

Координационная химия, 2021, T. 47, № 3, стр. 147-152
Квантово-химическое изучение строения и устойчивости димеров бис-дикетонатов меди(II)
А. Г. Стариков 1, А. А. Старикова 1, *, В. И. Минкин 1
1 Научно-исследовательский институт физической и органической химии
Южного федерального университета
Ростов-на-Дону, Россия
* E-mail: aastarikova@sfedu.ru
Поступила в редакцию 29.09.2020
После доработки 09.10.2020
Принята к публикации 13.10.2020
Аннотация
Выполнено квантово-химическое исследование димеров дикетонатов меди ([Cu(Acac)2]2 и [Cu(Acac)Hfac)]2) с использованием различных функционалов (B3LYP, TPSSh, PBE0 и B2PLYP) и базисных наборов (6-311++G(d,p) и Def2-TZVP), а также c учетом дисперсионных взаимодействий (D3BJ). Показано, что расчеты с применением функционалов B3LYP и TPSSh в сочетании с базисом Def2-TZVP дают наилучшее согласие с экспериментом. Установлено, что величины ошибок, обусловленных выбранными базисными наборами (BSSE), не вносят существенных изменений в энергии стабилизации рассмотренных систем. Полученные результаты позволяют заключить, что квантово-химическое изучение аналогичных ассоциатов переходных металлов следует проводить без включения эмпирических дисперсионных поправок D3BJ.
Координационные соединения переходных металлов с β-дикетонами привлекают повышенное внимание специалистов различных областей с момента их получения [1]. Они используются в качестве катализаторов ряда промышленно важных органических реакций, например изомеризации алкенов и эпоксидирования по Мукояма [2–4], являются прекурсорами для создания металлосодержащих покрытий [5, 6]. В последнее время возрастающий интерес к комплексам с β-дикетоновыми лигандами обусловлен возможностью получения моно- и полиядерных систем, демонстрирующих необычные магнитные свойства [7–13]. Недавно была показана перспективность соединений данного класса как мономолекулярных магнитов (single-molecule magnet – SMM) [11, 12, 14, 15]. Выявленная возможность механического и электрического манипулирования молекулярным магнетизмом позволяет найти им применение в устройствах наномехатроники и спинтроники [16]. Значительную группу магнитно-активных структур на основе бис-дикетонатов представляют комплексы меди с радикал-содержащими лигандами [17–19]. Для объяснения магнитного поведения таких гетеролигандных соединений привлекают методы квантовой химии, достоверность результатов которых во многом определяется типом использованного приближения. При изучении строения, механизмов реакций и объяснения физико-химических свойств комплексов переходных металлов, содержащих большое количество атомов (>100), наиболее часто применяют метод теории функционала плотности (DFT), однако необоснованные сочетания функционалов и базисных наборов могут приводить к получению некорректных данных [20].
Ранее мы выполнили квантово-химическое (DFT B3LYP/6-311++G(d,p)) изучение строения и энергетических характеристик олигомеров дикетонатов Co(II), Ni(II) и Zn(II) [21]. Также были рассмотрены неформирующие полиядерные структуры бис-дикетонаты Cu(II), димеры которых обнаружены только в кристаллах разнолигандных комплексов [22–24]. Показано, что расчетные схемы, учитывающие дисперсионные взаимодействия, существенно завышают устойчивость изученных координационных соединений [21]. Поскольку этот результат расходится с примерами успешного применения дисперсионных поправок при изучении комплексов, не содержащих переходные металлы с открытой электронной оболочкой [25, 26], в настоящей работе выполнено расширенное теоретическое исследование биядерных структур [Cu(Аcac)2]2 и [Cu(Аcac)(Рfac)]2 (Аcac = = ацетилацетонат, Нfac = гексафторацетилацетонат) с использованием четырех функционалов и двух базисных наборов, а также рассмотрено влияние дисперсионных поправок D3BJ [27], применяемых при изучении магнитно-активных соединений. Выбор в качестве объектов исследования димеров бис-дикетонатов меди обусловлен тем, что их образование не сопровождается значительным изменением геометрии мономеров, в отличие от аналогичных комплексов кобальта, никеля и цинка [21], вследствие чего на реакционном пути отсутствуют переходные состояния, способные оказать влияние на энергетические характеристики изучаемого процесса димеризации.
МЕТОДИКА РАСЧЕТОВ
Расчеты проводили с помощью программы Gaussian 16 [28] методом DFT с использованием функционалов (B3LYP [29], TPSSh [30], PBE0 [31] и B2PLYP [32]) и расширенных базисов 6‑311++G(d,p) и Def2-TZVP, комбинации которых успешно применяются при изучении комплексов переходных металлов [33–40]. Локализацию стационарных точек на поверхности потенциальной энергии (ППЭ) осуществляли путем полной оптимизации геометрии молекулярных структур с проверкой стабильности DFT волновой функции и расчетом силовых констант. Графические изображения молекулярных структур получали при помощи программы ChemCraft [41].
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Согласно кристаллохимическим данным, комплекс Cu(Аcac)(Нfac) формирует димеры, в которых расстояния между ионом меди и атомом кислорода соседних молекул составляют 2.70–2.72 Å [22, 23] (рис. 1). Эти величины указывают на достаточно слабые межмолекулярные взаимодействия, определяемые дисперсионными силами и эффектами упаковки. В кристалле Cu(Аcac)2 расстояние между молекулами комплекса, расположенными стопками, достигает 3.02–3.06 Å [42–44], что в совокупности с практически планарным строением бис-хелатов позволяет предположить формирование таких структур только за счет упаковки в кристалле.
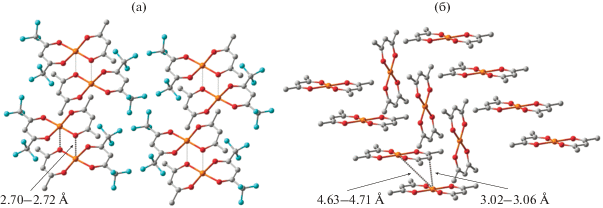
Как было показано ранее [21], расчет [Cu(Аcac)2]2 в приближении B3LYP/6-311++G(d,p) с использованием в качестве стартовой геометрии данных РСА приводит к структуре, в которой наименьшее расстояние между атомами двух молекул составляет 3.481 Å (рис. 2a), превышающее значения, полученные в ходе рентгенодифракционных исследований (3.02–3.06 Å) [42–44]. Данный факт можно объяснить тем, что при проведении расчетов эффекты кристаллической решетки не учитывались. Оптимизация геометрии [Cu(Аcac)2]2 с включением дисперсионной поправки (B3LYP/6-311++G(d,p) + D3BJ) приводит к ассоциату с нехарактерным для димеров бис-хелатов меди искажением молекул, расстояние между которыми меньше найденного в кристалле (рис. 2б).
Рис. 2.
Пространственное строение и геометрические характеристики димеров [Cu(Acac)2]2 и [Cu(Acac)(Hfac)]2, вычисленные без (a, в) и с включением дисперсионных поправок (б, г) соответственно.
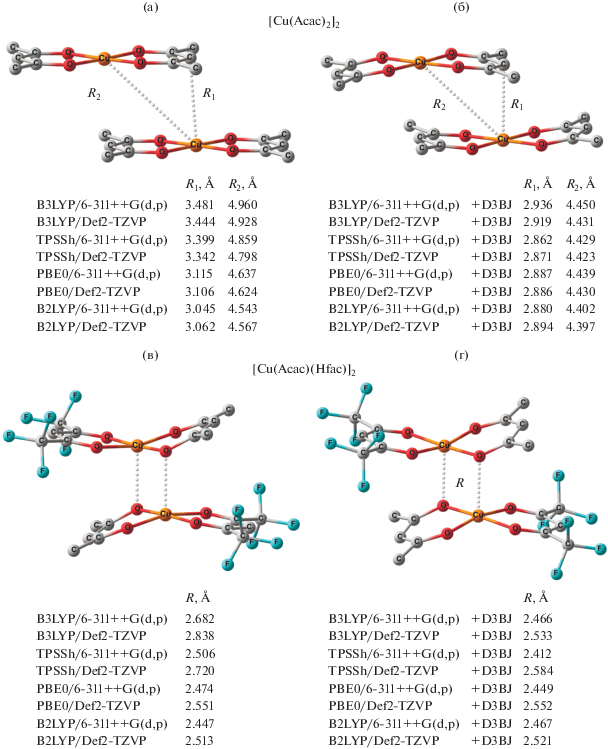
С целью получения ответа на вопрос, не является ли предсказанное искажение следствием использования приближения B3LYP/6-311++G(d,p), выполнены расчеты с применением функционалов TPSSh, PBE0 и B2PLYP и базисов 6-311++G(d,p) и Def2-TZVP. Представленные на рис. 2a результаты расчетов характеристических расстояний R1 и R2 согласуются с данными B3LYP [21]. Оптимизация геометрии без учета дисперсионных взаимодействий приводит к завышению расстояния между комплексами, а включение в расчетную схему дисперсионных поправок D3BJ сопровождается сокращением расстояний между молекулами и изгибом хелатных циклов (рис. 2б).
Отдельного внимания заслуживают результаты высокоуровневых расчетов с применением двойного гибридного функционала B2PLYP, включающего эффекты корреляции электронов. Как следует из рис. 2a, при использовании данного приближения локализована структура с укороченными по сравнению с найденными в результате РСА межатомными расстояниями Cu–Cu (рис. 1). Включение в расчетную схему дисперсионных поправок D3BJ способствует искажению бис-хелатов и сокращению межмолекулярных расстояний R1, R2 до значений, аналогичных полученным с использованием функционалов B3LYP, TPSSh и PBE0 в комбинации с D3BJ. Этот результат позволяет заключить, что, независимо от выбора функционала и базиса, применение упомянутых выше дисперсионных поправок приводит к некорректному воспроизведению экспериментальной геометрии комплекса Cu(Аcac)2.
Координационное соединение Cu(Аcac)(Нfac) – один из немногих бис-дикетонатов меди, имеющих димерное строение в кристалле [22, 23]; о существовании биядерных структур в растворе не сообщалось. Представленные на рис. 2в данные позволяют заключить, что функционал B3LYP в комбинации с базисами 6-311++G(d,p) и Def2-TZVP хорошо воспроизводит геометрию [Cu(Аcac)(Нfac)]2. Корректные результаты при использовании функционала TPSSh получены только в базисе Def2-TZVP, а применение PBE0 способствует существенному занижению межмолекулярного контакта Cu–O по сравнению с данными РСА (рис. 1). Проведение оптимизации геометрии посредством двойного гибридного функционала B2PLYP привело к димерам, в которых расстояние между ионом металла и атомом кислорода соседней молекулы оказалось короче найденного в эксперименте (рис. 2). Этот результат ставит под сомнение применимость данного приближения при квантово-химическом изучении подобных систем. Независимо от комбинации функционал/базис, включение в расчетную схему дисперсионной поправки D3BJ способствует значительному сокращению расстояния между молекулами [Cu(Аcac)(Нfac)]2 (рис. 2г) и, как следствие, переоценке его устойчивости.
Предсказываемая расчетами B3LYP и TPSSh невысокая энергия стабилизации (Eстаб) димера [Cu(Аcac)2]2 (2–4 ккал/моль, табл. 1) хорошо согласуется с нереализуемостью самоассоциата в растворе и газовой фазе. При использовании функционалов PBE0 и B2PLYP получены Eстаб, лежащие в интервале 6–11 ккал/моль. Эти значения могут быть обусловлены эффектами упаковки. Включение в расчетную схему дисперсионных поправок сопровождается не только искажением молекул и сокращением расстояния между бис-хелатами (рис. 2б), но и повышением энергии стабилизации до 20–23 ккал/моль (табл. 1). Такие величины характерны для молекулярных ассоциатов, стабилизированных водородными связями, но представляются завышенными в комплексах, расстояния между молекулами которых более 3 Å. Аналогичные результаты получены для [Cu(Аcac)- (Нfac)]2: ожидаемые небольшие значения энергий стабилизации предсказаны с помощью B3LYP и TPSSh; несколько выше величины, найденные с использованием функционалов PBE0 и B2PLYP, а дисперсионные поправки D3BJ завышают устойчивость димера (табл. 1).
Таблица 1.
Энергии стабилизации без учета (Eстаб) и с учетом энергии нулевых колебаний $\left( {E_{{{\text{стаб}}}}^{{{\text{ZPE}}}}} \right)$ (все значения приведены в ккал/моль) димеров [Cu(Acac)2]2 и [Cu(Acac)(Hfac)]2, вычисленные с помощью метода DFT
| Базисный набор | [Cu(Acac)2]2 | [Cu(Acac)(Hfac)]2 | ||
|---|---|---|---|---|
| Eстаб | $E_{{{\text{стаб}}}}^{{{\text{ZPE}}}}$ | Eстаб | $E_{{{\text{стаб}}}}^{{{\text{ZPE}}}}$ | |
| B3LYP | ||||
| 6-311++G(d,p)* | 3.1 | 2.8 | 2.7 | 2.6 |
| 6-311++G(d,p) + D3BJ | 23.3 | 22.2 | 22.2 | 21.1 |
| Def2-TZVP | 2.5 | 2.3 | 0.6 | 0.5 |
| Def2-TZVP + D3BJ | 23.1 | 22.0 | 19.5 | 18.7 |
| TPSSh | ||||
| 6-311++G(d,p) | 4.0 | 3.9 | 3.8 | 3.6 |
| 6-311++G(d,p) + D3BJ | 21.6 | 20.8 | 19.4 | 18.9 |
| Def2-TZVP | 3.3 | 3.1 | 0.9 | 0.8 |
| Def2-TZVP + D3BJ | 20.9 | 19.9 | 17.0 | 16.3 |
| PBE0 | ||||
| 6-311++G(d,p) | 7.2 | 7.1 | 7.0 | 6.8 |
| 6-311++G(d,p) + D3BJ | 21.5 | 20.6 | 20.3 | 19.7 |
| Def2-TZVP | 6.4 | 6.1 | 4.2 | 4.0 |
| Def2-TZVP + D3BJ | 20.6 | 19.7 | 17.1 | 16.4 |
| B2PLYP | ||||
| 6-311++G(d,p) | 11.3 | 12.6 | ||
| 6-311++G(d,p) + D3BJ | 23.4 | 23.8 | ||
| Def2-TZVP | 9.6 | 8.3 | ||
| Def2-TZVP + D3BJ | 21.5 | 19.3 | ||
* Результаты расчетов в приближении B3LYP/6-311++G(d,p) заимствованы из [21].
Для оценки влияния погрешности примененных базисов (6-311++G(d,p) и Def2-TZVP) на значения энергий стабилизации димеров вычислены ошибки суперпозиции базисных наборов (BSSE – Basis Set Superposition Error) [45]. Расчеты BSSE выполнены в приближениях B3LYP/6-311++G(d,p)/Def2-TZVP, обеспечивающих хорошее согласие с экспериментальными данными и большую вариабельность учета дисперсионных взаимодействий. В табл. 2 приведены результаты расчетов величин BSSE, полученных с использованием различных схем учета дисперсионных взаимодействий: CAM [46], D3BJ и их комбинации CAM + D3BJ. Вычисленная с применением данного подхода энергия комплексообразования практически совпадает с найденной без учета BSSE, а ошибки суперпозиции базисных наборов находятся в пределах 3 ккал/моль, за исключением расчета [Cu(Аcac)(Нfac)]2 в приближении B3LYP/6-311++G(d,p) с включением дисперсионных поправок, в котором погрешность доходит до 5 ккал/моль. Следовательно, результаты, полученные с помощью расчетов B3LYP, не содержат систематических ошибок, существенно влияющих на сформулированные выводы.
Таблица 2.
Энергии комплексообразования с учетом коррекции BSSE ($E_{{{\text{обр}}}}^{{{\text{BSSE}}}}$) и ошибки базисных наборов (BSSE) (все значения приведены в ккал/моль) димеров [Cu(Acac)2]2 и [Cu(Acac)(Hfac)]2, вычисленные при помощи метода DFT с использованием функционала B3LYP
| Базисный набор | $E_{{{\text{обр}}}}^{{{\text{BSSE}}}}$ | BSSE |
|---|---|---|
| [Cu(Acac)2]2 | ||
| *6-311++G(d,p) | 1.8 | 1.8 |
| 6-311++G(d,p) CAM-B3LYP | 5.5 | 2.3 |
| 6-311++G(d,p) + D3BJ | 22.4 | 2.7 |
| 6-311++G(d,p) CAM-B3LYP + D3BJ | 20.4 | 2.7 |
| Def2-TZVP | 1.5 | 1.6 |
| Def2-TZVP CAM-B3LYP | 5.4 | 1.5 |
| Def2-TZVP + D3BJ | 22.4 | 2.5 |
| Def2-TZVP CAM-B3LYP + D3BJ | 16.5 | 2.2 |
| [Cu(Acac)(Hfac)]2 | ||
| 6-311++G(d,p) | 2.2 | 2.4 |
| 6-311++G(d,p) CAM-B3LYP | 8.9 | 4.3 |
| 6-311++G(d,p) + D3BJ | 21.3 | 4.8 |
| 6-311++G(d,p) CAM-B3LYP + D3BJ | 21.1 | 4.9 |
| Def2-TZVP | 1.9 | 1.7 |
| Def2-TZVP CAM-B3LYP | 7.5 | 2.4 |
| Def2-TZVP + D3BJ | 21.1 | 2.7 |
| Def2-TZVP CAM-B3LYP + D3BJ | 20.2 | 2.7 |
* Результаты расчетов в базисе 6-311++G(d,p) заимствованы из [21].
Таким образом, посредством систематического DFT-исследования димеров дикетонатов меди [Cu(Аcac)2]2 и [Cu(Аcac)(Нfac)]2 установлено, что использование дисперсионных поправок приводит к переоценке устойчивости этих слабосвязанных ассоциатов и сопровождается значительным искажением их геометрий. Предсказанные с учетом эмпирических поправок D3BJ энергии стабилизации димеров бис-ацетилацетонатов меди (20–23 ккал/моль) позволяют ожидать существования достаточно прочных биядерных структур, которые не зафиксированы ни в растворе, ни в кристаллическом состоянии [42–44]. Изучение димеров с применением приближений B3LYP, TPSSh, PBE0 и B2PLYP, учитывающего эффекты корреляции электронов, показало, что функционалы B3LYP и TPSSh в сочетании с Def2-TZVP дают наилучшее согласие с экспериментом, близкие результаты получены при использовании базиса 6‑311++G(d,p). Согласно расчетам BSSE, величины ошибок, обусловленных суперпозицией базисных наборов, лежат в диапазоне 3–5 ккал/моль и не вносят существенных поправок в энергии стабилизации рассмотренных систем.
Авторы заявляют об отсутствии конфликта интересов.
Список литературы
Claisen L., Ehrhardt E.F. // Chem. Ber. 1889. V. 22. № 1. P. 1009.
New Pathways for Organic Synthesis – Practical Applications of Transition Metals / Eds. Colquhoun H.M., Holton J., Thompson D.J., Twigg M.V. N.Y.: Plenum, 1984. 454 p.
Wentzel B.B., Gosling P.A., Feiters M.C., Nolte R.J.M. // Dalton. Trans. 1998. № 13. P. 2241.
Baisch U., Poli R. // Polyhedron. 2008. V. 27. № 9–10. P. 2175.
Condorelli G.G., Malandrino G., Fragalà I.L. // Coord. Chem. Rev. 2007. V. 251. № 13–14. P. 1931.
Lieberman C.M., Filatov A.S., Wei Z. et al. // Chem. Sci. 2015. V. 6. № 5. P. 2835.
Aromí G., Gamez P., Reedijk J. // Coord. Chem. Rev. 2008. V. 252. № 8–9. P. 964.
Vigato P.A., Peruzzo V., Tamburini S. // Coord. Chem. Rev. 2009. V. 253. № 7–8. P. 1099.
Старикова А.А., Стариков А.Г., Минкин В.И. // Коорд. химия. 2015. Т. 41. № 8. С. 451 (Starikova A.A., Starikov A.G., Minkin V.I. // Russ. J. Coord. Chem. 2015. V. 41. № 8. P. 487). https://doi.org/10.1134/S1070328415080060
Gavrikov A.V., Koroteev P.S., Dobrokhotova Zh.V. et al. // Polyhedron. 2015. V. 102. P. 48.
Gavrikov A.V., Efimov N.N., Dobrokhotova Zh.V. et al. // Dalton Trans. 2017. V. 46. № 35. P. 11806.
Gavrikov A.V., Efimov N.N., Ilyukhin A.B. et al. // Dalton Trans. 2018. V. 47. № 17. P. 6199.
Илюхин А.Б., Гавриков А.В., Доброхотова Ж.В., Новоторцев В.М. // Журн. неорган. химии. 2018. Т. 63. № 9. P. 1161 (Ilyukhin A.B., Gavrikov A.V., Dobrokhotova Zh.V., Novotortsev V.M. // Russ. J. Inorg. Chem. 2018. V. 63. № 9. P. 1186). https://doi.org/10.1134/S003602361809005X
Palii A.V., Korchagin D.V., Yureva E.A. et al. // Inorg. Chem. 2016. V. 55. № 19. P. 9696.
Korchagin D.V., Palii A.V., Yureva E.A. et al. // Dalton Trans. 2017. V. 46. № 23. P. 7540.
Domanov O., Weschke E., Saito T. et al. // Nanoscale. 2019. V. 11. № 22. P. 10615.
Fedin M.V., Veber S.L., Bagryanskaya E.G., Ovcharenko V.I. // Coord. Chem. Rev. 2015. V. 289–290. P. 341.
Tumanov S.V., Veber S.L., Tolstikov S.E. et al. // Inorg. Chem. 2017. V. 56. № 19. P. 11729.
Tumanov S.V., Veber S.L., Tolstikov S.E. et al. // Dalton Trans. 2020. V. 49. № 18. P. 5851.
Minkin V.I., Starikov A.G., Starikova A.A. // Pure Appl. Chem. 2018. V. 90. № 5. P. 811.
Starikov A.G., Ivanov D.G., Starikova A.A., Minkin V.I. // Chem. Pap. 2018. V. 72. № 4. P. 829.
Байдина И.А., Стабников П.А., Игуменов И.К., Борисов С.В. // Коорд. химия. 1984. Т. 10. № 12. С. 1699.
Le Brun P.C., Lyon W.D., Kuska H.A. // Inorg. Chem. 1986. V. 25. № 17. P. 3106.
Байдина И.А., Громилов С.А. // Журн. структур. химии. 1991. Т. 32. № 3. С. 96 (Baidina I.A., Gromilov S.A. // J. Struct. Chem. 1991. V. 32. № 3. P. 395).
Hujo W., Grimme S. // J. Chem. Theory Comput. 2013. V. 9. № 1. P. 308.
Risthaus T., Grimme S. // J. Chem. Theory Comput. 2013. V. 9. № 3. P. 1580.
Grimme S., Ehrlich S., Goerigk L. // J. Comput. Chem. 2011. V. 32. № 7. P. 1456.
Frisch M.J., Trucks G.W., Schlegel H.B. et al. Gaussian 16. Revision C. 01. Wallingford: Gaussian, 2016.
Becke A.D. // J. Chem. Phys. 1993. V. 98. № 7. P. 5648.
Tao J.M., Perdew J.P., Staroverov V.N., Scuseria G.E. // Phys. Rev. Lett. 2003. V. 91. № 14. P. 146401.
Adamo C., Barone V. // J. Chem. Phys. 1999. V. 110. № 13. P. 6158.
Grimme S. // J. Chem. Phys. 2006. V. 124. № 3. P. 034108.
Weymuth T., Couzijn E.P.A., Chen P., Reiher M. // J. Chem. Theory Comput. 2014. V. 10. № 8. P. 3092.
Spin States in Biochemistry and Inorganic Chemistry: Influence on Structure and Reactivity / Eds. Swart M., Costas M. Chichester: John Wiley & Sons, 2016. 464 p.
Kepp K.P. // Inorg. Chem. 2016. V. 55. № 6. P. 2717.
Husch T., Freitag L., Reiher M. // J. Chem. Theory Comput. 2018. V. 14. № 5. P. 2456.
Ershova I.V., Smolyaninov I.V., Bogomyakov A.S. et al. // Dalton Trans. 2019. V. 48. № 28. P. 10723.
Tezgerevska T., Rousset E., Gable R.W. et al. // Dalton Trans. 2019. V. 48. № 31. P. 11674.
Старикова А.А., Чегерев М.Г., Стариков А.Г. // Коорд. химия. 2020. Т. 46. № 3. С. 172 (Starikova A.A., Chegerev M.G., Starikov A.G. // Russ. J. Coord. Chem. 2020. V. 46. № 3. P. 193). https://doi.org/10.1134/S1070328420030070
Gransbury G.K., Livesay B.N., Janetzki J.T. et al. // J. Am. Chem. Soc. 2020. V. 142. № 24. P. 10692.
Chemcraft. Version 1.8. 2014. http://www.chemcraftprog.com.
Старикова З.А., Шугам E.А. // Журн. структур. химии. 1969. Т. 10. № 2. С. 290 (Starikova Z.A., Shugam E.A. // J. Struct. Chem. 1969. V. 10. № 2. P. 267).
Vreshch V.D., Yang J.-H., Zhang H. et al. // Inorg. Chem. 2010. V. 49. № 18. P. 8430.
Rooydell R., Wang R.-C., Brahma S. et al. // Dalton Trans. 2015. V. 44. № 17. P. 7982.
van Duijneveldt F.B., van Duijneveldt-van de Rijdt J.G.C.M., van Lenthe J.H. // Chem. Rev. 1994. V. 94. № 7. P. 1873.
Yanai T., Tew D., Handy N. // Chem. Phys. Lett. 2004. V. 393. № 1–3. P. 51.
Дополнительные материалы отсутствуют.
Инструменты
Координационная химия