

Координационная химия, 2021, T. 47, № 3, стр. 182-190
Синтез и характеризация нового кластерного комплекса {Mo3S4} c хемилабильным фосфино-селеноэфирным лигандом
Н. Ю. Шмелев 1, 2, М. И. Гонгола 1, 2, С. Ф. Малышева 3, Н. А. Белогорлова 3, А. В. Артемьев 1, Я. С. Фоменко 1, В. Ю. Комаров 1, 2, К. В. Сопов 2, Н. Б. Компаньков 1, Д. Г. Шевень 1, М. Н. Соколов 1, 2, 4, А. Л. Гущин 1, *
1 Институт неорганической химии им. А.В. Николаева СО РАН
Новосибирск, Россия
2 Новосибирский государственный университет
Новосибирск, Россия
3 Иркутский институт химии им. А.Е. Фаворского СО РАН
Иркутск, Россия
4 Химический институт им. А.М. Бутлерова Казанского (Приволжского)
федерального университета
Казань, Россия
* E-mail: gushchin@niic.nsc.ru
Поступила в редакцию 03.09.2020
После доработки 30.09.2020
Принята к публикации 06.10.2020
Аннотация
Реакция [Mo3S4(Tu)8(H2O)]Cl4 · 4H2O (Tu = тиомочевина) с (PhCH2CH2)2PCH2CH2SeC5H11)(PSe) с последующей очисткой на хроматографической колонке из силикагеля с использованием насыщенного раствора KPF6 в ацетоне в качестве элюента приводит к образованию [Mo3S4Cl3(PSe)3]PF6 (I) с выходом 44%. Соединение охарактеризовано методами РСА, ЯМР 1H, 31P{1H}, 77Se и ИК-спектроскопии, ЭСП, циклической вольтамперометрии и масс-спектрометрии с распылением в электрическом поле. В растворе I при комнатной температуре наблюдается образование нескольких форм, отличающихся способом координации трех PSe-лигандов, которые могут присоединяться к молибдену через один (фосфор) или два (фосфор и селен) донорных атома. Такое поведение ранее не наблюдалось для схожих по строению соединений с PS-лигандами аналогичного типа. Комплекс I демонстрирует более высокую каталитическую активность, чем его аналог с PS-лигандом, в реакции восстановления нитробензола в анилин под действием дифенилсилана.
Хемилабильные лиганды занимают важное место в координационной химии и металлокомплексном катализе. Ключевая особенность этих полидендатных лигандов – наличие нескольких типов донорных атомов с различным сродством к конкретному металлическому центру, что способствует формированию координационно ненасыщенных металлических центров посредством обратимого расщепления связи металл–лиганд с более слабым донорным атомом во время каталитического цикла [1]. Контролируемая координация–декоординация лабильной донорной функции хемилабильного лиганда может быть использована и для активации малых молекул [2–13].
Реакционная способность хемилабильных лигандов в составе моноядерных соединений изучена достаточно подробно. При этом полиядерные и кластерные соединения с хемилабильными лигандами остаются относительно малоизученными [14–19]. Однако в последнем случае хемилабильные лиганды могут выполнять несколько функций: хелатирующую, мостиковую или монодентатную. Так, для хорошо изученных карбонильных трехъядерных кластеров осмия и рутения с тиоэфирными лигандами было показано существование кинетически выгодных продуктов с мостиковой функцией и термодинамически выгодных продуктов с хелатирующей функцией хемилабильного лиганда [20–24].
В ИНХ СО РАН проводятся систематические исследования в области трехъядерных кластеров молибдена и вольфрама с халькогенидными мостиками [25–36]. Недавние исследования производных {M3S4} (M = Mo, W) с N,N'-хелатирующими лигандами, такими как 2,2'-бипиридин, 1,10-фенантролин и их аналогами [37–45], показали перспективы применения таких соединений в гомогенном катализе [41, 46–49], для активации ацетиленов [50] и в бионеорганической химии [45].
В наших последних работах мы использовали бифункциональные фосфино-тиоэфиры (PS) с различными функциональными группами для координации к кластерам {M3S4} (M = Mo, W) [51, 52]. В результате получены катионные комплексы [M3S4Cl3(PS)3]+, в которых все три PS-лиганда бидентатно координируются к металлу. Эти катионные комплексы способны превращаться в нейтральные комплексы [M3S4Cl4(PS)2(PS*)], в которых один из бидентатных PS-лигандов становится P-монодентатным (PS*), в присутствии хлорид-ионов. Константы равновесия этих реакций, оцененные из данных ЯМР-спектроскопии, могут служить количественной характеристикой хемилабильного поведения этих лигандов, которое выражено более ярко в случае Mo3S4 по сравнению с W3S4. Менее донорные заместители (арильные вместо алкильных) у атома серы PS-лигандов также способствуют возрастанию хемилабильных свойств за счет уменьшения донорной способность серы. Повышенная лабильность связи M–S должна приводить к увеличению каталитической активности за счет создания координационной вакансии. Действительно, в соответствии с этой тенденцией комплекс [Mo3S4Cl3(PS1)3]+ с фенильным заместителем при атоме серы проявляет наиболее высокую каталитическую активность в реакции восстановления нитробензола до анилина под действием Ph2SiH2 [52]. Комплексы вольфрама имеют гораздо более низкую каталитическую активность в этом процессе [51].
Расширяя исследования в этом направлении, в настоящей работе мы сообщаем о получении нового кластерного комплекса молибдена с фосфино-селеноэфирным лигандом и влиянии природы донорного атома халькогена на хемилабильные и каталитические свойства.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Эксперименты по синтезу соединения [Mo3S4Cl3-(PSe)3]PF6 (I) проводили с использованием аргон-вакуумной линии. Исходные соединения [Mo3S4(Tu)8(H2O)]Cl4 ⋅ 4H2O и фосфино-селеноэфир (бис(2-фенилэтил)[2-(пентилселенил)этил]фосфин) (PSе) получали по опубликованным методикам [53, 54]. Гексафторофосфат калия (KPF6) получали из коммерческих источников и использовали без дополнительной очистки.
Элементный C,H,N-анализ выполняли на анализаторе EuroEA3000 Eurovector. ИК-спектры (4000–400 см–1) записывали на спектрофотометре Perkin-Elmer System 2000 FTIR с образцов, запрессованных в таблетки KBr. Спектры ЯМР 1H, 31P{1H} и 77Se записывали при комнатной температуре в растворах CD3CN и CD2Cl2 в 5 мм ампулах на спектрометре Bruker Avance 500 с частотами 500 (1H), 202.46 (31P) и 113.63 (77Se) МГц. Химические сдвиги относили по ТМС (1H), 85%-ная H3PO4 (31P) и 77Se ((CH3)2Se). ЭСП записывали на спектрофотометре Specord M40, Helios γ в диапазоне 200–900 нм в растворе CH3CN. Масс-спектрометрию с ионизацией электрораспылением проводили на масс-спектрометре Agilent, 6130 Quadrupole MS, 1260 infinity LC. Использовали следующие условия: напряжение на капилляре 2.0 кВ, газ-осушитель азот, 300 л/ч, 120–150°C. Наблюдаемое изотопное распределение каждого компонента хорошо согласовывалось с теоретическим распределением, рассчитанным с использованием программы MassLynx 4.1.
Циклические вольтамперные кривые записывали на электрохимическом анализаторе 797 VA Computrance (Metrohm, Switzerland). Все измерения проводили в трехэлектродной ячейке, состоящей из рабочего стеклоуглеродного электрода, платинового вспомогательного электрода и хлорсеребряного (Ag/AgCl) электрода сравнения, заполненного раствором KCl (с = 3 моль/л). Растворитель (CH3CN) дегазировали продуванием аргона в течение 1–2 мин перед каждой съемкой. Раствор Bu4NPF6 (с = 0.1 моль/л) использовали в качестве электролита. Концентрация исследуемого комплекса ~1 × 10–1 моль/л. Значения потенциалов (E1/2) определялись как 1/2(Ea + Eк), где Ea и Eк – анодные и катодные потенциалы соответственно.
Термогравиметрический анализ (ТГА) выполняли на NETZSCH thermobalance TG 209 F1 Iris в инертной атмосфере (He).
Синтез [Mo3S4Cl3(PSe)3]PF6 (I). Смесь [Mo3S4(Tu)8(H2O)]Cl4 · 4H2O (0.40 г, 0.32 ммоль), PSe (0.61 г, 1.1 ммоль) и CH3CN (40 мл) перемешивали при кипячении в течение 5 ч с обратным холодильником. После охлаждения до комнатной температуры реакционную смесь упаривали досуха. К твердому остатку добавляли CH2Cl2, нерастворимый осадок тиомочевины отделяли фильтрованием. Фильтрат наносили на хроматографическую колонку с силикагелем (Sigma Aldrich; pore size 60 Å), промывали CH2Cl2 и элюировали раствором KPF6 в ацетоне (10 мг/мл). Полученный раствор упаривали досуха, к твердому остатку добавляли CH2Cl2, не растворившийся осадок неорганических солей отделяли фильтрованием. На раствор наслаивали избыток н-гексана. В результате получили зеленый кристаллический продукт I · 2.5CH2Cl2. Выход 0.273 г (44%). ТГА: потеря массы (3.87%) в интервале от комнатной температуры до 205°C соответствует удалению одной сольватной молекулы хлористого метилена.
ИК (ν, см–1): 3359 ср, 3194 ср, 3060 ср, 3025 ср, 2954 cр, 2927 ср, 2857 ср, 2044 cл, 1624 ср, 1603 ср, 1496 cр, 1454 с, 1407 ср, 1288 ср,1208 ср, 1137 ср, 1029 сл, 998 ср, 957 сл, 839 с, 739 c, 698 c, 557 с, 495 ср, 432 ср.
ЯМР 31P{1H} (CD3CN; 25°C; δ, м.д.): 42.25 с., 42.08 с., 41.58 с., 41.45 с., 41.26 с., 41.19 с., –144.6 (сеп.). ЯМР 31P{1H} (CD2Cl2; 25°С; δ, м.д.): 40.2 c., 40.1 c., 39.9 c., 39.7 c., 39.4 c., 39.2 c., –144.4 (сеп.). ЯМР 1H (CD3CN; δ, м.д.): 6.67–7.44 (м., 30H, Ph); 2.74–2.87 (м., 12H, CH2Ph); 2.40–2.50 (м., 6H, SeCH2CH2P); 2.20 (м., 6H, SeCH2Bu); 1.68–1.85 (м., 18H, CH2PCH2); 1.54 (м., 6H, CH2Pr); 1.33–1.40 (м., 12H, (CH2)2Me); 0.92 (м., 9H, CH3). ЯМР 77Se (CD3CN; δ, м.д.): 309.7 c., 308.8 c., 267.4 c., 266.6 c., 264.2 c., 263.7 c.
ESI-MSI (+, CH3CN, m/z): 1781.9 ([Mo3S4Cl3(PSe)3]+).
UV/Vis (CH3CN; λ, нм (ε, л моль–1 см–1): 251 (14 153), 356 (7274), 633 (413).
Каталитические тесты. К предварительно деаэрированному раствору I (9.66 мг, 0.0050 ммоль) в смеси CH3CN–CD3CN (${{V}_{{{\text{C}}{{{\text{H}}}_{3}}{\text{CN}}}}}$ : ${{V}_{{{\text{C}}{{{\text{D}}}_{3}}{\text{CN}}}}}$ = 3 : 1, 2 мл) добавляли нитробензол (10 мкл, 0.097 ммоль) и Ph2SiH2 (3.5 экв.). Реакционную смесь перемешивали в течение 16 ч при комнатной температуре. Аликвоты, взятые из конечного раствора и исходного раствора до добавления Ph2SiH2, анализировали методом спектроскопии ЯМР 1H. До добавления Ph2SiH2 конверсия нитробензола не наблюдалась. После окончания реакции конверсия нитробезола 87%. Выход I 48%.
РCА. Монокристалл I был отобран из-под маточного раствора и быстро охлажден до 100 K в струе N2-криостата. Рентгенодифракционные данные измерены на автоматическом четырехкружном дифрактометре Bruker APEX Duo с двухкоординатным CCD-детектором (MoKα, λ = 0.71073 Å, графитовый монохроматор) путем ω-сканирования с экспозицией 240 с/°. Интегрирование и учет поглощения по интенсивностям эквивалентных отражений проведены с использованием пакета программ APEX2 [55]. Структура расшифрована с помощью программы SHELXT [56] и уточнена в Olex2 [57] с применением SHELXL [56]. Основные кристаллографические данные и детали уточнения приведены в табл. 1. Позиции неводородных атомов кластерного катиона и аниона ${\text{PF}}_{6}^{ - }$ получены при поиске первичной модели или из разностного синтеза и уточнены в анизотропном приближении. Углеводородные фрагменты с атомами углерода с высокой анизотропией атомных смещений или имеющие значительные пики остаточной электронной плотности уточнялись расщепленными на две конформации с наложением ограничений на валентные и угловые расстояния. Параметры атомных смещений для близкорасположенных атомов углерода из альтернативных конформаций принимались равными, либо фиксировались. Атомы водорода заданы геометрически и уточнены по модели “наездника”. Шесть наибольших пиков остаточной электронной 2.2–1.2 e/Å3 находятся вблизи атомов Mo и Se (см. Обсуждение). Нелокализованная электронная плотность в пустотах между ионами учтена с помощью процедуры Solvent Mask программы Olex2.
Таблица 1.
Кристаллографические параметры и детали уточнения структуры I
| Параметр | Значение |
|---|---|
| M | 2137.96 |
| T, K | 100(2) |
| Пр. гр. | P21/n |
| a, Å | 15.0505(11) |
| b, Å | 24.2362(18) |
| c, Å | 24.6111(16) |
| β, град | 105.547(3) |
| V, Å3 | 8648.8(11) |
| Z | 4 |
| ρ(выч.), г/см3 | 1.642 |
| μ, мм–1 | 2.161 |
| Размер кристалла, мм | 0.447 × 0.36 × 0.15 |
| Диапазон 2θ, град | 2.4–51.4 |
| Число измеренных/независимых рефлексов | 46 636/15 875 |
| Rint/Rσ | 0.0634/0.0910 |
| Число уточняемых параметров/ограничений | 702/213 |
| GООF | 1.062 |
| R1/wR2 для I ≥ 2σ(I) | 0.1484/0.3095 |
| R1/wR2 по всем данным | 0.2179/0.3511 |
| Остаточная электронная плотность (min/max), e Å–3 | –1.56/2.20 |
Координаты атомов и величины тепловых параметров депонированы в Кембриджском банке структурных данных (ССDС № 2016802; https:// www.ccdc.cam.ac.uk/structures/).
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Ранее мы показали, что комплексы [M3S4(Tu)8- (H2O)]4+ (M = Mo, W), содержащие координированные молекулы тиомочевины (Tu), весьма лабильны и легко вступают в реакции лигандного обмена с образованием различных производных {M3S4} [40, 50–52]. Так, при взаимодействии [M3S4(Tu)8(H2O)]4+ с фосфино-тиоэфирами PS1 и PS2, строение которых представлено на схеме 1 , были получены комплексы состава [M3S4Cl3(PS)3]+, в которых каждый атом металла связан с PS-лигандом по бидентатному типу.
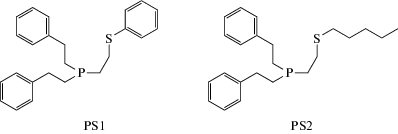
Схема 1 .
Одной из важных задач данной работы было выявить различия в свойствах кластерных соединений при замещении серы на селен в структуре лиганда. Для этого использовался фосфино-селеноэфир (PSe), представленный на схеме 1 . Предполагалось, что наличие более слабого донорного центра, селена вместо серы, может привести к более ярко выраженным хемилабильным свойствам.
Применяя описанный выше синтетический подход, мы синтезировали новый комплекс состава [Mo3S4Cl3(PSe)3]PF6 (I) с выходом 44% после очистки на хроматографической колонке из силикагеля и перекристаллизации из смеси дихлорметан–гексан (Vдихлорметан : Vгексан ≈ 1 : 5, схема 2 ). Умеренный выход продукта можно объяснить чувствительностью исходного фосфина к окислению, а также неизбежными потерями в процессах хроматографической очистки и перекристаллизации.
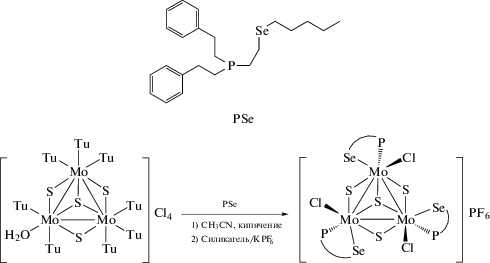
Схема 2 .
Отметим, что комплекс I – редкий пример координационного соединения, содержащего фосфино-селеноэфирный лиганд. В литературе имеется серия работ по комплексам d8-металлов (Ni, Pd, Pt), содержащим фосфино-селеноэфирные лиганды [58–61].
Монокристаллы соединения I получены в результате медленной диффузии гексана в раствор дихлорметана. Строение кластерного катиона [Mo3S4Cl3(PSe)3]+ представлено на рис. 1.
Рис. 1.
Упрощенная структурная формула кластерного катиона [Mo3S4Cl3(PSe)3]+ (а), пространственное строение [Mo3S4Cl3(PSe)3]+ в структуре I (б). Заместители при атомах фосфора и атомы водорода не показаны.
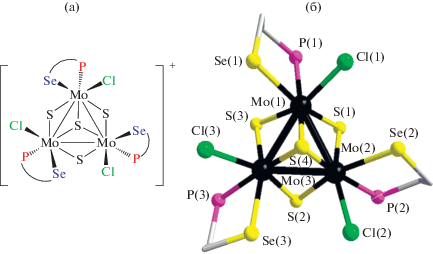
Основные длины связей приведены в табл. 2. Атомы молибдена и серы расположены таким образом, что образуется структура куба без одной вершины, типичная для данного класса соединений. Расстояния Mo–Mo и Mo–S и другие геометрические параметры находятся в соответствии с литературными данными для комплексов {Mo3S4} [62–64]. Каждый атом молибдена имеет искаженное октаэдрическое окружение, состоящее из трех мостиковых атомов серы, атома хлора и атомов фосфора и селена PSe-лиганда. Все атомы фосфора расположены в транс-положении по отношению к атому μ3-S. Такое пространственное расположение гетеробидентатного лиганда также наблюдается в комплексах {M3S4} с фосфино-тиоэфирами [52] и аминофосфинами [65, 66]. Обращает на себя внимание достаточно длинное расстояние Mo–Se (2.72–2.74 Å) по сравнению с обычной длиной связи Mo–Se (2.40–2.50 Å) между атомами металла и халькогенида кластерного ядра комплексов {Mo3Se4} [34, 67] и {Mo3Se7} [33, 44, 68–70]. Это объясняет более легкий разрыв связи Mo–Se и возможность существования нескольких форм в растворе (см. ниже). Наличие остаточной электронной плотности рядом с тяжелыми атомами (Mo, Se) свидетельствует в пользу наличия в структуре дополнительных (~5%) способов расположения катионов (возможно, изомерных) в кристаллической структуре I. Аномально высокие для 100 K значения параметров атомных смещений всех атомов, а также высокая анизотропия эллипсоидов или наличие явных расщеплений позиций атомов углерода, по-видимому, связаны с высокой конформационной подвижностью органических лигандов, а также с сильным разупорядочением сольватных молекул.
Таблица 2.
Основные длины связей (Å) в структуре I
| Связь | d, Å |
|---|---|
| Mo–Mo | 2.755(3), 2.762(2), 2.759(2) |
| Mo–μ2S | 2.304(5), 2.296(4), 2.326(5), 2.308(5), 2.303(8), 2.279(5) |
| Mo–μ3S | 2.338(6), 2.321(7), 2.344(6) |
| Mo–Cl | 2.453(4), 2.442(6), 2.437(8) |
| Mo–P | 2.544(5), 2.542(7), 2.546(5) |
| Mo–Se | 2.738(3), 2.722(2), 2.738(3) |
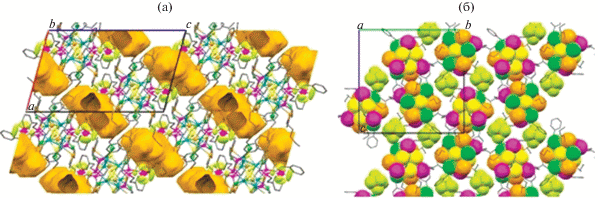
В кристаллической структуре кластерные катионы и анионы ${\text{PF}}_{6}^{ - }$ укладываются в слои, перпендикулярные направлению (1 0 –1) (диагонально векторам a и c решетки, рис. 2). В рамках слоя имеются пустоты малого объема (~30 Å3), недостаточные для размещения сольватных молекул CH2Cl2 или C6H14. Однако между слоями имеются полости объемом 524 Å3 (две полости на элементарную ячейку, 1/2 полости на формульную единицу), достаточным для размещения 3–6 молекул CH2Cl2. Оценка электронной плотности 228 e на полость, полученная с помощью процедуры Olex2 Solvent Mask, дает содержание сольватных молекул 2.7 CH2Cl2 на формульную единицу.
Рис. 2.
Проекция кристаллической структуры I вдоль оси b (а). Оранжевым показаны доступные для размещения сольватных молекул пустоты. Проекция слоя, перпендикулярного направлению (1 0 –1), вдоль оси a (б). Неуглеродные атомы показаны в ван-дер-ваальсовых сферах, атомы водорода не показаны.
В ИК-спектре соединения I обнаружены полосы валентных колебаний связей C–H при 2857–3060 cм–1 и валентных колебаний ароматических колец в интервале 1603–1624 cм–1. В области 1407–1496 см–1 проявляются деформационные колебания CH2 и CH2P. Характеристичная полоса колебаний фрагмента Mo3–µ3-S наблюдается при 432 см–1. Сильные полосы поглощения при 557 и 839 см–1 отнесены к колебаниям P–F аниона ${\text{PF}}_{{\text{6}}}^{--}.$
Электроспрей-масс-спектр (ESI-MS) соединения I в CH3CN показывает пик при m/z = 1781.9 с характерным изотопным распределением для [Mo3S4Cl3(PSe)3]+. В ЭСП зафиксирована слабая полоса при 633 нм (ε = 413 л моль–1 см–1), характеристичная для кластерного ядра {Mo3S4} [71].
Циклическая вольтамперограмма I в CH3CN демонстрирует несколько последовательных процессов восстановления, характерных для кластерного ядра {Mo3S4}. Первый процесс при –0.49 В (отн. Ag/AgCl) носит обратимый характер (ΔE ≈ ≈ 80 мВ), что согласуется с данными для [Mo3S4Cl3- (PS1)3]PF6 (Е1/2 = 0.44 В, ΔE = 80 мВ) [52]. Остальные процессы при –1.27 и 1.52 В полностью необратимы.
Необычная картина наблюдается в спектре ЯМР 31P{1H}. Вместо ожидаемого одного сигнала, как в спектрах [M3S4Cl3(PS)3]+ с фосфино-тиоэфирами, наблюдается три пары сигналов с близкими хим. сдвигами внутри каждой пары. Так, при комнатной температуре в CD3CN значения составляют 42.25 и 42.08, 41.58 и 41.55, 41.26 и 41.19 м.д. Это коррелирует с данными спектра ЯМР 77Se, в котором также обнаружены три группы сигналов: 309.7 и 308.8, 267.4 и 266.6, 264.2 и 263.7 м.д. Такое поведение мы объясняем присутствием в растворе нескольких форм, отличающихся типом координации PSe-лиганда. В исходном соединении все три PSe-лиганда находятся в хелатной форме и связаны с молибденом при помощи обоих донорных атомов фосфора и селена. Такая форма существует в кристалле. Мы предполагаем, что в растворе имеет место отщепление одного из концов хелатного цикла (того, что связан с молибденом более слабым донорным центром – селеном). В этом случае PSe-лиганд становится монодентатным и связанным с металлом только через атом фосфора. Поскольку в состав комплекса входят три PSe-лиганда, то возможно образование следующих форм в растворе: с одним, двумя или тремя монодентатными PSe. При этом освободившееся координационное может быть занято молекулой растворителя L (рис. 3).
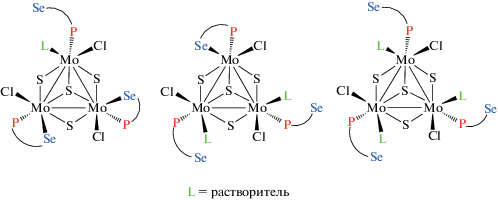
Эта ситуация уникальная, поскольку ранее полученные комплексы молибдена и вольфрама [M3S4Cl3(PS)3]+ с PS-лигандами устойчивы в растворе, в спектрах ЯМР всегда присутствует только один сигнал в характерной области [51, 52]. Хемилабильные свойства начинают проявляться только при взаимодействии [M3S4Cl3(PS)3]+ с Cl–. В результате происходит разрыв одной из связей Mo–SL, координация хлорид-иона и образование нейтрального комплекса [Mo3S4Cl4(PS)2(PS*)]. Методом ЯМР 31P{1H} были оценены константы этих равновесий. Интересно, что в случае вольфрама образование нейтральной формы практически не удается детектировать с помощью ЯМР, т.е. концентрации [W3S4Cl4(PS)2(PS*)] очень низкие.
(1)
$\begin{gathered} {{\left[ {{\text{M}}{{{\text{o}}}_{{\text{3}}}}{{{\text{S}}}_{{\text{4}}}}{\text{C}}{{{\text{l}}}_{{\text{3}}}}{{{\left( {{\text{PS}}} \right)}}_{{\text{3}}}}} \right]}^{ + }} + {\text{C}}{{{\text{l}}}^{--}} \rightleftarrows \\ \rightleftarrows \left[ {{\text{M}}{{{\text{o}}}_{{\text{3}}}}{{{\text{S}}}_{{\text{4}}}}{\text{C}}{{{\text{l}}}_{{\text{4}}}}{{{\left( {{\text{PS}}} \right)}}_{{\text{2}}}}\left( {{\text{PS*}}} \right)} \right]. \\ \end{gathered} $Cпектры ЯМР 31P{1H} I в CD3CN были записаны при разных температурах: –25, –15, 0, 25, 40, 55°C (рис. 4). Соответствующие значения хим. сдвигов (δP) наблюдаемых сигналов представлены в табл. 3. При комнатной температуре можно выделить три пары сигналов c δP = 42.25 и 42.08 (I группа); 41.58 и 41.45 (II группа); 41.26 и 41.19 м.д. (III группа). С понижением температуры все сигналы смещаются в область более слабого поля, при этом разница между хим. сдвигами сигналов в каждой группе уменьшается. При ‒25°C сигналы в I и II группах сливаются и проявляются в виде уширенных синглетов (42.38 и 41.64 м.д. соответственно). Разница между хим. сдвигами сигналов II группы составила всего 0.04 м.д. (0.13 м.д. при 25°C). При повышении температуры наблюдается обратный эффект: все сигналы смещаются в область более сильного поля и становятся более широкими, разница между хим. сдвигами сигналов в каждой группе увеличивается. При 55°C сигналы III группы сливаются в одну широкую линию (41.00 м.д.). Наблюдаемая картина в спектрах ЯМР весьма сложна, и на данный момент у нас нет однозначной ее интерпретации. Можно предположить, что в растворе соединения I могут протекать несколько параллельных процессов. Во-первых, как упоминалось выше, наиболее вероятна диссоциация одного или нескольких фосфино-селеноэфирных лигандов с разрывом связи Mo–Se с образованием структур, указанных на рис. 3. Во-вторых, возможны обменные процессы, при которых будут меняться местами селен и хлор и/или растворитель и хлор, чему способствует раскрытие хелатного цикла.
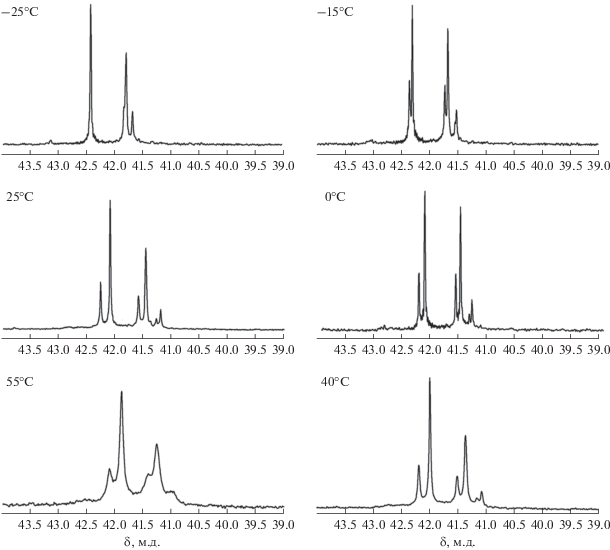
Таблица 3.
Cпектры ЯМР 31P{1H} I в CD3CN при различных температурах
| Температура, °C | Химические сдвиги, м.д.* |
|---|---|
| –25 | 42.38 (I гр.); 41.79, 41.75 (II гр.); 41.64 (III гр.) |
| –15 | 42.34, 42.29 (I гр.); 41.71, 41.66 (II гр.); 41.53, 41.51 (III гр.) |
| 0 | 42.26, 42.15 (I гр.); 41.60, 41.52 (II гр.); 41.37, 41.32 (III гр.) |
| 25 | 42.25, 42.08 (I гр.); 41.58, 41.45 (II гр.); 41.26, 41.19 (III гр.) |
| 40 | 42.17, 41.97 (I гр.); 41.49, 41.34 (II гр.); 41.14, 41.06 (III гр.) |
| 55 | 42.08, 41.87 (I гр.); 41.40, 41.25 (II гр.); 41.00 (III гр.) |
Ранее было установлено, что кластеры {Mo3S4} в различном координационном окружении высокоактивны в каталитическом превращении нитроаренов в соответствующие анилины [47, 48]. Аналогичные кластеры вольфрама обладают значительно меньшей активностью [51]. В данной работе комплекс I был протестирован в модельной реакции восстановления нитробензола с помощью Ph2SiH2 в ацетонитриле в мягких условиях (комнатная температура, атмосферное давление). Мониторинг этой реакции был выполнен с помощью спектроскопии ЯМР 1Н по отработанной методике [52]. Конверсия нитробензола в этом случае составила 87%, выход анилина 48%. Полученные значения заметно выше, чем для комплекса [Mo3S4Cl3(PS2)3]PF6 с тем же углеводородным заместителем при атоме серы (конверсия 79%, выход продукта 33%), но ниже, чем для [Mo3S4Cl3(PS1)3]PF6 с фенильным заместителем (99 и 83%). Таким образом, замена серы на селен в исходном фосфино-халькоэфире приводит к увеличению каталитической активности комплекса, что мы связываем с более ярковыраженными хемилабильными свойствами фосфино-халькоэфирных лигандов в [Mo3S4Cl3(PSе)3]PF6 по сравнению с [Mo3S4Cl3(PS2)3]PF6. Можно ожидать, что замена электронодонорного пентильного заместителя на электроноакцепторный фенильный при атоме селена приведет к значительному увеличению активности, возможно, даже превосходящей таковую для [Mo3S4Cl3(PS1)3](PF6).
Таким образом, в ходе данного исследования был выполнен синтез нового трехъядерного сульфидного кластерного комплекса [Mo3S4Cl3(PSe)3]PF6 с фосфино-селеноэфирным лигандом. Полученное соединение охарактеризовано набором физико-химических методов, в том числе установлена структура методом РСА. В растворе данного соединения при комнатной температуре наблюдалось образование нескольких форм, отличающихся по способу координации фосфино-селеноэфирных лигандов. Полученный комплекс проявил более высокую каталитическую активность в реакции восстановления нитробензола в анилин по сравнению со схожим соединением, содержащим фосфино-тиоэфирный лиганд.
Авторы заявляют об отсутствии конфликта интересов.
Список литературы
Hor T.S.A. // Acc. Chem. Res. 2007. № 65. P. 676.
Houk L.W. // J. Organomet. Chem. 1980. V. 192. № 1. P. C23.
Bader A., Lindner E. // Coord. Chem. Rev. 1991. V. 108. № 1. P. 27.
Bierenstiel M., Cross E.D. // Coord. Chem. Rev. 2011. V. 255. № 5–6. P. 574.
Braunstein P., Naud Â. // Angew. Chem. Int. Ed. 2001. V. 40. P. 680.
Espinet P., Soulantica K. // Coord. Chem. Rev. 1999. V. 193–195. P. 499.
Deckers P.J.W., Hessen B., Teuben J.H. // Angew. Chem. Int. Ed. 2001. V. 40. № 13. P. 2516.
Ros A., Estepa B., López-Rodríguez R. et al. // Angew. Chem. Int. Ed. 2011. V. 50. № 49. P. 11724.
Moxham G.L., Randell-Sly H.E., Brayshaw S.K. et al. // Angew. Chem. Int. Ed. 2006. V. 45. № 45. P. 7618.
Zhang W.H., Chien S.W., Hor T.S.A. // Coord. Chem. Rev. Elsevier B.V. 2011. V. 255. № 17–18. P. 1991.
Huynh H.V., Yeo C.H., Chew Y.X. // Organometallics. 2010. V. 29. № 6. P. 1479.
Jiménez M.V., Pérez-Torrente J.J., Bartolomé M.I. et al. // Organometallics. 2008. V. 27. № 2. P. 224.
Kuriyama M., Nagai K., Yamada K.-ichi et al. // J. Am. Chem. Soc. 2002. V. 124. № 30. P. 8932.
Braunstein P., Knorr M., Stern C. et al. // Coord. Chem. Rev. 1998. V. 180. P. 903.
Rasanen T.M., Jaaskelainen S., Pakkanen T.A. // J. Organomet. Chem. 1998. V. 553. P. 453.
King J.D., Monari M., Nordlander E. // J. Organomet. Chem. 1999. V. 573. P. 272.
Deeming A.J., Shinhmar M.K., Arce A.J. et al. // Dalton Trans. 1999. V. 3. P. 1153.
Tunik S.P., Koshevoy I.O., Poë A.J. et al. // Dalton Trans. 2003. P. 2457.
Pakkanen T.A. // Dalton Trans. 2004. V. 6. P. 2541.
Persson R., Monari M., Gobetto R. et al. // Organometallics. 2001. V. 20. № 20. P. 4150.
Hrovat D.A., Nordlander E., Richmond M.G. // Organometallics. 2012. V. 31. P. 6608.
Persson R., Stchedroff M.J., Uebersezig B. et al. // Organometallics. 2010. V. 29. № 10. P. 2223.
Persson R., Stchedroff M.J., Gobetto R. et al. // Eur. J. Inorg. Chem. 2013. № 13. P. 2447.
Mayberry D.D., Nesterov V.N., Richmond M.G. // Organometallics. 2019. V. 38. P. 2472.
Sokolov M.N., Fedin V.P., Sykes A.G. // Comprehensive Coordination Chemistry II. 2003. V. 4. P. 761.
Gushchin A.L., Hernandez-Molina R., Anyushin A.V. et al. // New J. Chem. 2016. V. 40. № 9. P. 7612.
Pino-Chamorro J.Á., Gushchin A.L., Fernández-Trujillo M.J. et al. // Chem. Eur. J. 2015. V. 21. № 7. P. 2835.
Morant-Giner M., Brotons-Alcázar I., Shmelev N.Y. et al. // Chem. Eur. J. 2020.
Гущин А.Л., Ларичева Ю.А., Соколов М.Н. и др. // Успехи химии. 2018. Т. 87. № 3. С. 670 (Gushchin A.L., Laricheva Y.A., Sokolov M.N. et al. // Russ. Chem. Rev. 2018. V. 87. № 7. P. 670).
Hernandez-Molina R., Gushchin A., González-Platas J. et al. // Dalton Trans. 2013. V. 42. № 42. P. 15016.
Федоров В.Е., Миронов Ю.В., Наумов Н.Г. и др. // Успехи химии. 2007. Т. 76. № 6. С. 529 (Fedorov V.E., Mironov Y. V, Naumov N.G. et al. // Russ. Chem. Rev. 2007. V. 76. № 6. P. 529).
Bustelo E., Gushchin A.L., Fernández-Trujillo M.J. et al. // Chem. Eur. J. 2015. V. 21. № 42. P. 14823.
Sokolov M.N., Gushchin A.L., Abramov P.A. et al. // In-org. Chem. 2007. V. 46. № 11. P. 4677.
Sokolov M.N., Gushchin A.L., Naumov D.Y. et al. // I-norg. Chem. 2005. V. 44. № 7. P. 2431.
Gushchin A.L., Llusar R., Vicent C. et al. // Eur. J. In-org. Chem. 2013. V. 2013. № 14. P. 2615.
Recatalá D., Llusar R., Gushchin A.L. et al. // ChemSusChem. 2015. V. 8. № 1. P. 148.
Laricheva Y.A., Gushchin A.L., Abramov P.A. et al. // Polyhedron. 2018. V. 154. P. 202.
Pino-Chamorro J.Á., Laricheva Y.A., Guillamón E. et al. // Inorg. Chem. 2016. V. 55. № 19. P. 9912.
Pino-Chamorro J.A., Laricheva Y.A., Guillamón E. et al. // New J. Chem. 2016. V. 40. № 9. P. 7872.
Gushchin A.L., Laricheva Y.A., Abramov P.A. et al. // Eur. J. Inorg. Chem. 2014. V. 2014. № 25. P. 4093.
Pedrajas E., Sorribes I., Gushchin A.L. et al. // ChemCatChem. 2017. V. 9. № 6. P. 1128.
Гущин А.Л., Ларичева Ю.А., Пирязев Д.А. и др. // Коорд. химия. 2014. Т. 40. № 1. С. 8 (Gushchin A.L., Laricheva Y.A., Piryazev D.A. et al. // Russ. J. Coord. Chem. 2014. V. 40. № 1. P. 5). https://doi.org/10.1134/S1070328414010023
Kryuchkova N.A., Syrokvashin M.M., Gushchin A.L. et al. // Spectrochim. Acta. A. 2018. V. 190. P. 347.
Gushchin A.L., Sokolov M.N., Peresypkina E.V. et al. // Eur. J. Inorg. Chem. 2008. V. 2008. № 25. P. 3964.
Dovydenko I.S., Laricheva Y.A., Korchagina K. V. et al. // J. Phys. Chem. B. 2019. V. 123. № 41. P. 8829.
Pedrajas E., Sorribes I., Junge K. et al. // Green Chem. 2017. V. 19. № 16. P. 3764.
Pedrajas E., Sorribes I., Junge K. et al. // ChemCatChem. 2015. V. 7. № 17. P. 2675.
Sorribes I., Wienhöfer G., Vicent C. et al. // Angew. Chem. Int. Ed. 2012. V. 51. № 31. P. 7794.
Safont V.S., Sorribes I., Andrés J. et al. // Phys. Chem. Chem. Phys. 2019. V. 21. № 31. P. 17221.
Pino-Chamorro J.A., Laricheva Y.A., Guillamón E. et al. // New J. Chem. 2016. V. 40. № 9. P. 7872.
Gushchin A.L., Shmelev N.Y., Malysheva S.F. et al. // Inorg. Chim. Acta. 2020. V. 508. № 1. P. 119645.
Gushchin A.L., Shmelev N.Y., Malysheva S.F. et al. // New J. Chem. 2018. V. 42. № 21. P. 17708.
Laricheva Y.A., Gushchin A.L., Abramov P.A. et al. // J. Struct. Chem. 2016. V. 57. № 5. P. 962.
Trofimov B.A., Gusarova N.K., Malysheva S.F. et al. // Synthesis. 2002. № 15. P. 2207.
APEX2 (version 1.08), SAINT (version 7.03), SAD-ABS (version 2. 11.), SHELXTL (version 6.12). Madison (WI, USA): Bruker AXS Inc., 2004.
Sheldrick G.M. // Acta Crrystallog. C. 2015. V. 71. P. 3.
Dolomanov O.V, Bourhis L.J., Gildea R.J. et al. // J. A-ppl. Cryst. 2009. V. 42. P. 339.
Zhou H., Sun H., Zheng T. et al. // Eur. J. Inorg. Chem. 2015. V. 2015. № 19. P. 3139.
Rosen M.S., Spokoyny A.M., MacHan C.W. et al. // In-org. Chem. 2011. V. 50. № 4. P. 1411.
Spokoyny A.M., Rosen M.S., Ulmann P.A. et al. // Inorg. Chem. 2010. V. 49. № 4. P. 1577.
Cunningham T.J., Elsegood M.R.J., Kelly P.F. et al. // Dalton Trans. 2010. V. 39. № 22. P. 5216.
Sokolov M.N., Gushchin A.L., Naumov D.Y. et al. // J. Cluster Sci. 2005. V. 16. № 3. P. 309.
Sokolov M.N., Gushchin A.L., Kovalenko K.A. et al. // Inorg. Chem. 2007. V. 46. № 6. P. 2115.
Beltrán T.F., Pino-Chamorro J.Á., Fernández-Trujillo M.J. et al. // Inorg. Chem. 2015. V. 54. № 2. P. 607.
Alfonso C., Feliz M., Safont V.S. et al. // Dalton. Trans. 2016. V. 45. № 18. P. 7829.
Fedin V.P., Sokolov M.N., Virovets A. V. et al. // Inorg. Chim. Acta. 1998. V. 269. № 2. P. 292.
Hernandez-Molina R., Dybtsev D.N., Fedin V.P. et al. // Inorg. Chem. 1998. V. 37. № 12. P. 2995.
Gushchin A.L., Sokolov M.N., Naumov N.Y. et al. // Russ. Chem. Bull. 2006. V. 55. № 11. P. 1966.
Gushchin A.L., Llusar R., Vicent C. et al. // Eur. J. I-norg. Chem. 2013. V. 2013. № 14. P. 1.
Fedin V.P., Sokolov M.N., Geras’ko O.A. et al. // Inorg. Chim. Acta. 1991. V. 187. № 1. P. 2615.
Varey J.E., Sykes A.G. // Dalton. Trans. 1993. V. 4. № 22. P. 3293.
Дополнительные материалы отсутствуют.
Инструменты
Координационная химия