

Координационная химия, 2020, T. 46, № 6, стр. 333-339
Новые гидриды галлия с дианионными аценафтен-1,2-дииминовыми лигандами
Т. С. Копцева 1, *, В. Г. Соколов 1, Е. В. Баранов 1, И. Л. Федюшкин 1
1 Институт металлоорганической химии им. Г.А. Разуваева РАН
Нижний Новгород, Россия
* E-mail: koptseva@iomc.ras.ru
Поступила в редакцию 23.12.2019
После доработки 27.01.2020
Принята к публикации 29.01.2020
Аннотация
Нагревание раствора соединения [(Dpp-bian)GaH2] (I) (Dpp-bian = 1,2-бис[(2,6-диизопропилфенил)имино]аценафтен) в пиридине приводит к элиминированию атома водорода и восстановлению дииминового лиганда до дианиона с образованием комплекса [(Dpp-bian)GaH(Py)] (II). При взаимодействии I с 1,3-диизопропил-4,5-диметилимидазол-2-илиденом (i-Pr2ImMe) происходит аналогичный процесс, а образующееся в результате реакции соединение представляет собой комплекс [(Dpp-bian)GaH(i-Pr2ImMe)] (III). Диамагнитные производные II и III охарактеризованы спектроскопией ЯМР 1Н. Молекулярная структура соединения II установлена методом РСА (СIF file CCDC № 1973549).
Соединения со связью Ga–H широко представлены в химии гидридов металлов 13-й группы [1–4]. Эти соединения вызывают значительный интерес с точки зрения их практического применения и фундаментальных аспектов. Например, летучие соединения на основе тригидрида галлия рассматриваются в качестве потенциальных прекурсоров для создания пленок или наночастиц GaN и GaAs в процессах MOCVD [2, 5–8]. Кроме того, дихлоргаллан Cl2GaH [9–11] и диалкилгалланы R2GaH [12–14] реагируют с ненасыщенными субстратами (кетонами, нитрилами, олефинами и алкинами) с образованием продуктов гидрогалирования [4, 10, 12, 15–29]. Достаточно подробно исследованы реакции гидрометаллирования замещенных алкинов дихлор- или диалкилгалланами, показано разнообразие путей реакций и образовавшихся галлийсодержащих продуктов [4, 17–29]. Следует отметить, что реакции гидрогаллирования в некоторых случаях протекают селективнее по сравнению с процессами гидроалюминирования [3, 4]. Важным классом гидридов галлия являются нейтральные комплексы, в которых фрагмент Ga–H хелатирован N,N-бидентатными лигандами [30–38]. Так, продемонстрирована возможность использования дикетиминатного производного [(Dpp-Nacnac)GaH(t-Bu)] (Dpp = 2,6-i-Pr2C6H3) в качестве катализатора восстановления CO2 до HC(O)OBpin (Рin = OCMe2CMe2O) или до MeOBpin, при использовании избытка HBРin (4,4,5,5-тетраметил-1,3,2-диоксаборолан) [37]. Таким образом, N,N-бидентатные лиганды способны расширить область применения гидридов галлия, позволяя создать молекулярные системы, альтернативные катализаторам на основе d-элементов.
Мы показали, что комплексы непереходных металлов с редокс-активными азотсодержащими N,N-хелатирующими лигандами проявляют необычную реакционную способность по отношению к различным классам соединений [39–46]. Например, дигаллан [(Dpp-bian)Ga–Ga(Dpp-bi-an)] (Dpp-bian = 1,2-бис[(2,6-диизопропилфенил)имино]аценафтен) демонстрирует дуализм переноса электрона в реакциях с рядом окислителей: в зависимости от субстрата электроны переносятся на последний либо с лигандов, либо со связи металл–металл [42, 43]. Подобно переходным металлам дигаллан способен к двухэлектронному окислительному присоединению органических галогенидов [47]. Кроме того, комплексы галлия и алюминия с дианионом Dpp-bian обратимо присоединяют алкины и еноны [39, 40, 44, 48]. В этих реакциях субстрат образует новые связи как с атомом металла, так и с редокс-активным лигандом. Очевидно, что применение редокс-активных лигандов в химии гидридов металлов может привести к металлокомплексам, которые также обладают нетипичной для гидридов реакционной способностью. При этом открываются перспективы создания эффективных, доступных и малотоксичных галлийгидридных реагентов или катализаторов для реакций восстановления и изомеризации, например ненасыщенных субстратов.
Недавно мы получили и охарактеризовали новые мономерные гидридные соединения галлия [(Dpp-bian)GaH(R)] (R = Cl, H, OSi(Ph)H2), содержащие редокс-активный лиганд Dpp-bian. Несмотря на то что эти соединения оказались инертны по отношению к некоторым алкинам и алкенам, реализованный процесс гидросилилирования бензальдегида фенилсиланом в присутствии каталитических количеств дигидрида [(Dpp-bian)GaH2] допускает возможность внедрения по фрагменту Ga–H субстратов с активированными кратными связями [49].
В настоящей работе мы сообщаем о синтезе, молекулярных структурах и спектральных характеристиках новых гидридов галлия с аценафтен-1,2-дииминовыми лигандами [(Dpp-bian)GaH(Py)] (II) и [(Dpp-bian)GaH(i-Pr2ImMe)] (III).
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Соединение [(Dpp-bian)GaH2] (I) [49] получено по известной методике. Соединения I–III чувствительны к кислороду и влаге, поэтому все манипуляции по их синтезу, выделению и идентификации выполняли в вакууме с использованием техники Шленка или в атмосфере азота (Glovebox M. Braun). Толуол и бензол (Aldrich) сушили и хранили над натрийбензофеноном, пиридин (Aldrich) сушили натрием и отбирали конденсацией в вакууме непосредственно перед использованием. Дейтеропиридин (Aldrich) сушили натрием, дейтеробензол (Aldrich) – над натрийбензофеноном. Растворители отбирали конденсацией в вакууме в ампулы для регистрации спектров ЯМР, содержащие образцы исследуемых соединений.
ИК-спектры получали на спектрометре ФСМ-1201 в области 3998–449 cм–1 (образцы в вазелиновом масле). Спектр ЯМР 1H записывали на спектрометрах Bruker DPX-200 (200 МГц) и Bruker Avance III (400 МГц). Элементный анализ выполняли сжиганием образцов в кислороде по методу Прегля.
Синтез [(Dpp-bian)GaH(Py)] (II). Раствор 0.73 г (1.27 ммоль) соединения I в пиридине (20 мл) нагревали 4 ч при 160°С. После замены пиридина на толуол полученный синий раствор концентрировали до объема 10 мл. В течение 24 ч при 10°С образуются синие кристаллы соединения II. Выход 0.73 г (77%).
Спектр ЯМР 1Н (200 МГц; C5D5N; 298 К; δ, м.д.; J, Гц): 7.42–7.22 (м., 9Н), 7.22–7.10 (м., 4Н), 7.00–6.88 (м., 2Н), 6.23 (д., 2Н, J = 6.8), 3.72 (септ., 4Н, J = 6.8), 1.13 (д., 12Н, J = 6.8), 0.88 (д., 12Н, J = = 6.8).
ИК-спектр (ν, cм–1): 1933 с, 1610 ср, 1593 сл, 1574 сл, 1514 с, 1364 сл, 1332 сл, 1316 сл, 1251 ср, 1211 сл, 1178 сл, 1158 сл, 1136 сл, 1105 сл, 1081 сл, 1065 ср, 1054 сл, 1045 ср, 1015 сл, 997 сл, 997 сл, 968 сл, 925 с, 903 ср, 813 ср, 806 ср, 767 с, 734 с, 695 с, 663 ср, 636 ср, 624 ср, 544 сл, 534 сл, 522 сл, 465 ср.
Синтез [(Dpp-bian)GaH(i-Pr2ImMe)] (III). Мето-д А. К раствору 0.40 г (0.69 ммоль) соединения I в бензоле (20 мл) добавляли 1,3-диизопропил-4,5-диметилимидазол-2-илиден (i-Pr2ImMe) (0.12 г, 0.69 ммоль). Смесь перемешивали 15 мин при 50°С. После охлаждения до комнатной температуры из реакционной смеси удалили растворитель, оставшийся мелкокристаллический осадок соединения III промывали гексаном (5 мл) и высушивали в вакууме. Выход 0.39 г (75%).
Метод Б. В ампуле для регистрации спектров ЯМР к раствору 0.02 г (0.02 ммоль) соединения II добавляли i-Pr2ImMe (0.004 г, 0.02 ммоль). Смесь перемешивали 5 мин. Спектр ЯМР 1Н соответствует продукту, полученному методом А.
Спектр ЯМР 1Н (400 МГц; C6D6; 298 К; δ, м.д.; J, Гц): 7.37–7.25 (м., 6Н), 7.07 (д., 2Н, J = 6.7), 6.89 (т., 2Н), 6.40 (д., 2Н, J = 6.7), 5.42 (септ., 2Н, J = = 7.0), 4.23 (септ., 2Н, J = 6.7), 3.67 (септ., 2Н, J = = 6.7), 1.48 (д., 6Н, J = 6.7), 1.46 (с., 6Н), 1.34 (д., 6Н, J = 6.7), 1.21 (д., 6Н, J = 6.7), 0.97 (д., 12Н, J = 7.0), 0.88 (д., 6Н, J = 6.7).
ИК-спектр (ν, cм–1): 1860 с, 1614 сл, 1589 сл, 1511 с, 1335 ср, 1312 сл, 1256 ср, 1208 сл, 1179 ср, 1133 сл, 1109 ср, 1076 сл, 1056 сл, 999 сл, 936 сл, 924 ср, 893 сл, 810 с, 762 с, 722 ср, 683 сл, 634 ср, 622 сл, 608 сл, 548 ср, 517 сл, 497 с.
РСА II. Пригодные для РСА кристаллы соединения II получали кристаллизацией из толуола. Выбранный для анализа кристалл покрывали минеральным маслом (Aldrich) и фиксировали на стеклянном капилляре, затем помещали в холодный поток азота дифрактометра Bruker D8 Quest. Экспериментальные наборы интенсивностей (МоKα-излучение, λ = 0.71073 Å, ω- и φ-сканирование) интегрированы по программе SAINT [50]. SADABS [51] использована для введения поправок на поглощение. Структура расшифрована методом “dual-space” по программе SHELXT [52]. Неводородные атомы уточнены полноматричным МНК по $F_{{hkl}}^{2}$ в анизотропном приближении с помощью программных пакетов SHELXTL [53]. Атомы водорода помещены в геометрически рассчитанные положения и уточнены в модели наездника. Исключение составляют атомы водорода на атоме галлия, найденные из разностного синтеза Фурье электронной плотности и уточненные изотропно. В ассиметричной части кристаллической ячейки II найдена сольватная молекула толуола в общем положении. Основные кристаллографические характеристики и параметры рентгеноструктурного эксперимента для II приведены в табл. 1, избранные длины связей и валентные углы – в табл. 2.
Таблица 1.
Кристаллографические данные, параметры эксперимента и уточнения структуры II
| Параметр | Значение |
|---|---|
| М | 742.66 |
| Сингония | Моноклинная |
| Пр. гр. | P21/n |
| Температура, К | 100(2) |
| a, Å | 10.5769(3) |
| b, Å | 25.2796(6) |
| c, Å | 15.4536(4) |
| β, град | 104.613(1) |
| V, Å3 | 3998.32(18) |
| Z | 4 |
| ρ(выч.), г/см3 | 1.234 |
| μ, мм–1 | 0.725 |
| F(000) | 1576 |
| Размер кристалла, мм | 0.27 × 0.17 × 0.04 |
| Область измерений по θ, град | 2.11–28.00 |
| Индексы областей | –13 ≤ h ≤ 13, –33 ≤ k ≤ 33, –20 ≤ l ≤ 18 |
| Измеренных отражений | 32 870 |
| Независимых отражений (Rint) | 9633 (0.0346) |
| Отражений с I > 2σ(I) | 8126 |
| Поправка на поглощение (max/min) | 0.9144/0.7898 |
| Данные/ограничения/параметры | 9633/0/482 |
| GOOF | 1.023 |
| R1, wR2 (I > 2σ(I)) | 0.0322, 0.0742 |
| R1, wR2 (по всем отражениям) | 0.0427, 0.0773 |
| Δρmax/Δρmin, e Å–3 | 0.402/–0.427 |
Таблица 2.
Основные длины связей (Å) и валентные углы (град) в соединении II
| II | |
|---|---|
| Связь | d, Å |
| Ga–N(1) | 1.8930(11) |
| Ga–N(2) | 1.9022(12) |
| Ga–N(3) | 2.0535(12) |
| Ga–H(1) | 1.460(19) |
| N(1)–C(1) | 1.3850(18) |
| N(2)–C(2) | 1.3991(17) |
| C(1)–C(2) | 1.3750(19) |
| Угол | ω, град |
| N(1)GaN(2) | 90.25(5) |
| N(1)GaN(3) | 106.88(5) |
| N(1)GaH(1) | 124.8(7) |
| N(2)GaH(1) | 127.6(8) |
| N(3)GaH(1) | 102.1(8) |
| N(2)GaN(3) | 102.55(5) |
Структура II зарегистрирована в Кембриджском банке структурных данных (CCDC № 1973549; ccdc.cam.ac.uk/getstructures).
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Исследования, проведенные нами в области гидридов металлов 13-й группы с редокс-активными лигандами показали, что гидриды алюминия могут быть стабилизированы как дианионом Dpp-bian, так и его анион-радикалом [54–56], в то время как все полученные производные галлия стабилизированы лишь анион-радикальным лигандом [49].
Синтез гидрида алюминия с дианионом Dpp-bian обменной реакцией [(Dpp-bian)Na2] с HAlCl2 приводит к образованию комплекса [(Dpp-b-ian)AlH(THF)] [55], тогда как аналогичная реакция [(Dpp-bian)Na2] с HGaCl2 дает дианионное производное [(Dpp-bian)Ga–Ga(Dpp-bian)] [57, 58]. Дигаллан также образуется в реакции смешанного галоген-гидридного комплекса [(Dpp-b-ian)Ga(H)Cl] с металлическим натрием, что указывает на нестабильность дианионного моногидрида галлия. Тем не менее ранее мы обнаружили, что галоген-производные галлия с дианионными дииминовыми лигандами [(Dpp-bian)Ga(I)Py] [42] и [(Dpp-bian)Ga(Cl)Py] [47] могут быть ста-билизированы таким сильным основанием Льюиса, как пиридин. Известный в литературе моногидрид галлия с диазадиеновым лигандом [{((Dpp)NCH)2}GaH(Mes2ImH)] (Mes2ImH = = 1,3-мезитилимидазол-2-илиден) [32], также содержит электронодонорную молекулу карбена, что говорит о возможности получения дианионного аценафтендииминового комплекса гидрида галлия этим же путем.
Действительно, нагревание раствора соединения I в пиридине приводит к элиминированию одного из атомов водорода и образованию комплекса [(Dpp-bian)GaH(Py)] (II) (cхема 1). Соединение II выделено в виде синих кристаллов с выходом 77%.
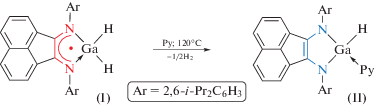
Схема 1 .
В ИК-спектре комплекса II колебания связи Ga–H проявляются в виде интенсивной полосы при 1933 см–1. Это значение довольно близко к таковым для соединений I (1897 и 1872 cм–1) и [(Dpp-bian)GaH(Cl)] (1939 cм–1) [49], но отличается от значения для диазадиенового гидрида [{((Dpp)NCH)2}GaH(Mes2ImH)] (1854 см–1) [32].
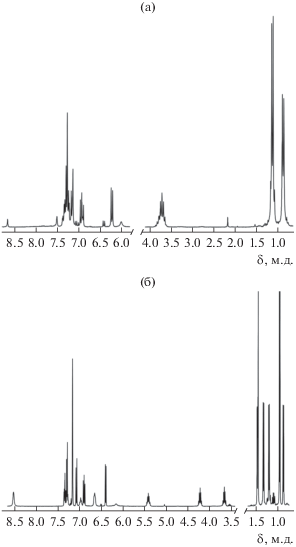
Спектр ЯМР 1H соединения II (рис. 1а) в дейтеропиридине содержит сигналы протонов метильных групп 2,6-i-Pr2-C6H3-заместителей (дублеты 0.88 (12Н) и 1.13 (12Н) м.д.), а также септет метиновых протонов (3.72 (4Н) м.д.) этих заместителей. Дублет при 6.23 (2Н) м.д., а также мультиплеты в диапазонах 6.88–7.00 (2Н), 7.10–7.22 (4Н) и 7.22–7.42 (9Н) м.д. относятся к ароматическим протонам нафталинового фрагмента, фенильных заместителей при атомах азота и координированного пиридина.
Аналогичная ситуация наблюдается и в реакции соединения I с карбеном i-Pr2ImMe (cхема 2). Реакция протекает в бензоле при 50°С в течение 15 мин, сопровождается изменением цвета раствора с коричневого на зеленый и приводит к образованию продукта [(Dpp-bian)GaH(i-Pr2ImMe)] (III). В ИК-спектре комплекса III колебания связи Ga–H проявляются в виде интенсивной полосы при 1860 см–1. Это значение близко к соответствующему значению для соединения [{((Dpp)NCH)2}GaH(Mes2ImH)] (1854 см–1) [32], однако заметно отличается от такового для соединений II (1933 см–1) и гидрида алюминия [(Dpp-bian)AlH(i-Pr2ImMe)] (1732 см–1) [59].
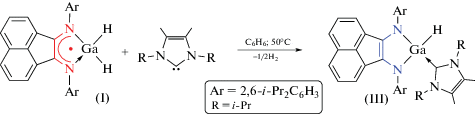
Схема 2 .
Спектр ЯМР 1H соединения III (рис. 1б) содержит сигналы несимметричного лиганда Dpp-bian: четыре попарно эквивалентных метиновых протона изопропильных групп в 2,6-i-Pr2C6H3-заместителей проявляются в виде септетов при 4.23 (2Н) и 3.67 (2Н) м.д. Протоны метильных групп изопропильных заместителей дают четыре дублета при 1.48 (6Н), 1.34 (6Н), 1.21 (6Н) и 0.88 (6Н) м.д. Септет и дублет протонов i-Pr-заместителей, а также синглет протонов метильных групп карбенового лиганда проявляются при 5.42 (2Н), 0.97 (12H) и 1.46 (6Н) м.д. соответственно. Ароматические протоны наблюдаются в виде двух дублетов (7.07 (2Н), 6.40 (2Н) м.д.), псевдотриплета при 6.89 (2Н) и мультиплета в диапазоне δ 7.37–7.25 (6Н) м.д.
Комплекс [(Dpp-bian)AlH(i-Pr2ImMe)], аналогичный соединению III, мы получили реакцией соединения [(Dpp-bian)AlH(THF)] с имидазол-2-илиденом путем замещения молекулы координированного THF на карбен [59]. Реакция соединения II с i-Pr2ImMe в ампуле для регистрации спектров ЯМР сопровождается изменением цвета раствора с синего на зеленый, полученный спектр аналогичен описанному выше и свидетельствует о замещении молекулы пиридина на NHC-лиганд, в результате чего образуется комплекс III и свободный пиридин (8.53 (2Н), 6.98 (1Н), 6.66 (2Н) м.д.) (рис. 1б).
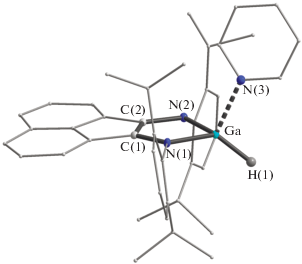
Согласно данным РСА, соединение II представляет собой мономерный четырехкоординационный комплекс галлия (рис. 2). Анализ длин связей в металлоцикле комплекса II указывает на дианионное состояние Dpp-bian лиганда в нем. Связи N(1)–C(1) 1.3850(18) и N(2)–C(2) 1.3991(17) Å удлинены по сравнению с соответствующими связями в комплексах I (N(1)–C(1) 1.3238(18), N(2)–C(2) 1.3291(18) Å) и [(Dpp-bian)Ga(H)Cl] (N(1)–C(1) 1.327(3), N(2)–C(2) 1.326(3) Å) с анион-радикалом Dpp-bian [49]. С другой стороны, расстояния Ga–N в комплексе II (1.8930(11) и 1.9022(12) Å) заметно меньше расстояний Ga–N в гидридах галлия (Ga–N(1) 1.9825(12), Ga–N(2) 1.9968(12) Å для I и Ga–N(1) 1.9581(17), Ga–N(2) 1.9519(17) Å для [(Dpp-bian)Ga(H)Cl]), что свидетельствует о более сильном связывании галлия с дианионом Dpp-bian по сравнению с его анион-радикалом. Связь Ga–H в комплексе II (1.460(19) Å) близка к соответствующим расстояниям в гидриде I (1.53(2) и 1.47(2) Å) и соединении [{((Dpp)NCH)2}GaH(Mes2ImH)] (1.498(16) Å) [32].
Рис. 2.
Молекулярная структура II. Тепловые эллипсоиды приведены с 30%-ной вероятностью. Атомы водорода, за исключением H(1), не показаны.
Таким образом, получены и охарактеризованы новые мономерные гидриды галлия [(Dpp-b-ian)-GaH(Py)] (II) и [(Dpp-bian)GaH(i-Pr2ImMe)] (III), содержащие редокс-активный лиганд Dpp-bian в дианионной форме. Полученные нами ранее гидриды галлия оказались инертными к некоторым непредельным соединениям, однако процесс восстановительного элиминирования атома водорода под действием донорных молекул, таких как пиридин и имидазол-2-илиден, вероятно, может быть использован для гидрирования подобных субстратов. При этом продемонстрирована возможность стабилизирования дианионного моногидрида галлия молекулой сильного основания Льюиса. В ближайшее время мы планируем исследовать реакционную способность новых комплексов II и III по отношению к некоторым органическим соединениям, а также малым молекулам, в частности CO и CO2. Это позволит определить как возможность использования данных производных в катализе реакций органического синтеза, так и расширить фундаментальные представления о химии комплексов металлов с редокс-активными лигандами.
Авторы заявляют об отсутствии конфликта интересов.
Список литературы
Downs A.J., Pulham C.R. // Chem. Soc. Rev. 1994. V. 23. № 3. P. 175.
Jegier J.A., Gladfelter W.L. // Coord. Chem. Rev. 2000. V. 206–207. P. 631.
Aldridge S., Downs A.J. // Chem. Rev. 2001. V. 101. № 11. P. 3305.
Uhl W. // Coord. Chem. Rev. 2008. V. 252. № 15–17. P. 1540.
Luo B., Gladfelter W.L. // Chem. Commun. 2000. № 10. P. 825.
Luo B., Young V.G., Gladfelter W.L. // Inorg. Chem. 2000. V. 39. № 8. P. 1705.
Cowley A.R., Downs A.J., Himmel H.J. et al. // Dalton Trans. 2005. № 9. P. 1591.
Luo B., Kucera B.E., Gladfelter W.L. // Chem. Commun. 2005. № 27. P. 3463.
Schmidbaur H., Findeiss W., Gast E. // Angew. Chem. Int. Ed. 1965. V. 4. № 2. P. 152.
Ohshita J., Schmidbaur H. // J. Organomet. Chem. 1993. V. 453. № 1. P. 7.
Nogai S., Schmidbaur H. // Inorg. Chem. 2002. V. 41. № 18. P. 4770.
Eisch J.J. // J. Am. Chem. Soc. 1962. V. 84. № 20. P. 3830.
Uhl W., Cuypers L., Graupner R. et al. // Z. Anorg. Allg. Chem. 2001. V. 627. № 4. P. 607.
Uhl W., Cuypers L., Geiseler G. et al. // Z. Anorg. Allg. Chem. 2002. V. 628. № 5. P. 1001.
Schmidbaur H., Klein H.F. // Angew. Chem. Int. Ed. 1966. V. 5. № 3. P. 312.
Schumann H., Hartmann U., Wassermann W. // Polyhedron. 1990. V. 9. № 2–3. P. 353.
Takami K., Mikami S., Yorimitsu H. et al. // J. Org. Chem. 2003. V. 68. № 17. P. 6627.
Uhl W., Breher W., Haddadpour S. et al. // Organometallics. 2005. V. 24. № 9. P. 2210.
Uhl W., Haddadpour S., Matar M. // Organometallics. 2006. V. 25. № 1. P. 159.
Uhl W., Bock H.R., Breher F. et al. // Organometallics. 2007. V. 26. № 9. P. 2363.
Uhl W., Claesener M., Haddadpour S. et al. // Dalton Trans. 2007. № 4. P. 417.
Uhl W., Claesener M. // Inorg. Chem. 2008. V. 47. № 11. P. 4463.
Uhl W., Claesener M., Haddadpour S. et al. // Z. Anorg. Allg. Chem. 2008. V. 634. № 15. P. 2889.
Uhl W., Claesener M. // Inorg. Chem. 2008. V. 47. № 2. P. 729.
Uhl W., Claesener M., Hepp A. et al. // Dalton Trans. 2009. № 47. P. 10550.
Uhl W., Hepp A., Westenberg H. et al. // Organometallics. 2010. V. 29. № 6. P. 1406.
Uhl W., Claesener M., Kovert D. et al. // Organometallics. 2011. V. 30. № 11. P. 3075.
Uhl W., Kovert D., Zemke S. et al. // Organometallics. 2011. V. 30. № 17. P. 4736.
Uhl W., Layh M., Rhotert I. et al. // Z. Naturforsch. 2013. V. 68. № 5–6. P. 503.
Schmidt E.S., Jockisch A., Schmidbaur H. // Dalton Trans. 2000. № 7. P. 1039.
Cole M.L., Jones C., Junk P.C. et al. // Chem. Eur. J. 2005. V. 11. № 15. P. 4482.
Jones C., Mills D.P., Rose R.P. // J. Organomet. Chem. 2006. V. 691. № 13. P. 3060.
Bonello O., Jones C., Stasch A. et al. // Organometallics. 2010. V. 29. № 21. P. 4914.
Singh S., Ahn H.J., Stasch A. et al. // Inorg. Chem. 2006. V. 45. № 4. P. 1853.
Seifert A., Scheid D., Linti G. et al. // Chem. Eur. J. 2009. V. 15. № 44. P. 12114.
Turner J., Abdalla J.A.B., Bates J.I. et al. // Chem. Sci. 2013. V. 4. № 11. P. 4245.
Abdalla J.A.B., Riddlestone I.M., Tirfoin R. et al. // Angew. Chem. Int. Ed. 2015. V. 54. № 17. P. 5098.
Herappe-Mejía E., Trujillo-Hernández K., Garduño-Jiménez J. et al. // Dalton Trans. 2015. № 44. P. 16894.
Fedushkin I.L., Nikipelov A.S., Lyssenko K.A. et al. // J. Am. Chem. Soc. 2010. V. 132. № 23. P. 7874.
Fedushkin I.L., Moskalev M.V., Lukoyanov A.N. et al. // Chem. Eur. J. 2012. V. 18. № 36. P. 11264.
Fedushkin I.L., Moskalev M.V., Baranov E.V. et al. // J. Organomet. Chem. 2013. V. 747. P. 235.
Fedushkin I.L., Skatova A.A., Dodonov V.A. et al. // Inorg. Chem. 2014. V. 53. № 10. P. 5159.
Fedushkin I.L., Nikipelov A.S., Skatova A.A. et al. // Eur. J. Inorg. Chem. 2009. № 25. P. 3742.
Fedushkin I.L., Kazarina O.V., Lukoyanov A.N. et al. // Organometallics. 2015. V. 34. № 8. P. 1498.
Moskalev M.V., Lukoyanov A.N., Baranov E.V. et al. // Dalton Trans. 2016. № 45. № 40. P. 15872.
Dodonov V.A., Skatova A.A., Cherkasov A.V. et al. // Russ. Chem. Bull., Int. Ed. 2016. V. 65. № 5. P. 1171.
Fedushkin I.L., Dodonov V.A., Skatova A.A. et al. // Chem. Eur. J. 2018. V. 24. № 8. P. 1877.
Fedushkin I.L., Nikipelov A.S., Morozov A.G. et al. // Chem. Eur. J. 2012. V. 18. № 1. P. 255.
Sokolov V.G., Koptseva T.S., Moskalev M.V. et al. // Inorg. Chem. 2017. V. 56. № 21. P. 13401.
SAINT. Version 8.38A. Data Reduction and Correction Program. Madison (WI, USA): Bruker AXS, 2017.
Sheldrick G.M. SADABS. Version 2016/2. Bruker/Siemens Area Detector Absorption Correction Program. Madison (WI, USA): Bruker AXS, 2016.
Sheldrick G.M. // Acta Crystallpgr. A. 2015. V. 71. P. 3.
Sheldrick G.M. SHELXTL. Version 6.14. Structure Determination Software Suite. Madison (WI, USA): Bruker AXS, 2003.
Sokolov V.G., Koptseva T.S., Moskalev M.V. et al. // Russ. Chem. Bull., Int. Ed. 2017. V. 66. № 9. P. 1569.
Sokolov V.G., Koptseva T.S., Dodonov V.A. et al. // Russ. Chem. Bull., Int. Ed. 2018. V. 67. № 12. P. 2164.
Sokolov V.G., Koptseva T.S., Moskalev M.V. et al. // Russ. J. Coord. Chem. 2019. V. 45. № 9. P. 637. https://doi.org/10.1134/S1070328419090070
Fedushkin I.L., Lukoyanov A.N., Tishkina A.N. et al. // Chemistry Eur. J. 2010. V. 16. № 25. P. 7563.
Fedushkin I.L., Lukoyanov A.N., Ketkov S.Y. et al. // Chem. Eur. J. 2007. V. 13. № 25. P. 7050.
Sokolov V.G., Koptseva T.S., Rumyantcev R.V. et al. // Organometallics. 2020. V. 39. № 1. P. 66.
Дополнительные материалы отсутствуют.
Инструменты
Координационная химия