ), равное 5, необычно мало по сравнению с комплексом II (КЧ 6), несмотря на больший размер катиона Sm2+ по сравнению с Sm3+. Среди соединений лантаноидов(II) известен только один аналогичный комплекс иттербия [YbII(Nacnac)(µ-I)(Thf)]2 (КЧ 5) [44]. Ион Yb2+ координирован двумя иодид-ионами и только одной молекулой Тhf, несмотря на то что комплекс кристаллизуется из этого растворителя. Поскольку ионный радиус Yb2+ заметно меньше такового для Sm2+ (1.08 и 1.22 Å соответственно, для КЧ 7 [41]), можно заключить, что молекула Тhf занимает больший объем в координационной сфере, чем иодид-анион. Молекулы в обеих кристаллических модификациях IIIa и IIIб ориентируются относительно ближайших соседей одинаково, а атом иода одного комплекса располагается между циклами двух молекул Тhf соседнего (рис. 1б). При этом образуются стопки, распространяющиеся в направлении [100 ] . Соседние молекулы в них смещены на вектор трансляции, который определяет минимальный параметр решетки a (в обоих случаях, см. табл. 3). Две модификации различаются относительным разворотом соседних стопок. Дикетиминатный лиганд в комплексе принимает характерную для него геометрию “лодки”, при этом атомы C(1), C(3), N(1), N(2) лежат практически в одной плоскости. Атом C(2) отклоняется от среднеквадратичной плоскости этих атомов на 0.120–0.144 Å, атом самария в том же направлении – на 0.958 и 0.961 Å, или 1.084 и 1.114 Å, в зависимости от модификации (нумерация атомов на рис. 1а). Длины связей Sm–I, Sm–N и Sm–O практически одинаковы для двух модификаций, при этом они существенно удлинены по сравнению с аналогичными связями в комплексе II (табл. 2), что однозначно подтверждает степень окисления самария +2 в комплексе III. Показательно также сравнение ИК спектров комплексов II и III. Характеристичные частоты колебаний сопряженной системы связей в β-дикетиминате чувствительны к иному лигандному окружению. Согласно предыдущим исследованиям [28], β-дикетиминатный комплекс Sm(III) имеет в спектре характеристичные пики, соответствующие валентным колебаниям связей C=C, C=N и C–CH3 в лиганде (1528, 1398, 1314, 1166, 924 см–1), а также колебаниям связи C–O в координированном Тhf (853 см–1). В спектре комплекса III наблюдаются заметные сдвиги пиков дикетиминатного лиганда в область больших энергий, за исключением пика при 1398 см–1 (1551, 1381, 1324, 1174, 934 см–1), пик координированного Тhf проявляется при 840 см–1. В спектре также присутствует полоса средней интенсивности при 1622 см–1, соответствующая образованию небольших количеств HNacnac в результате гидролиза при записи спектра.
Таблица 3.
Избранные длины связей (Å) в комплексах II [28] и IIIа
| Связь | IIб | IIIa (IIIб) |
|---|---|---|
| Sm–N(1) | 2.400(3) | 2.514(4), 2.507(4)в (2.523(5), 2.514(5)в) |
| Sm–N(2) | 2.515(4), 2.519(4)в (2.522(5), 2.521(5)в) | |
| Sm–O(1) | 2.536(3) | 2.552(4), 2.546(4)в (2.571(4), 2.544(4)в) |
| Sm–O(2) | 2.559(1), 2.557(4)в (2.570(4), 2.587(5)в) | |
| Sm–I | 3.0823(5), 3.0072(4) | 3.1536(5), 3.1582(5)в (3.1554(6), 3.1577(7)в) |
а Нумерация атомов приведена в соответствии с рис. 1. б Молекула комплекса лежит на кристаллографической плоскости симметрии, проходящей через атомы Sm и I. в Данные для второй независимой молекулы.
Рис. 1.
Молекулярная структура комплекса III по данным РСА (a); упаковка молекул III в кристалле (б); молекулярная структура комплекса IV по данным РСА (в). Тепловые эллипсоиды 50%-ной вероятности, атомы водорода не показаны, органические фрагменты приведены в упрощенном виде.
Образование комплекса Sm(II) из Sm(III) в реакции, протекающей в отсутствие очевидных сильных восстановителей, – довольно необычный факт, поскольку все соединения Sm(II) являются сильными восстановителями (так, E° для пары ${{{\text{SmI}}_{{\text{2}}}^{ + }} \mathord{\left/ {\vphantom {{{\text{SmI}}_{{\text{2}}}^{ + }} {{\text{Sm}}{{{\text{I}}}_{{\text{2}}}}}}} \right. \kern-0em} {{\text{Sm}}{{{\text{I}}}_{{\text{2}}}}}}$ в Тhf равен 1.41 В относительно Fc+/Fc [45]). Можно предположить, что восстановителем в реакции является металлический калий, который мог оставаться в образце K2Se после его синтеза из элементов. Для исключения такой возможности K2Se синтезировали разными способами с использованием в качестве реакционной среды жидкого аммиака или Тhf с нафталином в качестве переносчика электронов. В обоих случаях тщательно контролировалось отсутствие окрашивания конечных растворов, которое говорило бы о примеси избытка калия. Содержание селена в полученных образцах K2Se хорошо соответствовало теоретическому, и допустимые остаточные количества свободного калия не могли привести к образованию заметных количеств продукта восстановления III. Следовательно, влияние примеси элементарного калия можно исключить.
Возможность превращения Sm3+ в Sm2+ может также реализоваться в результате так называемого “стерически индуцированного восстановления” (sterically induced reduction, SIR [46]). В таких процессах один из анионных лигандов отдает электрон и уходит в виде нейтральной частицы (например, Cp*– – e → Cp*· → $0.{\text{5Cp}}_{2}^{*},$ Cp* = пентаметилциклопентадиен). В реакции происходит замещение уходящего лиганда на менее объемный или восстановление иона лантаноида, сопровождающееся увеличением ионного радиуса (Ln3+ → Ln2+). Любой вариант приводит к уменьшению стерического напряжения в координационной сфере. Бόльшая стерическая загруженность координационной сферы приводит к тому, что процесс восстановления становится более выгодным. Для комплексов с β-дикетиминатными лигандами подобное восстановительное действие лиганда описано для комплексов европия, обладающего наибольшим потенциалом Ln3+/Ln2+ из всех лантаноидов [37, 47]. Так, при взаимодействии EuCl3 с менее объемным анионом (LMe)– образуется комплекс [EuIII${\text{L}}_{{\text{3}}}^{{{\text{Me}}}}$] (LMe = {N(2-MeC6H4)- C(Me)}2CH), в то время как наличие более объемного аниона (LMe2)– приводит к стерически-индуцированному восстановлению европия и образованию [EuII(LMe2)2(Тhf)] (LMe2 = {N(2,6-Me2C6H3)- C(Me)}2CH). Следует отметить, что известен только один пример стерически-индуцированного восстановления Sm3+ до Sm2+, которое происходит при попытке внедрить три исключительно объемных и жестких пентафенилциклопентадиенильных лиганда (CpBIG = η5-C5(4-tBuC6H4)) в координационную сферу Sm3+ [48]. В результате восстановления за счет лиганда образуется устойчивый сэндвичевый комплекс [SmII${\text{Cp}}_{{\text{2}}}^{{{\text{BIG}}}}$], стабилизированный за счет взаимодействий фенильных колец соседних циклов между собой. Реализация подобного восстановительного процесса в изучаемой реакции потребовала бы промежуточного образования стерически напряженного комплекса. Таким комплексом может быть [Sm(Nacnac)2]+, который мог бы образоваться в процессе лигандного диспропорционирования (уравнение (3)). Протекание этого процесса может быть связанo с одновременным образованием нерастворимого селенида самария (уравнение (4)). Действительно, в отдельном эксперименте показано, что взаимодействие SmI3 и K2Se в Тhf постепенно приводит к полному осаждению самария в виде Sm2Se3.
(3)
$2{\text{Sm}}\left( {{\text{Nacnac}}} \right){{{\text{I}}}_{2}} \to \left[ {{\text{Sm}}{{{\left( {{\text{Nacnac}}} \right)}}_{2}}} \right]{\text{I}} + {\text{Sm}}{{{\text{I}}}_{3}},$(4)
$\begin{gathered} 4{\text{Sm}}\left( {{\text{Nacnac}}} \right){{{\text{I}}}_{2}} + 3{{{\text{K}}}_{{\text{2}}}}{\text{Se}} \to \\ \to {\text{S}}{{{\text{m}}}_{{\text{2}}}}{\text{S}}{{{\text{e}}}_{3}} + 2\left[ {{\text{Sm}}{{{\left( {{\text{Nacnac}}} \right)}}_{{\text{2}}}}} \right]{\text{I}} + 6{\text{KI}}. \\ \end{gathered} $В [42] показано, что аналогичный комплекс тулия [Tm(Nacnac)2]+ чрезвычайно неустойчив, и один из лигандов легко теряет протон метильной группы основной цепи в присутствии оснований. При этом образуется гетеролигандный комплекс [Tm(Nacnac)(Nacnac')] (Nacnac' = = CH(C(CH3)NDipp)(C(=CH2)NDipp)2–). Поскольку K2Se может играть роль основания, можно ожидать подобного превращения и в изучаемой реакции. Однако депротонирование лиганда должно привести к сохранению комплекса Sm(III), а не к восстановлению металла; кроме того, [Sm(Nacnac) (Nacnac')] не был обнаружен в реакционной смеси. Другим вероятным путем преобразования [Sm(Nacnac)2]+ может быть восстановление до нейтрального комплекса [SmII(Nacnac)2] (уравнение (5), Red = восстановитель), устойчивого в условиях реакции [39]. Можно предположить, что конечный комплекс III образуется в результате ещe одного процесса обмена лигандов в условиях реакции (уравнение (6)). Таким образом, суммарный процесс (уравнения (4)–(6)) можно представить как восстановление комплекса Sm(III) до Sm(II) с уходом одного иодидного лиганда, сопровождающeeся образованием селенида самария(III). Образование в ходе реакции твердой фазы, содержащей самарий и селен в значительных и сравнимых количествах, показано методом ЭДС. Такой суммарный процесс (возможная реакция представлена уравнением (7) будет приводить также к образованию комплекса [Sm(Nacnac)2] при соблюдении стабильности дикетиминатного лиганда в условиях реакции.
(5)
$\begin{gathered} \left[ {{\text{Sm}}{{{\left( {{\text{Nacnac}}} \right)}}_{2}}} \right]{\text{I}} + {\text{Red}} \to \\ \to \left[ {{\text{Sm}}{{{\left( {{\text{Nacnac}}} \right)}}_{2}}} \right] + {\text{Red}}{\kern 1pt} --{\kern 1pt} {\text{I,}} \\ \end{gathered} $(6)
$\begin{gathered} \left[ {{\text{Sm}}{{{\left( {{\text{Nacnac}}} \right)}}_{{\text{2}}}}} \right] + {\text{Sm(Nacnac}}){{{\text{I}}}_{2}} \to \\ \to {\text{Sm}}\left( {{\text{Nacnac}}} \right){\text{I}} + \left[ {{\text{Sm}}{{{\left( {{\text{Nacnac}}} \right)}}_{{\text{2}}}}} \right]{\text{I,}} \\ \end{gathered} $(7)
$\begin{gathered} ~6{\text{Sm}}\left( {{\text{Nacnac}}} \right){{{\text{I}}}_{2}} + 7{{{\text{K}}}_{{\text{2}}}}{\text{Se}} \to \\ \to {\text{S}}{{{\text{m}}}_{{\text{2}}}}{\text{S}}{{{\text{e}}}_{3}} + 2{\text{Sm}}\left( {{\text{Nacnac}}} \right){\text{I}} + \\ + \,\,2\left[ {{\text{Sm}}{{{\left( {{\text{Nacnac}}} \right)}}_{2}}} \right] + 10{\text{KI}} + 2{{{\text{K}}}_{{\text{2}}}}{\text{S}}{{{\text{e}}}_{2}}. \\ \end{gathered} $О возможности перераспределения лигандов между комплексами Sm(II) говорит тот факт, что при перекристаллизации отдельно полученного комплекса III из гексана наблюдается образование небольших количеств нерастворимого в этих условиях дииодида самария. Отдельные кристаллы этого соединения выделены также в некоторых случаях из раствора в Тhf после реакции II с K2Se. Вероятно, комплекс [Sm(Nacnac)2] обладает заметно большей растворимостью, что препятствует его кристаллизации.
В изучаемой системе возможны два восстановителя: селенид- и дикетиминат-анион. Их восстановительные потенциалы в условиях обсуждаемой реакции неизвестны, но возможность протекания окислительно-восстановительного процесса можно оценить на примере известных реакций. Так, была продемонстрирована возможность восстановления элементарного селена иодидом самария(II) с образованием ${\text{Se}}_{2}^{{2 - }}$ или Se2– в зависимости от стехиометрии восстановителя [49]. Для полного восстановления до Se2– необходим избыток SmI2, и даже в этом случае реакция идeт не до конца. Обеспечить полное протекание реакции можно только при добавлении гексаметилфосфатриамида, который увеличивает восстановительный потенциал SmI2 в растворе ТГФ. Наш эксперимент показал, что взаимодействие SmI2 с K2Se2 в ТГФ происходит очень медленно при повышенной температуре (при 70°С неполное протекание реакции становится заметным за ~15 сут). Таким образом, можно сделать вывод о том, что редокс-потенциал K2Se2/K2Se близок к таковому для ${\text{SmI}}_{2}^{ + }$/SmI2 в Тhf, и K2Se может быть восстановителем в изучаемой реакции. Напротив, мы считаем, что анион Nacnac– не может выступать восстановителем в этом процессе, так как в противном случае исходный комплекс [Sm(Nacnac)I2(Тhf)2] может подвергаться внутримолекулярному редокс-процессу, приводящему к образованию соединений Sm(II), чего не наблюдается даже при длительном нагревании растворов этого комплекса в процессе перекристаллизации [28]. Кроме того, при одноэлектронном окислении аниона Nacnac– должен образовываться димерный продукт (Nacnac)2 [50], но характерные сигналы этих соединений в спектрах ЯМР реакционного раствора отсутствуют. По всей видимости, роль дикетиминатного лиганда заключается в обеспечивании необходимой стерической загруженности иона Sm3+, поскольку в отсутствие дикетиминатного лиганда восстановление SmI3 при тех же условиях не происходит.
Предлагаемая схема процесса согласуется с от-сутствием реакции для комплексов неодима, который обладает существенно большим восстановительным потенциалом по сравнению с самарием [51]. Не до конца понятно видимое отсутствие взаимодействия комплекса II с другими (ди-)халькогенидами калия, кроме K2Se. Наблюдаемое отсутствие окислительно-восстановительных процессов в этих случаях можно объяснить: а) низкой растворимостью халькогенидов калия в условиях эксперимента, что снижает движущую силу и скорость реакции; б) недостаточно высоким восстановительным потенциалом некоторых халькогенидов по сравнению с Se2– (например, S2– или ${\text{Se}}_{2}^{{2 - }}$). Тем не менее нельзя исключать, что реакции восстановления могут идти и при взаимодействии с другими халькогенидами, но с меньшей скоростью.
Образующиеся в небольших количествах кристаллы иодида самария(II) были исследованы методом РСА. Структура содержит молекулярные комплексы [SmI2(Тhf)5] и включает также 1.25 молекулы сольватного Тhf на атом самария. Комплекс обладает практически правильным пентагонально-бипирамидальным строением, молекулы Тhf располагаются в экваториальной плоскости (рис. 1в). В элементарной ячейке содержатся четыре независимых молекулы комплекса, которые обладают близкими геометрическими параметрами (длины связей Sm–I 3.227(2)–3.266(2); Sm–O 2.546(11)–2.630(10) Å). Отметим, что попытки установления структуры SmI2(Тhf)5 · nТhf проводили ранее [52], однако из-за низкого качества полученных структурных данных авторам удалось локализовать лишь тяжелые атомы Sm и I и установить геометрию комплекса. R-фактор составил 15%, пр. гр. P21/n, параметры ячейки a = = 23.154(12) Å, b = 26.033(6) Å, c = 23.866(10) Å, β = 111.69(4)°, V = 13367(11) Å3, Z = 16. Точное количество сольватных молекул ранее установить также не удалось. Структура полученных нами кристаллов SmI2(Тhf)5 · 1.25Тhf была решена в 2 раза меньшей по объему ячейке как немероэдрический двойник в пр. гр. $P\bar {1}$, Z = 8. Удалось надежно локализовать как координированные, так и сольватные молекулы ТГФ. К сожалению, координаты атомов в предшествующей статье [52] и соответствующем структурном файле КБСД (ZEKXIW) не приведены, поэтому нельзя сказать, идентичны ли в действительности данные соединения, или они являются разными фазами.
Таким образом, реакции ионного обмена неприменимы для получения β-дикетиминатных комплексов лантаноидов с (поли-)халькогенид-анионами в координационной сфере. По-видимому, лучший синтетический метод для таких комплексов – реакции с участием окислительно-восстановительных превращений [18, 19]. Комплексы самария с N-донорными лигандами, согласно предлагаемой схеме взаимодействия II с K2Se, также способны участвовать в процессах стерически-индуцированного восстановления, которые были до сих пор известны в основном для циклопентадиенильных производных. Потенциально, возможность варьирования стерического влияния N-донорных лигандов в координационной сфере самария может привести к дальнейшему развитию химии окислительно-восстановительных процессов с участием комплексов этого элемента.
Авторы заявляют об отсутствии конфликта интересов.
Список литературы
Atwood D.A., Platt S.M., Cotton S.A., Harrowfield J.M. et al. The Rare Earth Elements: Fundamentals and Applications. Chichester: John Wiley & Sons, 2012.
Brennan J.G. Thiolates, Selenolates, and Tellurolates // The Rare Earth Elements: Fundamentals and Applications / Ed. Atwood D.A. Chichester: John Wiley & Sons, 2012. P. 215.
Kataoka T., Tsukahara Y., Hasegawa Y., Wada Y. // Chem. Commun. 2005. № 48. P. 6038.
Kar S., Boncher W.L., Olszewski D., et al. // J. Am. Chem. Soc. 2010. V. 132. № 40. P. 13960.
Selinsky R.S., Han J.H., Pe E.A.M., Guzei I.A. // J. Am. Chem. Soc. 2010. V. 132. № 21. P. 15997.
Ghosh A.B., Saha N., Sarkar A. et al. // RSC Adv. 2015. V. 5. № 124. P. 102818.
Groom C.R., Bruno I.J., Lightfoot M.P., Ward S.C. // Acta Crystallogr. B. 2016. V. 72. P. 171.
Zhou J. // Coord. Chem. Rev. 2016. V. 315. P. 112.
Evans W.J., Rabe G.W., Ziller J.W., Doedens R.J. // Inorg. Chem. 1994. V. 33. № 2. P. 2719.
Fieser M.E., Johnson C.W., Bates E. et al. // Organometallics. 2015. № 34. P. 4387.
Evans W.J., Rabe G.W., Ansari A., Ziller J.W. // Angew. Chem. 1994. V. 859. № 3. P. 3.
Evans W.J., Davis B.L., Champagne T.M., Ziller J.W. // PNAS. 2006. V. 103. № 34. P. 12678.
Zhou A.X., Hong J., Tian H., Zhang L. // Angew. Chem. Int. Ed. 2017. V. 57. № 4. P. 1062.
Werner D., Zhao X., Best S.P. et al. // Chem. Eur. J. 2017. V. 23. № 9. P. 2084.
Ma Y.Z., Bestgen S., Gamer M.T. et al. // Angew. Chem. Int. Ed. 2017. V. 56. № 43. P. 13249.
Kühling M., McDonald R., Liebing P., Hilfert L. et al. // Dalton Trans. 2016. V. 45. № 25. P. 10118.
Zhang F., Zhang J., Zhou X. // Inorg. Chem. 2017. V. 56. № 4. P. 2070.
Maron A.L., Han X., Xiang L. et al. // Chem. Eur. J. 2017. V. 23. № 59. P. 14728.
Knight L.K., Piers W.E., McDonald R. // Chem. Eur. J. 2000. V. 6. № 23. P. 4322.
Corbey J.F., Fang M., Ziller J.W., Evans W.J. // Inorg. Chem. 2014. V. 53. № 6. P. 3099.
Kornienko A.Y., Emge T.J., Brennan J.G. // J. Am. Chem. Soc. 2001. V. 123. № 48. P. 11933.
Huebner L., Kornienko A., Emge T.J., Brennan J.G. // Inorg. Chem. 2005. V. 44. № 14. P. 5118.
Coughlin E.J., Zeller M., Bart S.C. // Angew. Chem. Int. Ed. 2017. V. 56. № 40. P. 12142.
Fagin A.A., Kuznetsova O. V., Balashova T. V., Cherkasov A.V. et al. // Inorg. Chim. Acta. 2018. V. 469. P. 227.
Afonin M.Y., Sukhikh T.S., Konokhova A.Y., Konchenko S.N. // Russ. J. Coord. Chem. 2018. V. 44. № 2. P. 147. https://doi.org/10.1134/S107032841802001X
Jin G.-X., Cheng Y., Lin Y. // Organometallics. 2002. V. 18. № 6. P. 947.
Cheng Y., Jin G.-X., Shen Q., Lin Y. // J. Organomet. Chem. 2001. V. 631. P. 94.
Mironova O.A., Sukhikh T.S., Konchenko S.N., Pushkarevsky N.A. // Polyhedron. 2019. V. 159. P. 337.
Wayda A.L., Cheng S., Mukerji I. // J. Organomet. Chem. 1987. V. 330. P. C17.
Ярембаш Е.И., Елисеев А.А. Халькогениды редкоземельных элементов. Синтез и кристаллохимия. М.: Наука, 1975. 258 с.
Thompson D.P., Boudjouk P. // J. Org. Chem. 1988. V. 53. № 9. P. 2109.
Mossbauer F., Lss O. // Inorg. Chem. 1991. V. 30. № 11. P. 2540.
APEX2 (version 2.0). SAINT (version 8.18c), and SADABS (version 2.11). Mdison (WI, USA): Bruker Advanced X-ray Solutions, 2000–2012.
Sheldrick G.M. // Acta Crystallogr. A. 2015. V. 71. № 1. P. 3.
Sheldrick G.M. // Acta Crystallogr. C. 2015. V. 71. № 1. P. 3.
Dolomanov O. V., Bourhis L.J., Gildea R.J., Howard J.A.K. et al. // J. Appl. Crystallogr. 2009. V. 42. № 2. P. 339.
Evans W.J., Hughes L.A., Hanusa T.P. // J. Am. Chem. Soc. 1984. V. 106. № 15. P. 4270.
Harder S. // Angew. Chem. Int. Ed. 2004. V. 43. № 20. P. 2714.
Shen X., Zhang Y., Xue M., Shen Q. // Dalton Trans. 2012. V. 41. № 13. P. 3668.
Xue M., Zheng Y., Hong Y., Yao Y. et al. // Dalton Trans. 2015. V. 44. № 46. P. 20075.
Shannon R.D. // Acta Crystallogr. A. 1976. V. 32. P. 751.
Hitchcock P.B., Lappert M.F., Protchenko A. V. // Chem. Commun. 2005. V. 301. № 7. P. 951.
Lide D.L. CRC Handbook of Chemistry and Physics. Boca Raton: CRC Press, 2004.
Hitchcock P.B., Khvostov A. V., Lappert M.F., Protchenko A.V. // J. Chem. Soc. 2009. № 13. P. 2383.
Enemærke R.J., Daasbjerg K., Skrydstrup T. // Chem. Commun. 1999. № 4. P. 343.
Evans W.J., Davis B.L. // Chem. Rev. 2002. V. 102. № 6. P. 2119.
Jiao R., Shen X., Xue M., Zhang Y. et al. // Chem. Commun. 2010. V. 46. № 23. P. 4118.
Ruspic C., Moss J.R., Schürmann M., Harder S. // Angew. Chem. Int. Ed. 2008. V. 47. № 11. P. 2121.
Sekiguchi M., Tanaka H., Takami N., Ogawa A. et al. // Heteroat. Chem. 1991. V. 2. № 3. P. 427.
Shimokawa C., Itoh S. // Inorg. Chem. 2005. V. 44. № 9. P. 3010.
Morss L.R. // Chem. Rev. 1976. V. 76. № 6. P. 827.
Evans W.J., Gummersheimer T.S., Ziller J.W. // J. Am. Chem. Soc. 1995. V. 117. № 35. P. 8999.
Дополнительные материалы отсутствуют.
Инструменты
Координационная химия