

Координационная химия, 2020, T. 46, № 6, стр. 325-332
Компьютерное моделирование строения и магнитных свойств комплексов кобальта с N-замещенными пиридинофанами и функционализированными радикалами о-бензохинонами
В. И. Минкин 1, А. А. Старикова 1, *, М. Г. Чегерев 1, А. Г. Стариков 1
1 Научно-исследовательский институт физической и органической химии
Южного федерального университета
Ростов-на-Дону, Россия
* E-mail: alstar@ipoc.sfedu.ru
Поступила в редакцию 20.12.2019
После доработки 29.01.2020
Принята к публикации 31.01.2020
Аннотация
Методом теории функционала плотности (DFT UTPSSh/6-311++G(d,p)) изучено строение и рассчитаны энергетические и магнитные характеристики моноядерных комплексов кобальта с N,N'-диалкил-2,11-диаза[3.3]-(2,6)пиридинофанами и о-бензохиноновыми лигандами, содержащими стабильные радикалы (нитронилнитроксил и TEMPO). Установлено, что варьирование алкильных заместителей при атомах азота тетраазамакроцикла позволяет управлять устойчивостью электромерных форм комплексов и, как следствие, возможностью реализации механизмов магнитной бистабильности (спин-кроссовер и валентная таутомерия). Природа радикальной группы определяет силу обменных взаимодействий, в которых она участвует, но не оказывает влияния на обмен в редокс-активном фрагменте.
Поиск комплексов переходных металлов с открытой оболочкой, проявляющих один из наиболее значимых видов электронной бистабильности – вариабельность магнитных свойств, представляет собой перспективное направление химии и материаловедения. Такие магнитно-активные соединения могут найти практическое применение в качестве основы молекулярных переключателей, молекулярных магнитов, устройств памяти и спиновых кубитов [1–9]. Наиболее распространенными механизмами, приводящими к изменению магнитных характеристик в результате внешних воздействий (температуры, света, давления, электромагнитного поля и др.), являются спин-кроссовер (СКО), обусловленный электронной перегруппировкой внутри валентной оболочки иона металла между его низкоспиновым и высокоспиновым состояниями [4, 10], и валентная таутомерия (ВТ) – обратимый внутримолекулярный перенос электронов с участием иона переходного металла и координированного редокс-активного лиганда [11]. СКО широко изучен в комплексах железа [12, 13], но также встречается в соединениях кобальта [14], в то время как ВТ детально исследована в кобальтовых системах с о-бензохинонами (диоксоленами) [15–18].
Ранее был опубликован цикл работ, посвященных синтезу и исследованию катионных комплексов переходных металлов, включающих один редокс-активный лиганд и тетрадентатное макроциклическое азотсодержащее основание [19–31]. Этот структурный мотив успешно использовался для получения биядерных магнитно-активных соединений [32–40]. Построенные таким способом системы привлекают повышенный интерес благодаря их способности претерпевать как ВТ [19, 21–28], так и СКО [30, 31] в зависимости от природы тетраазамакроциклического лиганда. Недавно посредством теоретического и экспериментального исследования серии о-бензохиноновых комплексов кобальта с N,N'-диалкил-2,11-диаза[3.3]-(2,6)пиридинофанами (пиридинофанами) показано, что возможность протекания перегруппировок, приводящих к изменению спиновых состояний, и тип механизма этого процесса могут управляться заместителями при атомах азота макроцикла [41–43].
Металлокомплексы с о-хинонами, функционализированными радикальными фрагментами, привлекают особое внимание благодаря присутствию в них трех носителей спина: ион металла, анион-радикальная семихиноновая (SQ) форма лиганда и парамагнитный заместитель. Известны комплексы первого переходного ряда с о-хиноновыми лигандами, включающими нитронилнитроксильный радикал, связанный с диоксоленовым кольцом непосредственно либо через мостиковые группы [44–48]. Выполнены исследования, посвященные изучению гетероспиновых комплексов марганца, кобальта, никеля и меди с двумя редокс-активными лигандами, содержащими парамагнитную группу во вспомогательном азотсодержащем основании [49–52], а также соединений кобальта, включающих 3,6-ди-трет-бутил-о-бензохиноновые лиганды и 1,3,5-трифенилформазанатный анион [53]. Успешный синтез и установление строения комплекса железа(III) с катехолом (Cat), содержащим нитронилнитроксильный заместитель, и трис(2-пиридилметил)аминовым лигандом [48], а также многообразие химии стабильных радикалов [54, 55] открывают широкие перспективы для получения новых соединений, сконструированных подобным способом.
В настоящей работе с целью изучения особенностей магнитного поведения комплексов такого типа и выяснения влияния радикального заместителя на способность претерпевать механизмы ВТ и/или СКО мы провели квантовохимические расчеты геометрических, энергетических и магнитных характеристик пиридинофановых комплексов кобальта с о-бензохиноном, содержащим 4,4,5,5-тетраметил-4,5-дигидро-1H-имидазол-3-оксид-1-оксильный (нитронилнитроксильный, NN) и (2,2,6,6-тетраметилпиперидин-1-ил)оксильный (ТЕМРО) радикалы (соединения I и II соответственно).
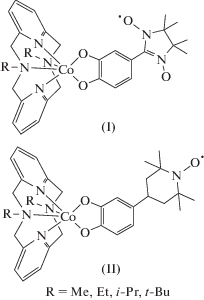
МЕТОДИКА РАСЧЕТОВ
Расчеты проводили с помощью программы Gaussian 16 [56] методом теории функционала плот-ности (DFT) с использованием функционала UTPSSh [57, 58] и расширенного базиса 6‑311++G(d,p), сочетание которых корректно воспроизводит энергетические и магнитные характеристики комплексов, проявляющих механизмы магнитной бистабильности [41–43, 59–65]. Согласно полученным в данном приближении результатам расчетов экспериментально изученных соединений кобальта, СКО реализуется при ΔEHS–LS < 6 ккал/моль, ВТ – при ΔEHS–LS < < 10 ккал/моль [43]. Ранее была показана необходимость учета внешнесферных противоионов при DFT-изучении комплексов переходных металлов с редокс-активными лигандами [66]. В этой связи в настоящем исследовании квантовохимические расчеты проведены для соединений, включающих гексафторфосфатный анион. Локализацию стационарных точек на поверхности потенциальной энергии (ППЭ) осуществляли путем полной оптимизации геометрии молекулярных структур с проверкой стабильности DFT волновой функции и расчетом силовых констант. Вычисление параметров обменного взаимодействия (J, см–1) осуществляли в рамках формализма “нарушенной симметрии” (broken symmetry, BS) [67] с использованием метода обобщенной проекции спина, предложенного Ямагучи [68]. Графические изображения молекулярных структур получали при помощи программы ChemCraft [69].
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
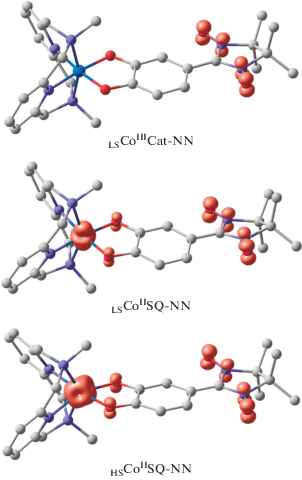
Основным состоянием комплекса I (R = Me) является низкоспиновый электронный изомер (электромер [70]) LSCoIIICat-NN, содержащий диамагнитные ион кобальта и катехолатную форму редокс-лиганда (табл. 1). Единственный неспаренный электрон в этой структуре локализован на радикальном NN-фрагменте (рис. 1). Найденный на квартетной ППЭ изомер LSCoIISQ-NN расположен выше низкоспиновой формы на 5.8 ккал/моль. Значительная дестабилизация структуры с высокоспиновым ионом кобальта HS-CoIISQ-NN (12.9 ккал/моль) указывает на ее термическую недостижимость. Следовательно, соединение I (R = Me) будет находиться в низкоспиновом состоянии в широком диапазоне температур, что согласуется с полученными ранее результатами в родственных соединениях [31, 41–43] и свидетельствует о незначительном влиянии нитронилнитроксильного радикала на энергетические характеристики комплекса. В то же время присутствие в молекуле NN-фрагмента обеспечивает парамагнетизм электромера LSCoIIICat-NN.
Таблица 1.
Спин (S), полная энергия без учета (Е) и с учетом энергии нулевых колебаний (EZPE), значение оператора квадрата спина (S2), относительная энергия без учета (ΔЕ) и с учетом энергии нулевых колебаний (ΔEZPE) в электромерах комплексов I и II (R = Me, Et, i-Pr, t-Bu), рассчитанные методом DFT UTPSSh/6-311++G(d,p)
| Электромер | S | Е | EZPE | S2 | Е | EZPE |
|---|---|---|---|---|---|---|
| ат. ед. | ккал/моль | |||||
| I (R = Me) | ||||||
| LSCoIIICat-NN | 1/2 | –4079.673906 | –4079.041809 | 0.794 | 0.0 | 0.0 |
| LSCoIISQ-NN | 3/2 | –4079.664656 | –4079.034870 | 3.807 | 5.8 | 4.4 |
| HSCoIISQ-NN | 5/2 | –4079.653341 | –4079.025625 | 8.803 | 12.9 | 10.2 |
| I (R = Et) | ||||||
| LSCoIIICat-NN | 1/2 | –4158.327737 | –4157.639282 | 0.794 | 0.0 | 0.0 |
| LSCoIISQ-NN | 3/2 | –4158.319071 | –4157.633135 | 3.807 | 5.4 | 3.9 |
| HSCoIISQ-NN | 5/2 | –4158.306655 | –4157.622437 | 8.802 | 13.2 | 10.6 |
| I (R = i-Pr) | ||||||
| LSCoIIICat-NN | 1/2 | –4236.977669 | –4236.233267 | 0.794 | 0.0 | 0.0 |
| LSCoIISQ-NN | 3/2 | –4236.972022 | –4236.229827 | 3.807 | 3.5 | 2.2 |
| HSCoIISQ-NN | 5/2 | –4236.962169 | –4236.221770 | 8.802 | 9.7 | 7.2 |
| I (R = t-Bu) | ||||||
| LSCoIIICat-NN | 1/2 | –4315.607707 | –4314.809305 | 0.795 | 2.8 | 3.8 |
| LSCoIISQ-NN | 3/2 | –4315.612097 | –4314.815432 | 3.807 | 0.0 | 0.0 |
| HSCoIISQ-NN | 5/2 | –4315.600592 | –4314.805657 | 8.802 | 7.2 | 6.1 |
| II (R = Me) | ||||||
| LSCoIIICat-TEMPO | 1/2 | –4028.961901 | –4028.270447 | 0.754 | 0.0 | 0.0 |
| LSCoIISQ-TEMPO | 3/2 | –4028.953434 | –4028.264375 | 3.771 | 5.3 | 3.8 |
| HSCoIISQ-TEMPO | 5/2 | –4028.941725 | –4028.254331 | 8.766 | 12.7 | 10.1 |
| II (R = Et) | ||||||
| LSCoIIICat-TEMPO | 1/2 | –4107.615549 | –4106.867821 | 0.754 | 0.0 | 0.0 |
| LSCoIISQ-TEMPO | 3/2 | –4107.607851 | –4106.862373 | 3.771 | 4.8 | 3.4 |
| HSCoIISQ-TEMPO | 5/2 | –4107.595461 | –4106.851488 | 8.766 | 12.6 | 10.2 |
| II (R = i-Pr) | ||||||
| LSCoIIICat-TEMPO | 1/2 | –4186.265312 | –4185.461613 | 0.754 | 0.0 | 0.0 |
| LSCoIISQ-TEMPO | 3/2 | –4186.260861 | –4185.459493 | 3.771 | 2.8 | 1.3 |
| HSCoIISQ-TEMPO | 5/2 | –4186.250585 | –4185.451177 | 8.765 | 9.2 | 6.5 |
| II (R = t-Bu) | ||||||
| LSCoIIICat-TEMPO | 1/2 | –4264.895380 | –4264.037976 | 0.754 | 3.4 | 4.2 |
| LSCoIISQ-TEMPO | 3/2 | –4264.900850 | –4264.044631 | 3.770 | 0.0 | 0.0 |
| HSCoIISQ-TEMPO | 5/2 | –4264.889273 | –4264.034735 | 8.766 | 7.3 | 6.2 |
Рис. 1.
Распределение спиновой плотности (cutoff = = 0.035 e/Å3) в электромерах комплекса I (R = Me), рассчитанное методом DFT UTPSSh/6-311++G(d,p). Здесь и на рис. 2, 3 атомы водорода опущены для ясности.
При изучении комплекса I (R = Et) получены сходные результаты (табл. 1): существенная дестабилизация высокоспиновой структуры HS-CoIISQ-NN относительно основного состояния LSCoIIICat-NN свидетельствует о нереализуемости перегруппировок, сопровождающихся изменением спинового состояния системы.
Переход к соединению I (R = i-Pr) сопровождается сокращением разности энергий между низкоспиновым и высокоспиновым изомерами до величины 9.7 (7.2 с учетом ZPE) ккал/моль, которая указывает на возможность протекания термически инициированной ВТ LSCoIIICat-NN ⇄ HSCoIISQ-NN (рис. 2). Такое поведение находится в согласии с теоретическими и экспериментальными данными, полученными для 3,5-ди-трет-бутил-о-бензохинонового комплекса кобальта с N-изопропилзамещенным пиридинофаном [41]. Изучение обменных взаимодействий в электромере HSCoIISQ-NN показывает умеренное антиферромагнитное связывание спинов неспаренных электронов иона металла с другими радикалами, в то же время обмен между SQ- и NN-фрагментами носит сильный ферромагнитный характер (табл. 2), что позволяет ожидать существование обсуждаемой структуры в высокоспиновом состоянии в широком диапазоне температур. Таким образом, комплекс I (R = i-Pr) способен претерпевать редокс-изомерные превращения, сопровождающиеся изменением спина системы (S) с 3/2 до 5/2.
Рис. 2.
Оптимизированные геометрии электромеров комплексов I (R = i-Pr, t-Bu), рассчитанные методом DFT UT-PSSh/6-311++G(d,p). Здесь и на рис. 3 длины связей даны в ангстремах.
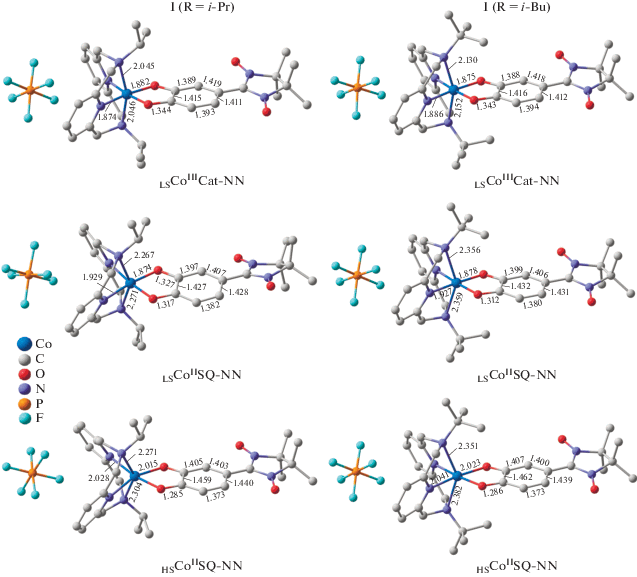
Таблица 2.
Спин (S) и параметр обменных взаимодействий (J, см–1) в электромерах LSCoIISQ-NN, HSCoIISQ-NN и LSCoIISQ-TEMPO, HSCoIISQ-TEMPO комплексов I и II (R = i-Pr, t-Bu), рассчитанные методом DFT UTPSSh/6-311++G(d,p)*
| Электромер | S | J1 | J2 | J3 |
|---|---|---|---|---|
| I (R = i-Pr) | ||||
| LSCoIISQ-NN | 3/2 | 835 | –71 | 529 |
| HSCoIISQ-NN | 5/2 | –34 | –59 | 898 |
| I (R = t-Bu) | ||||
| LSCoIISQ-NN | 3/2 | 832 | –47 | 438 |
| HSCoIISQ-NN | 5/2 | –51 | –14 | 610 |
| II (R = i-Pr) | ||||
| LSCoIISQ-TEMPO | 3/2 | 812 | –3 | 4 |
| HSCoIISQ-TEMPO | 5/2 | –48 | –2 | 3 |
| II (R = t-Bu) | ||||
| LSCoIISQ-TEMPO | 3/2 | 912 | –5 | 5 |
| HSCoIISQ-TEMPO | 5/2 | –82 | –5 | 2 |
Расчеты комплекса I (R = t-Bu) приводят к инверсии энергий изомеров LSCoIIICat-NN и LSCoIISQ-NN, в результате которой последний оказывается основным состоянием (табл. 1). Дестабилизация электромера, включающего низкоспиновый трехвалентный ион кобальта, обусловлена невозможностью формирования характерных для него коротких (не превышающих 2.05 Å) связей Сo–N. Это вызвано стерическими препятствиями, создаваемыми объемистыми трет-бутильными группами (рис. 2). Высокоспиновая форма HSCoIISQ-NN отстоит от основного состояния LSCoIISQ-NN на 7.2 (6.1 с учетом ZPE) ккал/моль, что делает вероятным СКО-процесс LSCoIISQ-NN ⇄ HSCoIISQ-NN и согласуется с полученными ранее данными по 3,5-ди-трет-бутил-о-бензохиноновому комплексу кобальта с N,N'-ди-трет-бутил-2,11-диаза[3.3]-(2,6)пиридинофаном [31, 41–43]. В электромере HSCoIISQ-NN, как и в рассмотренном выше высокоспиновом изомере комплекса I (R = = i-Pr), предсказаны умеренные антиферромагнитные обменные взаимодействия, в которые вовлечен ион металла, и сильное ферромагнитное связывание внутри лиганда (табл. 2). Следовательно, варьирование алкильных групп при атомах азота тетраазамакроцикла не приводит к изменению характера и силы обмена между парамагнитными центрами. Изомер LSCoIISQ-NN с низкоспиновым ионом кобальта характеризуется сильным ферромагнитным связыванием в редокс-активном фрагменте (J1 = 832 см–1), в то время как значения параметров J2 и J3, отвечающих за обмен СoII–NN и SQ–NN, близки к величинам, найденным для структуры HSCoIISQ-NN (табл. 2).
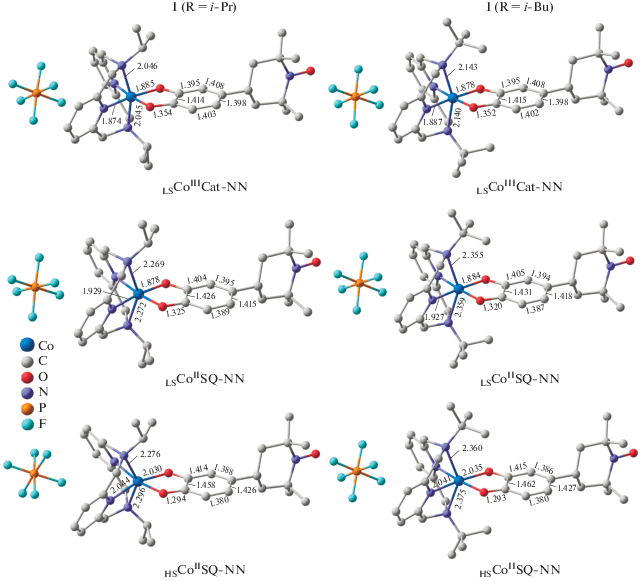
Из приведенных в табл. 1 результатов следует, что замена в о-бензохиноновом лиганде нитронилнитроксила на радикал ТЕМРО не оказывает существенного влияния на разность энергий между электромерами рассмотренных комплексов. Значительная дестабилизация высокоспиновых изомеров координационных соединений II (R = Me, Et) свидетельствует о нахождении обсуждаемых систем в низкоспиновом дублетном состоянии. Предсказанная величина энергетической щели между электромерами LSCoIIICat-TEMPO и HSCoIISQ-TEMPO комплекса II (R = i-Pr) (рис. 3) составляет 9.2 (6.5 с учетом ZPE) ккал/моль, что указывает на возможность протекания термически инициированной ВТ перегруппировки. В соединении II (R = t-Bu), содержащем трет-бутильные группы при атомах азота пиридинофанового лиганда, ожидается изменение магнитных свойств в результате СКО (табл. 1, рис. 3).
Рис. 3.
Оптимизированные геометрии электромеров комплексов II (R = i-Pr, t-Bu), рассчитанные методом DFT U-TPSSh/6-311++G(d,p).
Анализ значений параметра J1, приведенных в табл. 2, показывает сильное ферромагнитное связывание в паре LSCoII–SQ, в то же время высокоспиновый изомер способен проявлять умеренный антиферромагнитный обмен между ионом металла и семихиноном. Радикал TEMPO практически не взаимодействует с другими парамагнитными центрами (табл. 2). Примечательно, что характер обмена между ионом металла и SQ в соединениях I и II (R = i-Pr, t-Bu) аналогичен найденному в электромерах изученных ранее 3,5-ди-трет-бутил-о-бензохиноновых комплексов кобальта [31, 41–43]. Следовательно, рассмотренный способ введения радикала в семихинон не приводит к изменению обменных взаимодействий в редокс-активном фрагменте. В то же время природа радикальной группы (NN или TEMPO) определяет силу обменного связывания, в котором она участвует.
Таким образом, в результате проведенного компьютерного моделирования геометрических, энергетических и магнитных характеристик моноядерных комплексов кобальта с N,N'-диалкил-2,11-диаза[3.3]-(2,6)пиридинофанами и о-бензохиноновыми лигандами, содержащими нитронилнитроксил или TEMPO, установлено, что разность энергий между изомерами определяется объемностью алкильных заместителей в тетраазамакроцикле, но не зависит от наличия и типа радикальной группы. Соединения с N-метил- и N-этилзамещенными пиридинофанами будут находиться в низкоспиновом состоянии, включающем трехвалентный ион металла. В комплексах с N,N'-диизопропил-2,11-диаза[3.3]-(2,6)пиридинофаном ожидается термически инициированная валентно-таутомерная перегруппировка, а системы, содержащие трет-бутильные группы при атомах азота пиридинофанового основания, способны претерпевать спин-кроссовер. Преимуществом изученных соединений перед существующими аналогами, не содержащими радикальных заместителей [19–31, 41–43], является парамагнетизм всех электромеров, что расширяет потенциальные области применений соединений данного типа. Рациональный выбор радикала позволяет управлять силой обменных взаимодействий, в которые он вовлечен, при этом обмен между ионом кобальта и семихиноном остается неизменным. Использование при конструировании рассмотренных комплексов синтетически доступных структурных мотивов открывает возможности их получения.
Авторы заявляют об отсутствии конфликта интересов.
Список литературы
Sato O., Tao J., Zhang Y.Z. // Angew. Chem. Int. Ed. 2007. V. 46. № 13. P. 2152.
Molecular Switches / Eds. Feringa B.L., Browne W.R. Weinheim: Wiley-VCH, 2011. 824 p.
Aromí G., Aguilà D., Gamez P. et al. // Chem. Soc. Rev. 2012. V. 41. № 2. P. 537.
Spin-Crossover Materials: Properties and Applications / Ed. Halcrow M.A. Chichester: John Wiley&Sons, 2013. 564 p.
Sato O. // Nature Chem. 2016. V. 8. № 7. P. 644.
Senthil Kumar K., Ruben M. // Coord. Chem. Rev. 2017. V. 346. P. 176.
Lutsenko I.A., Kiskin M.A., Efimov N.N. et al. // Polyhedron. 2017. V. 137. P. 165.
Bazhina E.S., Aleksandrov G.G., Kiskin M.A. et al. // Eur. J. Inorg. Chem. 2018. № 47. P. 5075.
Lutsenko I.A., Kiskin M.A., Nelyubina Y.V. et al. // Polyhedron. 2019. V. 159. P. 426.
Spin Crossover in Transition Metal Compounds. I: Topics in Current Chemistry. V. 233 / Eds. Gütlich P., Goodwin H.A. Berlin–Heidelberg: Springer-Verlag, 2004.
Buchanan R.M., Pierpont C.G. // J. Am. Chem. Soc. 1980. V. 102. № 15. P. 4951.
Halcrow M.A. // Polyhedron. 2007. V. 26. № 14. P. 3523.
Harding D.J., Harding P., Phonsri W. // Coord. Chem. Rev. 2016. V. 313. P. 38.
Hayami S., Kato K., Komatsu Y. et al. // Dalton Trans. 2011. V. 40. № 10. P. 2167.
Pierpont C.G. // Coord. Chem. Rev. 2001. V. 216–217. P. 99.
Shultz D.A. Valence Tautomerism in Dioxolene Complexes of Cobalt // Magnetism: Molecules to Materials II / Eds. Miller J.S., Drillon M. Wiley, New York, 2001. P. 281.
Evangelio E., Ruiz-Molina D. // Eur. J. Inorg. Chem. 2005. № 15. P. 2957.
Tezgerevska T., Alley K.G., Boskovic C. // Coord. Chem. Rev. 2014. V. 268. P. 23.
Benelli C., Dei A., Gatteschi D., Pardi L. // Inorg. Chim. Acta. 1989. V. 163. № 1. P. 99.
Caneschi A., Dei A., Gatteschi D., Tangoulis V. // Inorg. Chem. 2002. V. 41. № 13. P. 3508.
Bencini A., Caneschi A., Carbonera C. et al. // J. Mol. Struct. 2003. V. 656. № 1–3. P. 141.
Beni A., Dei A., Rizzitano M., Sorace L. // Chem. Commun. 2007. № 21. P. 2160.
Beni A., Dei A., Laschi S. et al. // Chem. Eur. J. 2008. V. 14. № 6. P. 1804.
Dapporto P., Dei A., Poneti G., Sorace L. // Chem. Eur. J. 2008. V. 14. № 35. P. 10915.
Dei A., Feis A., Poneti G., Sorace L. // Inorg. Chim. Acta. 2008. V. 361. P. 3842.
Poneti G., Mannini M., Sorace L. et al. // Chem. Phys. Chem. 2009. V. 10. № 12. P. 2090.
Dei A., Sorace L. // Appl. Magn. Res. 2010. V. 38. № 2. P. 139.
Gransbury G.K., Boulon M.-E., Petrie S. et al. // Inorg. Chem. 2019. V. 58. № 7. P. 4230.
Старикова А.А., Чегерев М.Г., Стариков А.Г. // Коорд. химия. 2020. Т. 46. № 3. С. 172 (Starikova A.A., Chegerev M.G., Starikov A.G. // Russ. J. Coord. Chem. 2020. V. 46. № 3. P. 193).https://doi.org/10.1134/S1070328420030070
Simaan A.J., Boillot M.-L., Carrasco R. et al. // Chem. Eur. J. 2005. V. 11. № 6. P. 1779.
Graf M., Wolmershäuser G., Kelm H. et al. // Angew. Chem. Int. Ed. 2010. V. 49. № 5. P. 950.
Carbonera C., Dei A., Létard J.-F. et al. // Angew. Chem. Int. Ed. 2004. V. 43. № 24. P. 3136.
Tao J., Maruyama H., Sato O. // J. Am. Chem. Soc. 2006. V. 128. № 6. P. 1790.
Min K.S., DiPasquale A.G., Golen J.A. et al. // J. Am. Chem. Soc. 2007. V. 129. № 8. P. 2360.
Min K.S., DiPasquale A.G., Rheingold A.L. et al. // J. Am. Chem. Soc. 2009. V. 131. № 17. P. 6229.
Alley K.G., Poneti G., Aitken J.B. et al. // Inorg. Chem. 2012. V. 51. № 7. P. 3944.
Alley K.G., Poneti G., Robinson P.S.D. et al. // J. Am. Chem. Soc. 2013. V. 135. № 22. P. 8304.
Poneti G., Mannini M., Cortigiani B. et al. // Inorg. Chem. 2013. V. 52. № 20. P. 11798.
Mulyana Y., Alley K.G., Davies K.M. et al. // Dalton Trans. 2014. V. 43. № 6. P. 2499.
Madadi A., Itazaki M., Gable R.W. et al. // Eur. J. Inorg. Chem. 2015. № 30. P. 4991.
Стариков А.Г., Старикова А.А., Минкин В.И. // Д-окл. PАН. 2016. Т. 467. № 3. С. 300 (Starikov A.G., Starikova A.A., Minkin V.I. // Dokl. Chem. 2016. V. 467. № 1. Р. 83).
Starikova A.A., Chegerev M.G., Starikov A.G., Minkin V.I. // Comp. Theor. Chem. 2018. V. 1124. P. 15.
Tezgerevska T., Rousset E., Gable R.W. et al. // Dalton Trans. 2019. V. 48. № 31. P. 11674.
Shultz D.A., Bodnar S.H., Vostrikova K.E., Kampf J.W. // Inorg. Chem. 2000. V. 39. № 26. P. 6091.
Shultz D.A., Vostrikova K.E., Bodnar S.H. et al. // J. Am. Chem. Soc. 2003. V. 125. № 6. P. 1607.
Shultz D.A., Kumar R.K., Bin-Salamon S., Kirk M.L. // Polyhedron. 2005. V. 25. № 16–17. P. 2876.
Kirk M.L., Shultz D.A. // Coord. Chem. Rev. 2013. V. 257. № 1. P. 218.
Tichnell C.R., Shultz D.A., Popescu C.V. et al. // Inorg. Chem. 2015. V. 54. № 9. P. 4466.
Bubnov M., Cherkasova A., Teplova I. et al. // Polyhedron. 2016. V. 119. P. 317.
Bubnov M.P., Teplova I.A., Kopylova E.A. et al. // Inorg. Chem. 2017. V. 56. № 5. P. 2426.
Zolotukhin A.A., Bubnov M.P., Arapova A.V. et al. // Inorg. Chem. 2017. V. 56. № 24. P. 14751.
Черкасова А.В., Кожанов К.А., Золотухин А.А. и др. // Коорд. химия. 2019. Т. 45. № 7. С. 404 (Cherkasova A.V., Kozhanov K.A, Zolotukhin A.A. et al. // Russ. J. Coord. Chem. 2019. V. 45. № 7. P. 489).https://doi.org/10.1134/S1070328419070029
Protasenko N.A., Poddel’sky A.I., Bogomyakov A.S. et al. // Inorg. Chim. Acta. 2019. V. 489. P. 1.
Третьяков Е.В., Овчаренко В.И. // Успехи химии. 2009. Т. 78. № 11. С. 1051 (Tretyakov E.V., Ovcharenko V.I. // Russ. Chem. Rev. 2009. V. 78. № 11. P. 971).
Stable Radicals: Fundamental and Applied Aspects of Odd-electron Compounds / Ed. Hicks R. Wiley, Chichester, 2011. 588 p.
Frisch M.J., Trucks G.W., Schlegel H.B. et al. Gaussian 16. Revision A. 03. Wallingford: Gaussian, 2016.
Tao J.M., Perdew J.P., Staroverov V.N., Scuseria G.E. // Phys. Rev. Lett. 2003. V. 91. № 14. P. 146401.
Staroverov V.N., Scuseria G.E., Tao J., Perdew J.P. // J. Chem. Phys. 2003. V. 119. № 23. P. 12129.
Bannwarth A., Schmidt S.O., Peters G. et al. // Eur. J. Inorg. Chem. 2012. № 16. P. 2776.
Cirera J., Paesani F. // Inorg. Chem. 2012. V. 51. № 15. P. 8194.
Старикова А.А., Минкин В.И. // Коорд. химия. 2018. Т. 44. № 4. С. 229 (Starikova A.A., Minkin. V.I. // Russ. J. Coord. Chem. 2018. V. 44. № 8. Р. 483).https://doi.org/10.1134/S1070328418080079
Стариков А.Г., Старикова А.А., Чегерев М.Г., Минкин В.И. // Коорд. химия. 2019. Т. 45. № 2. С. 92 (Starikov A.G., Starikova A.A., Chegerev M.G., Minkin V.I. // Russ. J. Coord. Chem. 2019. V. 45. № 2. Р. 105). https://doi.org/10.1134/S1070328419020088
Cirera J., Via-Nadal M., Ruiz E. // Inorg. Chem. 2018. V. 57. № 22. P. 14097.
Старикова А.А., Метелица Е.А., Минкин В.И. // Коорд. химия. 2019. Т. 45. № 6. С. 350 (Starikova A.A., Metelitsa E.A., Minkin V.I. // Russ. J. Coord. Chem. 2019. V. 45. № 6. P. 411).https://doi.org/10.1134/S1070328419060095
Стариков А.Г., Чегерев М.Г., Старикова А.А., Минкин В.И. // Коорд. химия. 2019. Т. 45. № 10. С. 579 (Starikov A.G., Chegerev M.G., Starikova A.A., Minkin V.I. // Russ. J. Coord. Chem. 2019. V. 45. № 1. P. 675).https://doi.org/10.1134/S1070328419090082
Noodleman L. // J. Chem. Phys. 1981. V. 74. № 10. P. 5737.
Shoji M., Koizumi K., Kitagawa Y. et al. // Chem. Phys. Lett. 2006. V. 432. № 1–3. P. 343.
Chemcraft. Version 1.7. 2013: http://www.chemcraftprog.com.
Minkin V.I., Starikov A.G., Starikova A.A. // Pure Appl. Chem. 2018. V. 90. № 5. P. 811.
Bally T. // Nature Chem. 2010. V. 2. № 3. P. 165.
Дополнительные материалы отсутствуют.
Инструменты
Координационная химия