

Координационная химия, 2021, T. 47, № 2, стр. 81-91
Синтетические подходы к новым редокс-активным карбеновым лигандам
И. А. Никовский 1, К. А. Спиридонов 1, 2, А. А. Павлов 1, Ю. В. Нелюбина 1, 3, К. М. Карнаух 1, 4, А. В. Полежаев 1, 3, *
1 Институт элементоорганических соединений им. А.Н. Несмеянова РАН
Москва, Россия
2 Московский государственный университет им. М.В. Ломоносова
Москва, Россия
3 Московский государственный технический университет им. Н.Э. Баумана
Москва, Россия
4 Московский физико-технический институт
(национальный исследовательский университет)
Долгопрудный, Россия
* E-mail: avp@emtc.ru
Поступила в редакцию 02.05.2020
После доработки 21.05.2020
Принята к публикации 25.05.2020
Аннотация
Создание новых редокс-переключаемых молекул требует разработки простых и эффективных синтетических подходов. В настоящей работе показана возможность орто-литирования ферроценилкарб(арил)иминов (Ia–Iв) с последующей реакцией с кетонами с образованием 1,2-дизамещенных ферроценов (IIa–IIв). Последние, в свою очередь, могут быть подвергнуты циклизации под действием триметилсилилтрифлата с образованием катионных предшественников ферроценсодержащих N-гетероциклических карбенов (IIIa–IIIc), в которых гетероцикл аннелирован с одним из циклопентадиенильных колец ферроцена. Обработка IIIa–IIIв основанием в присутствии источника родия позволила получить карбеновые комплексы родия (IVa, IVб), в которых карбеновый лиганд по своим электронодонорным свойствам близок к циклическим алкиламинокарбенам. Соединения Iб и IVa исследованы методом РСА (CIF files CCDC № 2000413 и 2000414 cоответственно).
Создание современных “умных” материалов требует внедрения в их молекулярную структуру различных функциональных групп, способных предсказуемо изменять свое состояние при определенном направленном внешнем воздействии (так называемых молекулярных переключателей) [1–4]. Одним из наиболее простых способов такого переключения является обратимое изменение степени окисления одного из атомов в молекуле под действием электрического потенциала или окислителя/восстановителя [5]. Редокс-переключение может быть использовано в катализе [6], наномедицине [7], при создании химических логических элементов [2], сенсоров [8, 9], датчиков [10] или оптических элементов [8], в спинтронике [11, 12] и при создании других “умных” материалов [13]. Редокс-переключаемый катализ представляет собой атомно-экономный метод, в котором катализатор может существовать в нескольких каталитически активных состояниях с различной реакционной способностью, переключаемых с помощью окисления/восстановления лиганда. Поскольку эти состояния происходят от одного предшественника, стоимость химического синтеза снижается [6, 14].
В последние двадцать лет N-гетероциклические карбены постепенно заменяют традиционные фосфиновые лиганды благодаря уникальному сочетанию свойств [15, 16], включающих простоту получения, нечувствительность предшественников лигандов и комплексов к кислороду и влаге воздуха, прочное связывание с металлом [17], что позволяет проводить катализ в воздушной атмосфере или при высоких температурах и давлениях, если это необходимо [18, 19]. К недостаткам таких лигандов следует отнести относительно узкий диапазон электронодонорной способности [20], а также создание умеренной (по сравнению с фосфинами) стерической нагрузки вокруг атома металла [21, 22]. Одними из наиболее перспективных лигандов являются циклические алкил(амино) карбены (CAACs) [23], которые обладают большей донорной способностью, чем традиционные имидазолиевые и имидазолиновые карбены [24].
Ранее мы разработали методологию получения предшественников ферроценсодержащих N-гетероциклических карбенов [25], а также ферроценсодержащих пинцентных комплексов [26–28]. В настоящей работе мы использовали этот синтетический подход к N-гетероциклическим карбенам и их комплексам, содержащим аннелированный ферроценильный фрагмент, для последующего их использования в редокс-переключаемом катализе.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Все операции, связанные с синтезом комплексов, выполняли в среде аргона в сосудах Шленка с использованием аргон/вакуумной линии или в атмосфере сухого азота в перчаточном боксе МBraun с использованием коммерчески доступных органических растворителей и реагентов. 2,6-Диизопропиланилин, ферроценкарбоксальдегид, бензофенон, бутиллитий, пиридин (Sigma-Aldrich) использовали без дальнейшей очистки. Растворители очищали перегонкой над натрием с бензофеноном или гидридом кальция. N-(2,6-Диметилфенил)-1-ферроценилметиленимин (Iа) и N-(2,6-диизопропилфенил)-1-ферроценилметиленимин (Iв) синтезировали по ранее описанной методике [25]. Спектры ЯМР 1H, 13C и 19F регистрировали на спектрометрах Bruker Avance 400 и 600 (с рабочими частотами для протонов 400 и 600.22 МГц соответственно). Анализ на углерод, азот и водород проводили на микроанализаторе Carlo Erba, модель 1106.
Синтез N-(2,6-диэтилфенил)-1-ферроценилметиленимина (Iб). Ферроценилкарбоксальдегид (2 г, 10.75 ммоль) и 2,6-диэтиланилин (1.77 мл, 10.75 ммоль) растворяли в толуоле (70 мл) и добавляли 0.5 г молекулярных сит 4 Å. Смесь кипятили с обратным холодильником в течение 8 ч. Затем раствор охлаждали до комнатной температуры и фильтровали, толуол упаривали на роторном испарителе. Темно-красное масло перекристаллизовывали из петролейного эфира два раза c получением красных кристаллов. Выход 2.63 г (71%).
ЯМР 1H (CD3CN; 400 МГц; δ, м.д.): 8.27 (с., 1H, CH=N), 7.28 (д., 2H, JHH= 7.6 Гц, ArH), 7.21 (т., 1H, JHH= 7.6 Гц, ArH), 4.98 (с., 2H, C5H4), 4.68 (с., 2H, C5H4), 4.48 (с., 5H, C5H5), 4.27 (к., 4H, JHH = 7.5 Гц, CH(CH3)2), 1.39 (т., 6H, JHH= 6.8 Гц, CH(CH3)2). ЯМР 13C (CD3CN; 100 МГц; δ, м.д.): 162.42; 150.73; 133.18; 126.09; 123.59; 70.85; 69.06; 68.69; 24.62; 14.80.
Общая методика синтеза ферроценил дифенилметанолов. В колбу Шленка объемом 50 мл помещали соответствующий ферроценилимин (1.34 ммоль) и растворяли в 20 мл THF. Раствор охлаждали до –78°С и добавляли по каплям 589 мкл BuLi (1.1 экв., 2.5 М раствор в н-гексане). Смесь медленно нагревали до –20°C. В процессе нагревания образовывался красный осадок продукта литирования. Затем смесь охлаждали до –78°С и добавляли раствор бензофенона (1.5 ммоль) в 5 мл THF по каплям. Охлаждающую баню убирали, смесь нагревали до комнатной температуры и перемешивали в течение 1 ч. Затем добавляли несколько капель дистиллированной воды для гидролиза соли лития и растворитель удаляли на роторном испарителе. Образовавшийся красный смолистый остаток растворяли в дихлорметане, отфильтровывали, упаривали и перекристаллизовывали из н-гексана при –10°С.
(2-((2,6-Диметилфенилимино)метил)ферроценил)дифенилметанол (IIа) [25]. Выход 0.511 г (65%). ЯМР 1H (CD3CN; 400 МГц; δ, м.д.): 8.40 (с., 1H, OH), 7.96 (с., 1H, CH=N), 7.48 (д., 2H, JHH= 7.7 Гц, ArH), 7.26 (т., 2H, JHH= 7.3 Гц, ArH), 7.17 (т., 1H, JHH= 7.7 Гц, ArH), 7.03 (c., 5H, ArH), 6.84 (д., 2H, J JHH= 7.3 Гц, ArH), 6.75 (т., 1H, JHH= = 7.3 Гц, ArH), 4.69 (с., 1H, C5H3), 4.42 (с., 1H, C5H3), 4.26 (с., 5H, C5H5), 3.81 (с., 1H, C5H3), 1.65 (с., 6H, CH3). ЯМР 13C (CD3CN; 100 МГц; δ, м.д.): 167.64, 127.86, 127.82, 127.45, 127.19, 126.91, 126.5, 126.34, 123.87, 75.79, 74.64, 70.14, 69.10, 17.35.
(2-((2,6-Диэтилфенилимино)метил)ферроценил)дифенилметанол (IIб). Выход 0.430 г (61%). ЯМР 1H (CDCl3; 400 МГц; δ, м.д.): 8.50 (с., 1H, OH), 7.94 (с., 1H, CH=N), 7.56 (д., 2H, JHH= 7.5 Гц, ArH), 7.35 (т., 2H, JHH= 7.6 Гц, ArH), 7.27 (т., 1H, JHH= 7.5 Гц, ArH), 7.14 (c., 5H, ArH), 6.98 (c., 3H, ArH), 4.56 (с., 1H, C5H3), 4.42 (с., 1H, C5H3), 4.36 (с., 5H, C5H5), 3.95 (с., 1H, C5H3), 2.04 (к., 4H, JHH = 7.6 Гц, 2CH2), 0.86 (т., 6H, JHH= 7.6 Гц, CH3). ЯМР 13C (CDCl3; 100 МГц; δ, м.д.): 166.46, 127.89, 127.56, 127.38, 127.10, 126.59, 126.52, 125.82, 124.31, 77.12, 76.05, 74.75, 74.28,70.17, 69.00, 24.10, 14.82.
(2-((2,6-Диизопропилфенилимино)метил)ферроценил)дифенилметанол (IIв) [25]. Выход 0.740 г (72%). ЯМР 1H (CD3CN; 400 МГц; δ, м.д.): 8.28 (с., 1H, OH), 7.95 (с., 1H, CH=N), 7.45 (д., 2H, JHH = 7.75 Гц, ArH), 7.26 (т., 2H, JHH= 7.37 Гц, ArH), 7.26 (т., 1H, JHH= 7.75 Гц, ArH), 7.07 (м., 3H, ArH), 7.00 (т., 2H, JHH= 3.53 Гц, ArH), 6.93 (м., 3H, ArH), 4.74 (с., 1H, C5H3), 4.46 (с., 1H, C5H3), 4.25 (с., 5H, C5H5), 3.86 (с., 1H, C5H3), 2.82 (м., 2H, CH(CH3)2), 0.86 (д., 12H, CH(CH3)2). ЯМР 13C (CD3CN; 100 МГц; δ, м.д.): 167.32, 127.88, 127.34, 127.27, 126.86, 126.56, 126.48, 124.54, 122.65, 75.72, 74.95, 69.97, 69.29, 27.07, 23.64, 22.62.
Синтез (2-((2,6-диизопропилфенилимино)метил)ферроценил)дифенилэтанола (IIIг). В колбу Шленка объемом 50 мл помещали N-(2,6-диизопропилфенил)-1-ферроценилметиленимин (0.372 г, 0.67 ммоль) и 20 мл THF. Раствор охлаждали до ‒78°С и добавляли по каплям 294 мкл BuLi (1.1 экв., 2.5 М раствор в н-гексане). Смесь медленно нагревали до –20°C. В процессе нагревания образовывался красный осадок продукта литирования. Затем смесь охлаждали до –78°С и добавляли раствор 1,1-дифенилоксирана (0.147 г, 0.75 ммоль) в 5 мл THF по каплям. Охлаждающую баню убирали, смесь нагревали до комнатной температуры и перемешивали в течение 1 ч. Затем добавляли несколько капель дистиллированной воды для гидролиза соли лития и растворитель удаляли на роторном испарителе. Образовавшийся красный смолистый остаток растворяли в дихлорметане, отфильтровывали и упаривали. Полученный продукт очищали с помощью колоночной хроматографии (элюент: толуол–гексан (1 : 1)). Выход 0.017 г (13%). ЯМР 1H (CD3CN; 400 МГц; δ, м.д.): 8.25 (с., 1H, CH=N), 7.83 (д., 1Н, ArH), 7.64 (д., 2H, ArH), 7.56 (т., 1H, ArH), 7.51 (д., 2H, ArH), 7.29–7.32 (м., 5H, ArH), 7.23 (д., 2H, ArH), 4.62 (c., 1H, C5H3), 4.29 (с., 5H, C5H5), 4.25 (с., 1H, C5H3), 3.98 (с., 1H, C5H3), 3.96 (с., 2H, CH2), 3.12 (септ., 2H, JHH= 6.9 Гц, CH), 1.28 (д., 12H, JHH= 6.9 Гц, CH(CH3)2), 1.14 (д., 12H, JHH= 6.9 Гц, CH(CH3)2).
Общая методика синтеза имидазолиниевых предшественников карбенов. В перчаточном боксе в 20 мл виале растворяли соответствующий дифенилметанол (1.8 ммоль) в 5 мл CH2Cl2 и добавляли 0.5 мл пиридина. Полученный раствор охлаждали до –78°С и добавляли ангидрид трифторметанcульфоновой кислоты (82 мкл, 0.486 ммоль) по каплям. Реакционную смесь перемешивали в течение 40 мин при комнатной температуре. При перемешивании цвет реакционной смеси медленно менялся с темно-красного до ярко-фиолетового. Затем к полученному раствору добавляли 15 мл н-гексана, в результате выпадал осадок. Раствор декантировали, осадок промывали н-гексаном и растворяли в 5 мл CH2Cl2. Полученный раствор охлаждали до –78°С, отфильтровывали от осадка пиридиновой соли, добавляли 15 мл н-гексана и декантировали раствор. Полученный ярко-фиолетовый осадок высушивали в высоком вакууме.
6-(2,6-Диметилфенил)-5,5-дифенил-5H-пирроло[3,4-а]ферроцен-6-иум трифторметансульфонат (IIIa). Выход: 0.734 г (45%). ЯМР 1H (aцетон-d6; 600 МГц; δ, м.д.): 10.10 (с., 1H, CH = N), 7.77 (д., 2H JHH= 7.4 Гц, o-Ph'), 7.64 (т., 1H, JHH= 7.4 Гц, p‑Ph'), 7.55 (т., 2H, JHH= 7.4 Гц, m-Ph'), 7.44 (т., 1H, JHH= 7.6 Гц, p-PhMe2), 7.39 (т., 1H, JHH= 7.8 Гц, p-Ph), 7.29 (т., 2H, JHH= 7.8 Гц, m-Ph), 7.20–7.17 (м., 2H, m-PhMe2), 6.90 (д., 2H, JHH= 7.8 Гц, o‑Ph), 5.65 (с., 2H, C5H3), 5.42 (с., 1H, C5H3), 4.7 (с., 5H, C5H5), 1.85 (с., 1H, Me), 1.45 (с., 1H, Me). ЯМР 13C (aцетон-d6; 150.9 МГц; δ, м.д.): 181.27, 137.80, 137.46, 135.41, 131.90, 131.07, 130.20, 130.09, 129.89, 128.96, 128.82, 127.92, 127.64, 127.33, 102.90, 88.91, 81.94, 80.80, 73.26, 71.33, 66.38, 18.50, 18.38. ЯМР 19F (aцетон-d6, 376 МГц; δ, м.д.): = –78.67 (с., 3F, TfO).
6-(2,6-Диэтилфенил)-5,5-дифенил-5H-пирроло[3,4-а]ферроцен-6-иум трифторметансульфонат (IIIб). Выход 0.569 г (48%). ЯМР 1H (CD2Cl2; 400 МГц; δ, м.д.): 9.76 (с., 1H, CH=N), 7.60–7.51 (м., 3H, Ph), 7.48 (т., 1H, JHH= 7.7 Гц, Ph(Et)2), 7.16 (т., 2H, JHH= 7.8 Гц, Ph), 7.33–7.28 (м., 2H, Ph(Et)2), 7.20–7.14 (м., 4H, Ph), 6.71 (д., 1H, JHH= = 7.7 Гц, Ph), 5.63 (с., 1H, C5H3), 5.43 (с., 1H, C5H3), 5.04 (с., 1H, C5H3), 4.58 (с., 5H, C5H5), 2.15 (сек., 1H, JHH= 7.4 Гц, Et), 1.87 (сек., 1H, JHH= 7.7 Гц, Et), 1.67 (сек., 1H, JHH= 7.7 Гц, Et), 1.54 (сек., 1H, JHH= 7.4 Гц, Et), 0.92 (т, 3H, JHH= 7.7 Гц, Et), 0.63 (т, 3H, JHH= 7.4 Гц, Et). ЯМР 13C (CD2Cl2: 100 МГц; δ, м.д.): 180.53, 143.03, 140.64, 137.01, 135.55, 131.40, 130.05, 129.92, 128.75, 128.64, 127.96, 127.75, 126.62, 102.43, 89.55, 81.78, 80.79, 73.20, 70.85, 66.87, 24.37; 13.97, 13.59. ЯМР 19F (CD2Cl2; 376 МГц; δ, м.д.): –78.62 (с., 3F, TfO).
6-(2,6-Диизопропилфенил)-5,5-дифенил-5H-пирроло[3,4-а]ферроцен-6-иум трифторметан-сульфонат (IIIв). Выход 0.668 г (54%). ЯМР 1H (CD2Cl2; 600 МГц; δ, м.д.): 9.89 (с., 1H, CH=N), 7.53–7.44 (м., 6H, Ph), 7.25 (т., 2H, JHH= 7.9 Гц, Ph(iPr)2), 7.16 (т., 3H, JHH= 8.4 Гц, Ph), 6.65 (д., 2H, JHH= 7.9 Гц, Ph(iPr)2), 5.69 (с., 1H, C5H3) 5.40 (с., 1H, C5H3), 4.90 (с., 1H, C5H3), 4.62 (с., 5H, C5H5), 2.44 (п., 1H, JHH= 5.9 Гц, CH(CH3)2), 2.24 (п., 1H, JHH= 6.6 Гц, CH(CH3)2), 1.05 (д.д., 6H, JHH= = 5.9 Гц, JHH= 83 Гц, CH(CH3)2), 0.32 (д.д., 6H, JHH= 6.6 Гц, JHH= 25.5 Гц, CH(CH3)2). ЯМР 13C (CD2Cl2; 150.9 МГц; δ, м.д.): 181.04; 148.78, 146.17, 137.77, 133.33, 132.40, 131.27, 130.81, 130.32, 129.18, 128.65, 126.34, 125.28, 102.41, 90.07, 81.97, 81.19, 69.97, 67.26, 53.47, 29.91, 29.25, 26.80, 25.24, 21.46, 20.54. ЯМР 19F (CD2Cl2; 376 МГц; δ, м.д.): –78.64 (с., 3F, TfO).
Общая методика синтеза карбеновых комплексов родия. В перчаточном боксе в 20 мл виале смешивали бис(триметилсилил) амид лития (85 мг, 0.51 ммоль), циклооктадиен родий хлорид димер (200 мг, 0.51 ммоль) и соответствующий имидазолиниевый предшественник карбена (0.25 ммоль). После этого смесь сухих веществ охлаждали до –78°С и добавляли к ней THF. Полученный раствор перемешивали при –78°С в течение 30 мин, а затем нагревали до комнатной температуры и перемешивали еще 30 мин. Полученный комплекс очищали колоночной хроматографией (элюент: петролейный эфир–этил ацетат (5 : 1)).
Хлорциклооктадиен [2-(2,6-диметилфенил)-3,3-дифенил-ферросо[c]пиррол-2-илиден]родий (IVа). Выход 41 мг (23%). ЯМР 1H (CDCl3; 600 МГц; δ, м.д.): 7.65 (с., 2H, o-Ph), 7.43 (т., 1H, p-Ph/p-PhMe2), 7.30 (т., 2H, JHH= 7.4 Гц, m-Ph), 7.23 (т., 1H, JHH= 7.4 Гц, p-Ph/p-PhMe2), 7.05–7.12 (м., 4H, m-Ph + m-PhMe2), 6.97 (д., 2H, JHH= 7.6 Гц, o-Ph), 6.83 (д., 1H, JHH= 7.4 Гц, p-Ph), 5.51 (с., 1H, C5H3), 5.13 (м., 1Н, C8H12), 4.89 (м., 1Н, C8H12), 5.79 (с., 1H, C5H3), 4.36 (с., 5H, C5H5), 4.26 (м., 1Н, C8H12), 4.18 (с., 1H, C5H3), 2.96 (м., 1Н, C8H12), 2.64 (м., 1Н, C8H12), 2.28 (м., 1Н, C8H12), 2.12 (м., 1Н, C8H12), 1.93 (м., 1Н, C8H12), 1.86–1.78 (м., 2Н, C8H12), 1.53 (м., 2Н, C8H12). ЯМР 13C (CDCl3; 150.9 МГц; δ, м.д.): 252.74 (d., JCRh= 43.3 Гц), 141.75, 141.33, 139.04, 138.16, 134.05, 131.32, 129.98, 129.41, 129.70, 128.66, 127.91, 127.73, 127.42, 126.84, 100.37 (д., JCRh= 5.7 Гц), 99.88 (д., JCRh= 5.7 Гц), 98.64, 96.17, 88.36, 75.94, 70.59, 69.67 (JCRh= = 14.4 Гц), 67.85 (JCRh= 14.4 Гц); 66.80, 62.67, 34.61, 31.70, 31.55, 29.40, 27.59, 22.77, 21.33, 18.96.
Хлорциклооктадиен [2-(2,6-диэтилфенил)-3,3-дифенил-ферросо[c]пиррол-2-илиден]родий (IVб). Выход 35 мг (19%). ЯМР 1H (CDCl3; 600 МГц; δ, м.д.): 7.36–7.46 (м., 2Н, Ph), 7.37 (т., 2H, JHH= 7.1 Гц, Ph ), 7.29–7.33 (м., 2H, Ph), 7.07 (д., 2H, JHH= 6.5 Гц, Ph), 7.04 (т., 2H, JHH= 7.7 Гц, Ph), 6.95–6.98 (м., 3H, Ph), 5.50 (с., 1H, C5H3), 5.14 (м., 1Н, C8H12), 4.92 (м., 1Н, C8H12), 4.78 (с., 1H, C5H3), 4.38 (с., 5H, C5H5), 4.18 (м., 1Н, C8H12), 3.68 (с., 5H, C5H5), 2.85 (сек., 1H, JHH= 7.8 Гц, Et), 2.64 (м., 1Н, C8H12), 2.24 (м., 1Н, C8H12), 2.13 (м., 1Н, C8H12), 1.93 (м., 4Н, C8H12 + 2Et), 1.50 (м., 2Н, C8H12), 0.70 (т., 3H, JHH= 7,3 Гц, Et), 0.63 (т., 3H, JHH= 7.3 Гц, Et). ЯМР 13C (CDCl3; 150.9 МГц; δ, м.д.): 252.92 (д., JCRh= 43.3 Гц), 143.86, 143.29, 140.94, 140.27, 134.10, 131.26, 129.26, 128.76, 128.71, 128.65, 127.75, 127.59, 126.87, 123.63, 100.27 (д., JCRh= 5.1 Гц); 99.79 (д., JCRh= 5.6 Гц); 98.33, 96.03, 88.56, 75.86, 70.29, 69.95 (JCRh= 13.6 Гц), 67.86 (JCRh= 14.8 Гц), 66.71, 62.46, 34.67, 31.62, 31.39, 29.45, 28.04, 27.35, 25.36, 24.13, 22.69, 14.95, 14.18, 11.77.
РСА монокристаллов Iб и IVa, полученных медленным испарением на воздухе из раствора в гексане (Iб) и из его смеси с хлористым метиленом в соотношении 1 : 1 (IVa), проведен на дифрактометре Bruker APEX2 CCD (MoKα-излучение, графитовый монохроматор, ω-сканирование). Структуры расшифрованы с использованием программы ShelXT [29] и уточнены МНК в анизотропном полноматричном приближении по $F_{{hkl}}^{{\text{2}}}$ с помощью программы Olex2 [30]. Положения атомов водорода рассчитаны геометрически, и они уточнены в изотропном приближении по модели наездника. Основные кристаллографические данные и параметры уточнения представлены в табл. 1.
Таблица 1.
Основные кристаллографические данные и параметры уточнения для структур Iб и IVa
| Параметр | Значение | |
|---|---|---|
| Iб | IVa | |
| М | 345.25 | 771.02 |
| T, K | 120 | 120 |
| Сингония | Ромбическая | Триклинная |
| Пр. гр. | Pbca | P1 |
| Z | 16 | 2 |
| a, Å | 9.8084(8) | 10.060(4) |
| b, Å | 15.2180(12) | 10.963(4) |
| c, Å | 45.882(4) | 17.157(7) |
| α, град | 90.00 | 88.353(9) |
| β, град | 90.00 | 81.334(9) |
| γ, град | 90.00 | 67.954(8) |
| V, Å3 | 6848.5(10) | 1733.1(12) |
| ρ(выч.), г см–3 | 1.339 | 1.478 |
| μ, см–1 | 8.80 | 10.04 |
| F(000) | 2912 | 798 |
| 2θmax, град | 54 | 54 |
| Число измеренных отражений | 61778 | 18483 |
| Число независимых отражений (Rint) | 7485 (0.1102) | 7537 (0.1505) |
| Число отражений с I > 2σ(I) | 5362 | 3939 |
| Количество уточняемых параметров | 419 | 427 |
| R1, wR2 (I > 2σ(I)) | 0.0530, 0.0917 | 0.0673, 0.1207 |
| R1, wR2 (все данные) | 0.0861, 0.1043 | 0.1508, 0.1547 |
| GOOF | 1.084 | 0.953 |
| Δρmax/Δρmin, e Å–3 | 0.498/–0.655 | 0.987/–0.885 |
Структурные данные для соединений Iб и IVa депонированы в Кембриджском банке структурных данных (CCDC № 2000413 и 2000414; http:// www.ccdc.cam.ac.uk/).
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
В литературе известен ряд карбенов и карбеновых комплексов, содержащих ферроценильный фрагмент [31, 32]. Следует отметить, что в большинстве подобных соединений ферроценилметильный заместитель находится на атоме азота и отделен от карбенового атома углерода несколькими несопряженными связями [31]. В результате окисление/восстановление ферроценильного фрагмента практически не оказывает влияние на электронодонорную способность лиганда, что делает подобный синтетический дизайн неудачным для редокс-переключаемых систем, в которых требуется прямое влияние редокс-фрагмента на электронодонорную способность лиганда. Ранее мы разработали синтетически простой путь получения предшественников карбенового лиганда [25, 33, 34], в котором гетероциклическое кольцо аннелировано с металлоценовым фрагментом, что потенциально позволяет непосредственно влиять на карбеновый центр.
Показано [25], что бутиллитий способен эффективно литировать ферроценимины в орто-положение к иминогруппе без сколько-нибудь заметного присоединения по электрофильному атому углерода иминогруппы. В настоящей работе мы расширили спектр используемых иминов производным диэтиланилина для лучшей растворимости карбеновых комплексов в органических растворителях. Для этого мы синтезировали серию N-(2,6-диалкилфенил)-1-ферроценилметилениминов (Iа–Iв) по предложенной ранее методике. Полученные соединения детально охарактеризованы при помощи набора физико-химических методов, включая РСА. В частности, подтверждена структура соединения Iб (рис. 1), содержащего в кристалле две симметрически-независимые молекулы комплекса, которые почти не различались по своим геометрическим параметрам (табл. 2). В обоих случаях фрагмент –С(H)=N– практически лежал в плоскости циклопентадиенильного кольца, на что указывали значения угла между соответствующими плоскостями до 11°. Для сравнения угол между двумя циклопентадиенильными лигандами ферроценильного фрагмента в двух симметрически-независимых молекулах комплекса составлял 3.93(14)° и 3.68(14)°. Напротив, 2,6-диметилзамещенная фенильная группа оказалась развернутой относительно плоскости фрагмента –С(H)=N– на 68.82(10)° и 78.65(10)° соответственно ввиду наличия у нее объемных этильных заместителей в орто-положениях фенильного кольца. Это, по-видимому, и привело к тому, что в кристалле Iб отсутствуют стекинг-взаимодействия, образования которых можно было ожидать между ароматическими фрагментами различных типов (циклопентадиенильными лигандами, фенильными группами и даже фрагментами –С(H)=N–), входящими в состав указанного комплекса.

Таблица 2.
Основные геометрические параметры соединения Iб по данным PCA при 120 К*
| Параметр | Значение |
|---|---|
| Fe–C(Cp), Å | 2.037(3)–2.058(3) |
| α, град | 3.93(14), 3.68(14) |
| β, град | 11.00(12), 7.72(12) |
| γ, град | 68.82(10), 78.65(10) |
Все полученные имины успешно орто-литированы по ранее описанной методике [25] и введены в реакцию с бензофеноном с образованием 1,2-дизамещенных фероценов (схема 1 ).

Схема 1.
Выход соединения IIб вследствие его лучшей растворимости оказался ниже по сравнению с IIа и IIс, что затрудняло его выделение в индивидуальном виде с помощью кристаллизации. Подобные ферроценилдифенилметанолы IIa–IIв способны вступать вo внутримолекулярную реакцию циклизации при генерировании карбокатиона протонированием гидроксильной группы с последующим элиминированием воды. В результате образуется аннелированное с циклопентадиенилом прирролиевое кольцо (схема 2 ). Полученный цикл, хотя он содержит пять атомов, сильно напряжен, в том числе за счет отталкивания между объемными заместителями в арильном кольце и незамещенным циклопентадиенильным фрагментом. Это отражается на устойчивости предшественников карбенов и делает их склонными к гидролизу и, возможно, на устойчивости самих комплексов. Для устранения напряжения в цикле нами предложен подход к его расширению на один атом углерода, используя в качестве электрофила в реакции с литиевой солью 1,1-дифенилоксиран.
Однако мы не добились высоких выходов продукта IIIг (максимум 13%) и высокой конверсии реакции, что в первую очередь связано с низкой реакционной способностью 1,1-дифенилоксирана по отношению к нуклеофилам. Попытки устранить эту проблему путем добавления солей меди и различных хелатирующих реагентов также не привели к успеху.
Для циклизации полученных ферроценилдифенилметанолов в пирролиевые соли мы использовали ангидрид трифторметансульфоновой кислоты в присутствии пиридина при –78°С (схема 2 ).
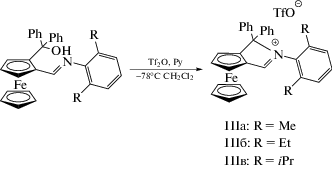
Схема 2 .
Использование в качестве основания таких реагентов, как гидрид натрия, бутиллитий или бис(триметилсилил) амид лития, также позволяет получить целевой продукт, однако выделить его при использовании данного синтетического протокола бывает затруднительно вследствие протекания большего количества побочных реакций. Использование в качестве основания пиридина приводит к образованию трифторметансульфоната пиридиния в качестве побочного продукта, который плохо растворяется в хлористом метилене и может быть отделен фильтрованием. Полученные соединения IIIa–IIIв быстро гидролизуются влагой воздуха и плохо устойчивы в растворах даже в инертных условиях. Об их образовании в качестве единственного продукта реакции свидетельствуют данные спектроскопии ЯМР. Так, в спектрах ЯМР 1H присутствовал сигнал единственного протона пирролиевого кольца при 9.76–10.10 м.д., а в спектрах ЯМР 13С – сигнал соединенного с ним атома углерода в спектре при 180.53–181.27 м.д, что значительно отличается от сигналов имино группы предшественника (7.94–7.96 и 166.46–167.64 м.д. в спектрах 1Н и 13С соответственно). При этом сигнал гидроксильного протона пропадал. Орто-заместители в арильном фрагменте становились неэквивалентными из-за невозможности вращения вокруг связи СAr–N, что проявлялось в виде двойного набора сигналов в спектрах ЯМР (1.85 и 1.45 м.д. для IIIа). В спектре ЯМР 19F наблюдался синглет при –79.5 м.д., подтверждавший присутствие свободного трифлат-аниона. Данные элементного анализа согласуются с предложенной структурой продуктов реакции.
Полученные пирролиевые соли мы использовали для генерации карбенов in situ и последующей реакции с родиевым комплексом [Rh(Cod)2Cl]2 (cхема 3). В качествe оснований выбирали трет-бутилат калия и бис(триметилсилил)амид лития. В обоих случаях удалось выделить конечный продукт для предшественников, содержащих метильные и этильные заместители. Для соединения IIIc целевой продукт выделить не удалось, что, по всей видимости, связано со стерическими затруднениями, которые испытывает данный комплекс.

Схема 3 .
Стадию металлирования мы оптимизировали по количеству молей основания, в результате чего удалось увеличить выход продукта с 7 до 23% для IVa. При этом использование двух молей основания и двух молей исходного диенового комплекса даeт максимальный выход. Дальнейшее увеличение количества основания не приводило к увеличению выхода целевого продукта. Устойчивые на воздухе карбеновые комплексы охарактеризованы при помощи спектроскопии ЯМР. Так, в протонном спектре ЯМР продуктов реакции IVa, IVб отсутствует сигнал протона пирролиевого кольца (для исходного соединения 9.76–10.10 м.д.). Все протоны циклооктадиенового кольца становятся неэквивалентными и проявляются в широком диапазоне спектра (1.5–4.92 м.д. для IVб). В спектрах ЯМР 13С для атомов углерода, связанных непосредственно с атомом родия, наблюдаются дублеты с константами спин-спинового взаимодействия 5 и 13 Гц. Сигнал карбенового атома углерода проявляется в виде дублета при 252 м.д. с 1JCRh ≈ 41 Гц, что соответствует литературным данным для схожих соединений родия [35]. Орто-заместители в арильном фрагменте остаются неэквивалентными. В случае IVb неэквивалентными становятся и протоны этильной группы CH2, что проявляется в виде четырех сигналов в спектрах ЯМР 1Н (1.50–1.93 м.д.).
Оценку донорной способности карбеновых лигандов можно произвести, сравнивая химические сдвиги карбенового углеродного атома с таковыми для уже известных соединений (схема 4 ).

Схема 4 .
Из подобного анализа видно, что сигнал в спектре ЯМР 13С для карбенового углерода в полученных комплексах представляет собой дублет с δС = 250 м.д. (1JCRh ≈ 41 Гц), что значительно выше соответствующих значений для родиевых комплексов с фрагментом NHC (δС ≈ 190 м.д.) и ниже по сравнению с CAACs (δС ≈ 278 м.д.) [23, 24, 35].
Структура полученных комплексов родия дополнительно подтверждена на примере соединения IVa (рис. 2), закристаллизованного с одной молекулой гексана, который использовался для получения монокристаллов подходящего качества медленным испарением из смеси гексана с хлористым метиленом. Согласно данным его РСА ферроценильный фрагмент в IVa имеет характерное строение (табл. 3) с расстояниями Fe–С, лежащими в диапазоне 2.027(7)–2.071(7) Å, и углом между соответствующими ароматическими плоскостями, равным 3.316(5)°. Аннелированный пиррольный цикл при этом оказывается практически плоским, о чем свидетельствует среднее значение отклонения атомов из его среднеквадратичной плоскости, составляющее всего 0.07(1) Å. 2,6-Диметилзамещенная фенильная группа повернута относительно указанной плоскости на 85.1(3)°, что можно объяснить стерическим эффектом объемных метильных заместителей. Координационное окружение атома родия, образованное атомом углерода пиррола, хлорид-анионом и циклооктадиеновым лигандом, имеет форму, напоминающую тригональную призму (табл. 3). Количественно это можно описать при помощи так называемых “мер симметрии” [36], характеризующих отклонение формы координационного полиэдра от идеальной тригональной призмы S(TP-6). Чем эти значения меньше, тем лучше форма полиэдра описывается соответствующим многогранником. В полученном нами комплексе IVa величина тригонально-призматической S(TP-6) “меры симметрии”, оцененная на основе рентгенодифракционных данных с использованием программы Shape 2.1 [36], составляет 10.080. Для сравнения соответствующие величины отклонений формы полиэдра от других идеальных многогранников с шестью вершинами (например, пентагональной пирамиды или октаэдра), роль которых выполняют четыре атома углерода циклооктадиенового лиганда, атом углерода пиррола и хлорид-анион, лежат в интервале 17.822–25.243. При этом угол, образованный атомом азота пиррола, карбеновым атомом углерода и атомом углерода связанного с ним циклопентадиенильного кольца (рис. 2), в полученном комплексе IVa (105.1(6)°) меньше, чем в случае CAACs (106.5°) [23, 34], как и соответствующая длина связи C(1)–C(2) (1.457(10) Å против 1.516 Å для CAACs). Последняя также оказывается меньше, чем в аналогичных комплексах родия (1.473 Å), где вместо ферроцена содержится ароматическое кольцо [35]). Также стоит отметить, что, несмотря на наличие в комплексе IVa большого числа ароматических фрагментов, потенциально способных к образованию стекинг-взаимодействий, в кристалле такие взаимодействия отсутствуют.
Рис. 2.
Общий вид комплекса IVa в представлении атомов эллипсоидами тепловых колебаний (p = 50%). Сольватная молекула гексана и атомы водорода не показаны для ясности.
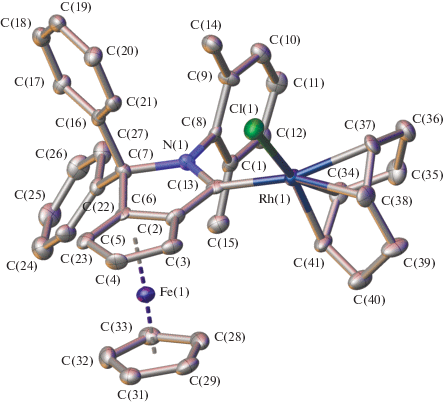
Таблица 3.
Основные геометрические параметры комплекса IVa по данным РСА при 120 К*
| Параметр | Значение |
|---|---|
| Fe–C(Cp), Å | 2.027(7)–2.071(7) |
| Rh–C(1), Å | 2.012(7) |
| Rh–Cl(1), Å | 2.372(2) |
| Rh–Cl(34), Å | 2.129(7) |
| Rh–C(37), Å | 2.205(7) |
| Rh–C(38), Å | 2.208(7) |
| Rh–C(41), Å | 2.106(7) |
| С(1)–С(2), Å | 1.457(10) |
| N(1)C(1)C(2), град | 105.1(6) |
| α, град | 3.316(5) |
| β, град | 85.1(3) |
| S(TP-6) | 10.080 |
| S(PPY-6) | 17.822 |
| S(OC-6) | 25.243 |
* Атомы C(Cp) соответствуют атомам углерода циклопентадиенильных фрагментов; α – угол между их среднеквадратичными плоскостями, β – угол поворота 2,6-диметилзамещенной фенильной группы относительно плоскости пиррола. S(TP-6), S(PPY-6) и S(OC-6) – отклонения формы полиэдра атома родия от идеальной тригональной призмы (TP-6), идеальной пентагональной пирамиды (PPY-6) и идеального октаэдра (OC-6) соответственно.
Таким образом, было показано, что литирование арилиминов ферроценкарбальдегида Ia–Iв происходит в орто-положение к иминогруппе с образованием литиеевой соли, способной к взаимодействию с бензофеноном с образованием 1,2-дизамещенных ферроценов IIa–IIв. Последние подвергаются циклизации с образованием катионных комплексов – предшественников ферроценсодержащих карбенов IIIa–IIIв. Под действием основания и в присутствии [Rh(Сod)Cl]2 данные соединения образуют комплекс родия с новым ферроценсодержащим N-гетероциклическим карбеном. Анализ структурных данных и сдвигов сигналов карбеновых атомов углерода в спектрах ЯМР 13С показывает, что электронодонорная способность карбеновых лигандов в синтезированных комплексах занимает промежуточное положение между традиционными имидазолиевыми N-гетероциклическими карбенами и циклическими алкил(амино) карбенами CAACs.
Авторы заявляют об отсутствии конфликта интересов.
Список литературы
Canary J.W. // Chem. Soc. Rev. 2009. V. 38. № 3. P. 747.
Molecular and Supramolecular Information Processing: From Molecular Switches to Unconventional Computing / Ed. Katz E. Weinheim Chichester: Wiley-VCH; John Wiley distributor, 2012.
Sivaev I. // Molecules. 2017. V. 22. № 12. P. 2201.
Molecular Switches / Eds. Feringa B.L., Browne W.R. Weinheim (Germany): Wiley-VCH, 2011.
Wang X., Song S., Zhang H. // Chem. Soc. Rev. 2020. V. 49. № 3. P. 736.
Blanco V., Leigh D.A., Marcos V. // Chem. Soc. Rev. 2015. V. 44. № 15. P. 5341.
Sims C.M., Hanna S K., Heller D.A. et al. // Nanoscale. 2017. V. 9. № 40. P. 15226.
Al-Kutubi H., Zafarani H.R., Rassaei L., Mathwig K. // Eur. Polym. J. 2016. V. 83. P. 478.
Wu T.-H., Hsu Y.-Y., Lin S.-Y. // Small. 2012. V. 8. № 13. P. 2099.
Sarkar S., Dutta S., Chakrabarti S. et al. // ACS Appl. Mater. Interfaces. 2014. V. 6. № 9. P. 6308.
Nikovskiy I., Polezhaev A., Novikov V. et al. // Chem. Eur. J. V. 26. № 25. P. 5629.
Pavlov A.A., Aleshin D.Y., Nikovskiy I.A. et al. // Eur. J. Inorg. Chem. 2019. V. 2019. № 23. P. 2819.
Gallei M., Rüttiger C. // Chem. Eur. J. 2018. V. 24. № 40. P. 10006.
Wei J., Diaconescu P.L. // Accounts Chem. Res. 2019. V. 52. № 2. P. 415.
Hopkinson M.N., Richter C., Schedler M., Glorius F. // Nature. 2014. V. 510. № 7506. P. 485.
Bourissou D., Guerret O., Gabbaï F.P., Bertrand G. // Chem. Rev. 2000. V. 100. № 1. P. 39.
Jacobsen H., Correa A., Poater A. et al. // Coord. Chem. Rev. 2009. V. 253. № 5. P. 687.
Crudden C.M., Allen D.P. // Coord. Chem. Rev. 2004. V. 248. № 21. P. 2247.
Crabtree R.H. // Coord. Chem. Rev. 2013. V. 257. № 3. P. 755.
Huynh H.V. // Chem. Rev. 2018. V. 118. № 19. P. 9457.
Díez-González S., Nolan S.P. // Coord. Chem. Rev. 2007. V. 251. № 5. P. 874.
Gusev D.G. // Organometallics. 2009. V. 28. № 22. P. 6458.
Melaimi M., Soleilhavoup M., Bertrand G. // Angew. Chem. Int. Ed. 2010. V. 49. № 47. P. 8810.
Lavallo V., Canac Y., Präsang C. et al. // Angew. Chem. Int. Ed. 2005. V. 44. № 35. P. 5705.
Nikovskiy I.A., Spiridonov K.A., Zakharova D.V. et al. // Inorg. Chim. Acta. 2019. V. 495. P. 118976.
Polezhaev A.V., Ezernitskaya M.G., Koridze A.A. // Inorg. Chim. Acta. 2019. V. 496. P. 118844.
Koridze A.A., Polezhaev A.V., Safronov S.V. et al. // Organometallics. 2010. V. 29. № 19. P. 4360.
Polezhaev A.V., Liss C.J., Telser J. et al. // Chem. Eur. J. 2018. V. 24. № 6. P. 1330.
Sheldrick G. // Acta Crystallogr. A. 2015. V. 71. № 1. P. 3.
Dolomanov O.V., Bourhis L.J., Gildea R. J. et al. // J. Appl. Crystallogr. 2009. V. 42. № 2. P. 339.
Peris E. // Chem. Rev. 2018. V. 118. № 19. P. 9988.
Siemeling U. // Eur. J. Inorg. Chem. 2012. V. 2012. № 22. P. 3523.
Takagaki W., Yasue R., Yoshida K. // Bull. Chem. Soc. Jpn. 2020. V. 93. № 2. P. 200.
Yoshida K., Yasue R. // Chemistry. 2018. V. 24. № 70. P. 18575.
Rao B., Tang H., Zeng X. et al. // Angew. Chem. Int. Ed. 2015. V. 54. № 49. P. 14915.
Alvarez S. // Chem. Rev. 2015. V. 115. № 24. P. 13447.
Дополнительные материалы отсутствуют.
Инструменты
Координационная химия