

Координационная химия, 2021, T. 47, № 1, стр. 13-20
Высокоспиновый комплекс кобальта(II) с рекордной анизотропией магнитной восприимчивости по данным парамагнитной спектроскопии ЯМР
Я. А. Панкратова 1, 2, Ю. В. Нелюбина 1, В. В. Новиков 1, А. А. Павлов 1, *
1 Институт элементоорганических соединений им. А.Н. Несмеянова РАН
Москва, Россия
2 Московский государственный университет им. М.В. Ломоносова
Москва, Россия
* E-mail: pavlov@ineos.ac.ru
Поступила в редакцию 11.04.2020
После доработки 14.05.2020
Принята к публикации 19.05.2020
Аннотация
Получен и охарактеризован тетраэдрический комплекс кобальта(II) [CoL2](HNEt3)2 (I), где L – 1,2-бис(метансульфонамидо)бензол, проявляющий свойства мономолекулярного магнита. С помощью парамагнитной спектроскопии ЯМР определены параметры электронной структуры комплекса I, полностью воспроизводящие результаты менее доступных методов исследования мономолекулярных магнитов. Оцененное для I значение аксиальной анизотропии магнитной восприимчивости (Δχакс = 34.5 × 10–32 м3 при 20°C) является рекордным среди всех комплексов переходных металлов, изученных методом ЯМР, что открывает широкие возможности для использования комплекса I в качестве парамагнитной метки для структурной биологии или сдвигающего агента и даже сенсора температуры для медицинской диагностики. Полученные данные также свидетельствуют о преимуществах парамагнитной спектроскопии ЯМР как метода исследования магнитных свойств и электронной структуры высокоанизотропных комплексов переходных металлов – предшественников многих функциональных материалов.
Парамагнитные комплексы металлов, демонстрирующие высокую магнитную анизотропию, имеют ряд перспективных практических применений в областях молекулярной электроники [1], магнитно-резонансной томографии (МРТ) [2] и структурной биологии [3]. Благодаря своим свойствам некоторые из этих комплексов, испытав воздействие внешнего постоянного магнитного поля, могут сохранять приобретенную в поле намагниченность на молекулярном уровне, т.е. вести себя подобно классическим магнитам [4, 5]. Такие соединения, которые принято называть мономолекулярными магнитами (МММ), в будущем могут лечь в основу устройств хранения информации на молекулярном уровне [6]. Подобное поведение МММ объясняется значительной магнитной анизотропией, приводящей к расщеплению электронных уровней (так называемое расщепление в нулевом поле). В результате релаксация намагниченности связана с преодолением энергетического барьера (1):
где D – энергия расщепления в нулевом поле, S – полный электронный спин.В случае, если барьер U оказывается много больше тепловой энергии kT, наблюдается длительное сохранение намагниченности при условии отсутствия побочных механизмов (таких как квантовое туннелирование, прямой и Рамановский механизмы) (2):
где τ – время релаксации намагниченности.На данный момент существует большое разнообразие методов изучения МММ, главный из которых – магнитометрия. Указанный метод в динамической вариации (в переменном магнитном поле) позволяет напрямую оценить времена релаксации намагниченности, а моделирование данных магнитометрии в постоянном поле дает возможность более подробно изучить причины наблюдаемых свойств путем нахождения параметров электронной структуры (g-тензора, тензора расщепления в нулевом поле и др.). Однако процесс определения этих параметров подразумевает моделирование экспериментальных данных некоторой моделью, которая не всегда оказывается уникальной, что может приводить к избыточной параметризации модели и, как следствие, к искаженным результатам. Комбинирование разных методов изучения электронной структуры парамагнитных комплексов металлов, таких как оптическая спектроскопия [7], магнитный круговой дихроизм [8] или спектроскопия ЭПР [9], позволяет получить более достоверные результаты [10, 11].
Спектроскопия ЯМР, традиционно используемая для установления строения диамагнитных соединений, редко применяется для исследования парамагнитных координационных комплексов ввиду сложности регистрации и интерпретации данных. Однако в последние годы подходы спектроскопии ЯМР для исследования магнитных свойств и электронной структуры именно таких соединений активно развиваются нами [12] и другими научными группами [13–16].
В настоящей работе мы синтезировали тетраэдрический комплекс кобальта(II) [CoL2](HNEt3)2 (I), где L – 1,2-бис(метансульфонамидо)бензол (схема 1 ), проявляющий свойства МММ [17]. Указанный комплекс выделен в чистом виде, а его состав и строение однозначно подтверждены данными спектроскопии ЯМР и РСА.
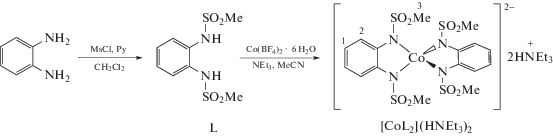
Схема 1 .
Методом парамагнитной спектроскопии ЯМР определены параметры электронной структуры I, которые хорошо согласуются с результатами других методов исследования, полученными ранее. Оцененное значение аксиальной анизотропии магнитной восприимчивости (Δχакс = 34.5 × 10–32 м3 при 20°C) для комплекса I рекордное среди всех комплексов переходных металлов, изученных методом ЯМР. Это открывает широкие возможности для его использования в качестве сдвигающего агента или даже сенсора температуры в МРТ, а также в качестве парамагнитной метки для исследования пространственной структуры биологических макромолекул.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Синтез комплекса I и лиганда L проводили на воздухе с использованием коммерчески доступных органических растворителей и реагентов. Элементный анализ на углерод, азот и водород проводили на микроанализаторе Carlo Erba, модель 1106.
Синтез лиганда L проводили по оптимизированной методике, ранее опубликованной в [18]. К кипящему раствору о-фенилендиамина (200 мг, 1.85 ммоль) в хлористом метилене (4 мл) по каплям добавляли раствор смеси пиридина (0.4 мл, 5.55 ммоль) и метансульфонилхлорида (0.3 мл, 4.07 ммоль). Реакционную смесь кипятили в течение 3.5 ч, затем медленно охлаждали в течение 12 ч. Образовавшийся светло-розовый осадок отфильтровывали, промывали хлористым метиленом, остатки растворителя удаляли на масляном насосе. Выход светло-розового мелкокристаллического продукта 228 мг (35%).
ЯМР 1H (CD3CN, 300.15 МГц, 18°C, δ, м.д.): 2.98 (с., 6H, Me), 7.33 (м., 2H, Ar–H), 7.49 (м., 2H, Ar–H), 7.57 (уш.с., 2H, NH).
Синтез [CoL2](HNEt3)2(I) проводили по оптимизированной методике, ранее опубликованной в [17]. К суспензии L (124 мг, 0.47 ммоль) в ацетонитриле (3 мл) добавляли триэтиламин (130 мкл, 1.88 ммоль). Через 2 мин к полученному раствору добавляли раствор [Co(BF4)] · 6H2O (80 мг, 0.23 ммоль) в ацетонитриле (0.5 мл). Образовавшийся фиолетовый раствор оставляли на ночь при интенсивном перемешивании. После окончания реакции к реакционной смеси добавляли толуол (4 мл) и продукт кристаллизовали при пониженной температуре (4°С). Вывод малинового кристаллического продукта 45 мг (26%).
ЯМР 1H (CD3CN, 600.22 МГц, 18°C, δ, м.д.): 112.37 (уш.с., 4H, 2), 86.41 (уш.с., 4H, 1), –7.10 (уш.с., 18H, CH3(Et)), –8.24 (уш.с., 12H, CH2(Et)), –30.41 (уш.с., 12H, 3), –37.02 (уш.с., 2H, NH).
Спектры ЯМР регистрировали для растворов в ацетонитриле-d3 на спектрометрах Bruker Avance 300 и 600 (Ларморова частота по протонам 300.15 и 600.22 МГц соответственно). Значения химических сдвигов (δ, м.д.) в спектрах ЯМР определяли относительно остаточного сигнала растворителя (ЯМР 1H 1.94 м.д.). Спектры комплекса I регистрировали с использованием следующих параметров: диапазон спектра 300 м.д., время регистрации 0.5 с, длительность релаксационной задержки 0.5 с, длительность импульса 6.5 мкс, количество накоплений 32. Для повышения соотношения сигнал/шум полученные спады свободной индукции обрабатывали путем экспоненциального взвешивания с коэффициентом до 7 Гц.
РСА I проведен на дифрактометре Bruker APEX2 CCD (MoKα-излучение, графитовый монохроматор, ω-сканирование). Структура расшифрована с использованием программы ShelXT [19] и уточнена в полноматричном МНК с помощью программы Olex2 [20] в анизотропном приближении по $F_{{hkl}}^{2}$. Атомы водорода NH-групп локализованы из разностных Фурье синтезов электронной плотности, положения остальных атомов водорода рассчитаны геометрически, и все они уточнены в изотропном приближении по модели наездника. Основные кристаллографические данные и параметры уточнения представлены в табл. 1.
Таблица 1.
Основные кристаллографические данные и параметры уточнения структуры [CoL2](HNEt3)2
| Параметр | Значение |
|---|---|
| М | 787.92 |
| T, K | 120 |
| Сингония | Ромбическая |
| Пр. гр. | P212121 |
| Z | 4 |
| a, Å | 12.1858(6) |
| b, Å | 16.0022(8) |
| c, Å | 18.3286(9) |
| V, Å3 | 3574.1(3) |
| ρ(выч.), г см–3 | 1.464 |
| μ, см–1 | 7.69 |
| F(000) | 1668 |
| 2θmax, град | 60 |
| Число измеренных отражений | 38 231 |
| Число независимых отражений (Rint) | 10 870 (0.0740) |
| Число отражений с I > 2σ(I) | 8726 |
| Количество уточняемых параметров | 434 |
| R1, wR2 (I > 2σ(I)) | 0.0439, 0.0826 |
| R1, wR2 (все данные) | 0.0623, 0.0900 |
| GOOF | 0.997 |
| Δρmax/Δρmin, e Å–3 | 0.411/–0.339 |
Координаты атомов и другие параметры структуры I депонированы в Кембриджском банке структурных данных (CCDC № 1995722; http:// www.ccdc.cam.ac.uk/).
Квантово-химические расчеты для комплекса I проводили с помощью программного пакета ORCA, v. 4 [21] в рамках теории функционала плотности (DFT) [22]. Геометрию комплекса оптимизировали без наложения симметрийных ограничений с использованием негибридного функционала PBE [23] и базисного набора def2-TZVP [24]. В качестве начального приближения использовали его структуру, установленную при помощи РСА. Эффекты сольватации учитывали в рамках модели CPCM программного пакета ORCA, v. 4. В качестве растворителя выбирали ацетонитрил, в растворе которого регистрировали спектры ЯМР. На основе полученной геометрии комплекса рассчитывали значения g-тензора и констант сверхтонкого взаимодействия для протонов с использованием гибридного функционала PBE0 [25] и базисного набора def2-TZVP [24]. Далее рассчитывали величины контактного сдвига по следующему выражению:
где gизо – изотропное значение электронного g-тензора, μB – магнетон Бора, ħ – постоянная Планка, γI – гиромагнитное отношение для протона, k – постоянная Больцмана, S – спиновое квантовое число, равное 3/2 для высокоспиновых комплексов кобальта(II), Т – температура. Размерности всех величин выражены в системе СИ.РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Комплекс [CoL2](HNEt3)2 (I) получали с умеренным выходом по оптимизированной методике, ранее опубликованной в [17] (cхема 1), при комнатной температуре. При попытках кристаллизовать его из реакционной смеси диэтиловым эфиром происходило формирование осадка в виде масла серого цвета. Однако смешивание одной части реакционной смеси с одной частью толуола с последующим выдерживанием полученного раствора в течение 48 ч при температуре 4°С приводило к образованию крупных малиновых монокристаллов (1–2 мм) комплекса I, природа которого однозначно подтверждена результатами РСА (pис. 1). Согласно полученным данным, молекулярная и кристаллическая структура I при 120 К (см. Экспериментальную часть) близко воспроизводит установленную ранее [17] при 100 К. В ней анионный комплекс [CoL2]2– связан с двумя катионами HN${\text{Et}}_{3}^{ + }$ за счет N–H…O водородных связей (N…O 2.829(4) и 2.725(4) Å, NHO 160.3(2)° и 160.7(2)°), образованных атомами кислорода его двух SO2-групп. В комплексе I ион кобальта(II) находится в высокоспиновом состоянии в окружении четырех атомов азота двух лигандов (Co–N 2.004(3)–2.018(3) Å), формирующих вокруг него практически идеальное тетраэдрическое окружение. Количественно его можно охарактеризовать при помощи так называемых “мер симметрии” (или continuous symmetry measures в англоязычной литературе [26]), описывающих отклонение формы координационного полиэдра CoN4 от идеального тетраэдра S(T-4). Чем это значение меньше, тем лучше форма полиэдра описывается указанным многогранником. В случае исследуемого комплекса I соответствующее значение S(T-4), оцененное на основе рентгенодифракционных данных при 120 К, составляет 6.268. Для сравнения величина отклонения от идеального плоского квадрата S(SP-4) (так называемая плоскоквадратная “мера симметрии”) достигает 23.219, что указывает на тетраэдрическое окружение иона кобальта(II) в комплексе I.
Для определения состава и строения комплекса I в растворе, а также описания его магнитных свойств и, соответственно, его потенциала в качестве сдвигающего агента в МРТ или парамагнитной метки для структурной биологии мы использовали спектроскопию ЯМР. Одним из традиционных способов изучения парамагнитных соединений при помощи спектроскопии ЯМР является метод Эванса [27]. В основе этого метода лежит эффект сдвига положения резонанса в спектре ЯМР любого ядра, находящегося в растворе парамагнитного вещества. Как правило, выбираются ядра, принадлежащие молекулам растворителя или соединения сравнения, специально добавленного в раствор. Величина наблюдаемого сдвига пропорциональна изотропному значению магнитной восприимчивости (χМ) изучаемого парамагнитного комплекса:
(4)
${{{\chi }}_{M}} = \frac{{\Delta \delta M}}{{{{\nu }_{0}}{{S}_{f}}m}} - {\chi }_{M}^{{{\text{диа}}}},$(5)
${{\chi }_{{{\text{изо}}}}} = {{({{\chi }_{{||}}} + 2{{\chi }_{ \bot }})} \mathord{\left/ {\vphantom {{({{\chi }_{{||}}} + 2{{\chi }_{ \bot }})} 3}} \right. \kern-0em} 3},$Вместе с тем ядра, входящие в состав парамагнитного комплекса, демонстрируют больший сдвиг в спектрах ЯМР, который принято называть парамагнитным (δпар):
(6)
${{\delta }_{{{\text{общ}}}}} = {{\delta }_{{{\text{диа}}}}} + {{\delta }_{{{\text{пар}}}}} = {{\delta }_{{{\text{диа}}}}} + {{\delta }_{{\text{к}}}} + {{\delta }_{{{\text{пк}}}}},$Сдвиг δпар, в свою очередь, принято разделять на контактный (δк), обусловленный наличием спиновой плотности на ядре, и псевдоконтактный (δпк). Поскольку экспериментально определить распределение спиновой плотности в молекуле весьма затруднительно, величину контактного сдвига обычно рассчитывают методами квантовой химии. Так как в основе псевдоконтактного сдвига лежит дипольное взаимодействие магнитных моментов ядра и неспаренного электрона, величина этого сдвига зависит от взаимного расположения взаимодействующих частиц. В качестве центра локализации неспаренных электронов выбирают парамагнитный ион, а координаты ядер определяют по данным РСА и/или квантово-химического расчета с оптимизацией геометрии молекулы.
(7)
${{\delta }_{{{\text{пк}}}}} = \frac{1}{{12\pi {{r}^{3}}}}\left[ {\Delta {{\chi }_{{{\text{акс}}}}}\left( {3{{{\cos }}^{2}}\theta - 1} \right)} \right],$Аксиальная анизотропия магнитной восприимчивости, выражаемая как разница между собственными значения соответствующего тензора (Δχакс= χ|| – χ⊥), является важной характеристикой электронной структуры молекулы. Во-первых, величина анизотропии сама по себе характеризует перспективность соединения в качестве парамагнитной метки и сдвигающего агента или сенсора температуры в МРТ, поскольку для этих целей наиболее целесообразно выбирать системы с наибольшей анизотропией. Во-вторых, анизотропия Δχакс вместе с изотропным значением χизо, которое может быть получено методом Эванса, полностью характеризует тензор магнитной восприимчивости χ, который непосредственно связан с электронными уровнями энергии через уравнение ван-Флека:
(8)
${{\chi }_{a}} = \frac{{{{N}_{A}}kT}}{{10}}\frac{{{{\partial }^{2}}}}{{\partial B_{a}^{2}}}\ln \left( {\mathop \sum \limits_i {{e}^{{ - \frac{{{{\psi }_{i}}|\hat {\mathcal{H}}|{{\psi }_{i}}}}{{kT}}}}}} \right),$(9)
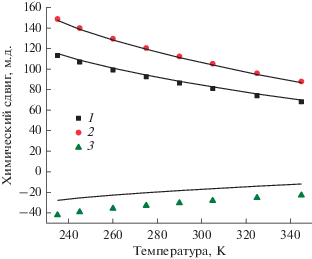
$\hat {\mathcal{H}} = \frac{D}{\hbar }\left( {S_{z}^{2} - \frac{{{{S}^{2}}}}{3}} \right) + \frac{{{{{\mu }}_{{\text{B}}}}}}{\hbar }gB\hat {S},$Для выбранного нами комплекса кобальта I спектры ЯМР 1Н регистрировали в диапазоне 235–345 К (pис. 2). Наблюдаемые значения химического сдвига для ядер 1 и 2 (соответствующая нумерация приведена на cхеме 1) моделировали уравнениями (6) и (7). В качестве δдиа использовали спектры ЯМР для свободного лиганда L, δк рассчитывали методами квантовой химии (см. Экспериментальную часть). Для всех температур было достигнуто отличное совпадение экспериментальных сдвигов ядер 1 и 2 с расчeтными (pис. 3), характеризуемое высоким значением критерия R2 > 0.99, что позволило получить достоверные значения анизотропии магнитной восприимчивости (Δχакс). Сходимость экспериментальных сдвигов с расчетными для протонов метильной группы ожидаемо оказалась несколько хуже. Подобное расхождение, которое обсуждалось ранее [28, 29], связано с конформационной лабильностью координационных соединений в растворе. При комнатной температуре соответствующее значение Δχакс для комплекса I составило 34.5 × 10–32 м3, что является абсолютным рекордом среди комплексов переходных металлов, изученных методом ЯМР. В частности, для большинства комплексов кобальта(II) – наиболее анизотропного иона переходных металлов – это значение лежит в интервале 3–7 × 10–32 м3 [30], для наиболее анизотропных комплексов, известных к настоящему моменту, оно достигает 20–25 × 10–32 м3 [31]. Столь большая магнитная анизотропия позволяет рассматривать комплекс I в качестве перспективной парамагнитной метки для структурной биологии или сдвигающего агента и даже сенсора температуры в МРТ. Это связано с тем, что бóльшая магнитная анизотропия парамагнитного соединения приводит к появлению более сильных парамагнитных сдвигов в спектре ЯМР, что позволяет исследовать макромолекулы бóльшего размера и легче детектировать сигнал в МРТ даже при низкой концентрации агента, а в случае сенсора – более точно определять температуру in vivo. При этом следует заметить, что многие магнитно-анизотропные комплексы химически нестабильны в растворе. Однако при использовании спектроскопии ЯМР химическая стабильность комплекса в данном фазовом состоянии подтверждается де-факто, что является важным преимуществом метода.
Рис. 1.
Общий вид комплекса I в представлении атомов эллипсоидами тепловых колебаний (p = 30%). Атомы водорода за исключением принадлежащих NH группам не показаны.
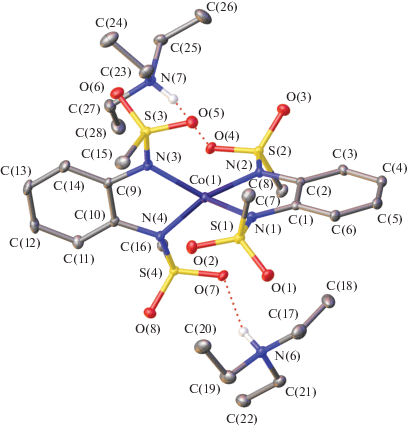
Рис. 2.
Спектры ЯМР 1Н комплекса I (600.22 МГц, CD3CN) в температурном диапазоне 235–345 К. Нумерация ядер приведена на cхеме 1.
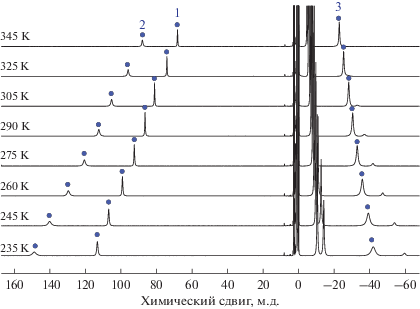
Рис. 3.
Температурная зависимость химических сдвигов в спектрах ЯМР 1Н для ядер 1–3 комплекса I (600.22 МГц, CD3CN). Экспериментальные значения показаны точками, расчeтные – сплошными линиями.
Применение метода Эванса позволило найти изотропное значение магнитной восприимчивости (χизо) для комплекса I в диапазоне 235–345 К. Полученные данные моделировали уравнением ван-Флека (8) вместе с температурной зависимостью анизотропии магнитной восприимчивости Δχакс с применением спин-гамильтониана (9), что дало следующие параметры электронной структуры: g⊥ = 2.49 ± 0.07, g|| = 3.13 ± 0.10, D = –140 ± ± 30 см–1 (рис. 4). Указанный разброс значений вызван как недостаточно высокой точностью квантово-химического расчета контактных сдвигов, так и погрешностью измерения методом Эванса [32, 33]. Полученные значения находятся в соответствии со значением энергии второго Крамерсова дублета 230 см–1, оцененного по данным дальней ИК-спектроскопии в магнитном поле [17], что соответствует энергии расщепления в нулевом поле |D| = 115 см–1.
Рис. 4.
Температурная зависимость Δχакс для комплекса I (a) и его температурная зависимость χизо, полученная методом Эванса (б). Сплошные линии соответствует одновременному моделированию данных спин-гамильтонианом с параметрами g⊥ = 2.49, g|| = 3.13, D = –140 см–1.
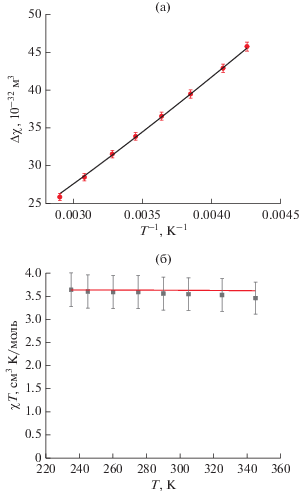
Таким образом, мы синтезировали тетраэдрический комплекс кобальта(II) (I), проявляющий свойства мономолекулярного магнита. Структура комплекса I подтверждена методом РСА, а при помощи парамагнитной спектроскопии ЯМР изучены его магнитные свойства в растворе. В частности, значение Δχакс при комнатной температуре для него составила 34.5 × 10–32 м3, что является рекордным среди всех известных комплексов переходных металлов, изученных методом ЯМР. Это позволяет рассматривать комплекс I в качестве перспективной парамагнитной метки для структурной биологии, а также сдвигающего агента и сенсора температуры в МРТ. Значения изотропной магнитной восприимчивости, измеренные параллельно методом Эванса, вместе с анизотропией магнитной восприимчивости, оцененной в ходе анализа наблюдаемых химических сдвигов в спектрах ЯМР, были смоделированы уравнением Ван-Флека в рамках формализма спин-гамильтониана. Полученные параметры электронной структуры (g⊥= 2.49 ± 0.07, g|| = 3.13 ± 0.10, D = –140 ± ± 30 см–1) находятся в полном соответствии с данными менее доступных методов исследования мономолекулярных магнитов, таких как дальняя ИК-спектроскопия в магнитном поле и магнитный круговой дихроизм. Полученные результаты также демонстрируют перспективность парамагнитной спектроскопии ЯМР как метода исследования магнитных свойств и электронной структуры высоко-анизотропных комплексов переходных металлов, являющихся предшественники многих функциональных материалов.
Авторы заявляют об отсутствии конфликта интересов.
Список литературы
Winpenny R.E. // Ang. Chem. Int. Ed. 2008. V. 47. № 42. P. 7992.
Sinharay S., Pagel M.D. // Ann. Rev. Anal. Chem. 2016. V. 9. P. 95.
Yagi H., Pilla K.B., Maleckis A. et al. // Structure. 2013. V. 21. № 6. P. 883.
Sessoli R., Gatteschi D., Caneschi A. et al. // Nature. 1993. V. 365. № 6442. P. 141.
Sessoli R., Powell A.K. // Coord. Chem. Rev. 2009. V. 253. № 19–20. P. 2328.
Frost J. M., Harriman K. L., Murugesu M. // Chem. Sci. 2016. V. 7. № 4. P. 2470.
Chia Y., Tay M. // Dalton Trans. 2014. V. 43. № 35. P. 13159.
Larrabee J.A., Alessi C.M., Asiedu E. et al. // J. Am. Chem. Soc. 1997. V. 119. № 18. P. 4182.
Palacios M.A., Nehrkorn J., Suturina E.A. et al. // Chem. Eur. J. 2017. V. 23. № 48. P. 11649.
Ishikawa N. // J. Phys. Chem. A. 2003. V. 107. № 30. P. 5831.
Pavlov A.A., Nehrkorn J., Pankratova Y.A. et al. // Phys. Chem. Chem. Phys. 2019. V. 21. № 16. P. 8201.
Novikov V.V., Pavlov A.A., Nelyubina Y.V. et al. // J. Am. Chem. Soc. 2015. V. 137. № 31. P. 9792.
Polovkova M.A., Martynov A.G., Birin K.P. et al. // Inorg. Chem. 2016. V. 55. № 18. P. 9258.
Damjanovic M., Katoh K., Yamashita M., Enders M. // J. Am. Chem. Soc. 2013. V. 135. № 38. P. 14349.
Hiller M., Krieg S., Ishikawa N., Enders M. // Inorg. Chem. 2017. V. 56. № 24. P. 15285.
Suturina E.A., Mason K., Botta M. et al. // Dalton Trans. 2019. V. 48. № 23. P. 8400.
Rechkemmer Y., Breitgoff F.D., Van Der Meer M. et al. // Nat. Commun. 2016. V. 7. № 1. P. 1.
Wu Z., Wen K., Zhang J., Zhang W. // Org. Lett. 2017. V. 19. № 11. P. 2813.
Sheldrick G.M. // Acta Crystallogr. A. 2008. V. 64. P. 112.
Dolomanov O.V., Bourhis L.J., Gildea R.J. et al. // J. Appl. Cryst. 2009. V. 42. P. 339.
Neese F. // WIREs Comput. Mol. Sci. 2018. V. 8. № 1. P. e1327.
Runge E., Gross E.K.U. // Phys. Rev. Lett. 1984. V. 52. № 12. P. 997.
Perdew J.P., Burke K., Ernzerhof M. // Phys. Rev. Lett. 1996. V. 77. № 18. P. 3865.
Weigend F., Ahlrichs R. // Phys. Chem. Chem. Phys. 2005. V. 7. № 18. P. 3297.
Adamo C., Barone V. // J. Chem. Phys. 1999. V. 110. № 13. P. 6158.
Alvarez S. // Chem. Rev. 2015. V. 115. № 24. P. 13447.
Schubert E.M. // J. Chem. Educ. 1992. V. 69. № 1. P. 62.
Comba P., Enders M., Großhauser M. et al. // Dalton Trans. 2017. V. 46. № 1. P. 138.
Damjanović M., Samuel P.P., Roesky H.W., Enders M. // Dalton Trans. 2017. V. 46. № 16. P. 5159.
Bertini I., Luchinat C., Parigi G., Pierattelli R. // ChemBioChem. 2005. V. 6. № 9. P. 1536.
Pavlov A.A., Savkina S.A., Belov A.S. et al. // ACS Omega. 2018. V. 3. № 5. P. 4941.
Ostfeld D., Cohen I.A. // J. Chem. Educ. 1972. V. 49. № 12. P. 829.
Yatsunyk L.A., Walker F.A. // Inorg. Chem. 2004. V. 43. № 2. P. 757.
Дополнительные материалы отсутствуют.
Инструменты
Координационная химия