

Координационная химия, 2021, T. 47, № 8, стр. 499-504
Синтез и структура (Bu4N)[Mo5OI13]: первый пятиядерный иодидный кластер молибдена
М. А. Михайлов 1, *, Т. С. Сухих 1, Д. Г. Шевень 1, М. Н. Соколов 1, 2, 3
1 Институт неорганической химии им. А.В. Николаева СО РАН
Новосибирск, Россия
2 Новосибирский национальный исследовательский государственный университет
Новосибирск, Россия
3 Казанский (Приволжский) федеральный университет
Казань, Россия
* E-mail: mikhajlovmaks@yandex.ru
Поступила в редакцию 16.02.2021
После доработки 16.03.2021
Принята к публикации 17.03.2021
Аннотация
При нагревании смеси LiI–I2–Mo в интервале температур 300–400°С c последующей экстракцией продукта получены первый иодидный кластер молибдена с металлостовом в виде тетрагональный пирамиды состава (Bu4N)[Mo5OI13] ⋅ THF (I) и сокристаллизат (Bu4N){[Mo5OI13]0.9[Mo6I14]0.1} ⋅ THF (II). Комплексы I и II изучены методом РСА (CIF files CCDC № 2063029 (I), 2063030 (II)). Атомы молибдена в [Mo5OI13]– образуют квадратную пирамиду c расстояниями Mo–Mo 2.67 Å между базальными атомами, Mo–Mo 2.72 Å между апикальным и базальным атомами молибдена. К основанию пирамиды координирован атом кислорода (Mo–μ4-O 2.10 Å). Кластерный анион [Mo5OI13]– можно представить как октаэдрический кластерный анион [Mo6I14]2–, в котором место фрагмента {MoI}– (d0, 6e) занято изоэлектронным атомом кислорода (s2p4, 6e). В структуре (II) кластерные анионы [Mo5OI13]– и [Mo6I14]2– занимают близкие атомные позиции.
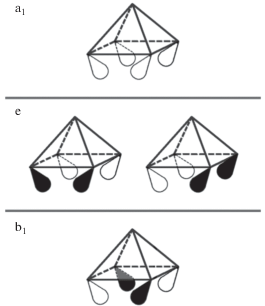
Характерной особенностью низших галогенидов молибдена и вольфрама является образование устойчивых кластерных группировок со связями металл–металл различной кратности [1]. Наиболее изучены октаэдрические кластеры, содержащие кластерные ядра {M6X8}4+ (M = Mo, W; X = Cl, Br, I) [2]. Активный интерес к ним в настоящее время вызван способностью этих кластеров проявлять яркую красную фосфоресценцию, что может найти практическое применение в различных областях [3]. Как правило, структуры металлоостовов кластеров низшей нуклеарности (3–5 атомов металла) могут быть формально выведены из октаэдра путем удаления одной (квадратная пирамида), двух (“бабочка” или квадрат) и трех вершин (треугольник) (рис. 1), хотя известны и тетраэдрические кластеры [4, 5]. Пятиядерные кластеры со структурой квадратной пирамиды представляют особый интерес как возможные предшественники гетерометаллических октаэдрических кластеров типа {M5M'X8}4+ (M' = гетероатом), особенно в случае X = I, поскольку именно иодидные кластерные комплексы молибдена [Mo6I8L6]z, L – терминальные лиганды, z – заряд координационной сферы, демонстрируют наибольшие квантовые выходы и времена жизни [3]. Можно ожидать, что замена одного из атомов молибдена на гетерометалл сможет увеличить диапазон количественных характеристик фосфоресценции (максимум длины волны эмиссии, квантовый выход, время жизни). В то время как для вольфрама описаны пятиядерные иодидные кластеры как в составе бинарных фаз [5], так и в виде анионных комплексов [W5I13]–/2– и [W5(µ4-C)I13]– [6] для молибдена были получены лишь хлоридный и бромидный кластеры [Mo5X13]2– (X = Cl, Br) [7–10].
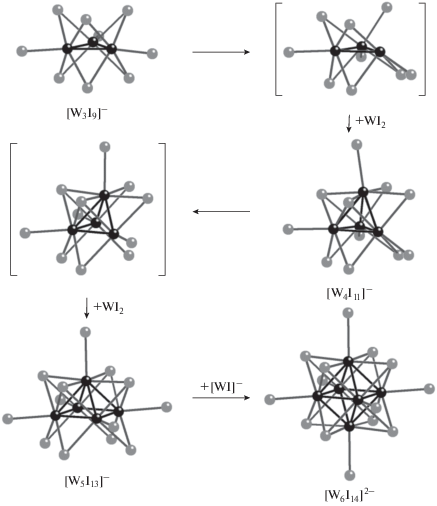
В настоящей работе мы получили первый пример иодидного кластера молибдена (Bu4N)[Mo5OI13] ⋅ ⋅ THF со структурой металлоостова в виде квадратной пирамиды, изученной методом рентгеноструктурного анализа.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Органические растворители тетрагидрофуран, ацетонитрил и этиловый спирт очищали по стандартным процедурам, гексан (“ос. ч.”) использовали без специальной очистки, как и диэтиловый эфир. Исходные реагенты Mo, I2, LiI, Bu4NI со степенью чистоты не ниже “ч. д. а.” приобретали из коммерческих источников. Высокотемпературные синтезы проводили в откачанных и запаянных стеклянных ампулах. После вскрытия ампул все манипуляции осуществляли на воздухе. Безводный LiI хранили в сухом боксе.
ИК-спектры записывали на спектрометрах VERTEX 80 (600–100 см–1) и SCIMITAR FTS 2000 (4000–400 см–1). Спектры КР записывали на спектрометре LabRAM HR Evolution (Horiba, Japan) с линией 632.8 нм He–Ne-лазера. Энергодисперсионную рентгеновскую спектроскопию выполняли на спектрометре Hitachi Tabletop Microscope TM-3000, снабженном системой QUANTAX 70. Элементный анализ выполняли в Аналитической лаборатории ИНХ СО РАН на CHNS-анализаторе Euro EA 3000.
Масс-спектрометрические данные получали на жидкостном хроматографе – масс спектрометре (LC-MC) фирмы Agilent (6130 Quadrupole MS, 1260 infinity LC). Анализ проводили в диапазоне (m/z) 350–3000 как для положительно, так и для отрицательно заряженных ионов в режиме SCAN. В качестве источника ионизации использовали электрораспыление. Осушителем служил поток газообразного азота, температура 350°C, скорость потока 7 л/мин; давление на распылителе (азот) 60 фунтов на кв. дюйм; напряжение на капилляре 4000 В. С целью сохранить слабо связанные формы в масс-спектрах, напряжение на фрагменторе было нулевым во всех экспериментах. Раствор исследуемого соединения (5 мкл) в дейтерированном ацетонитриле с концентрацией ~10–4 г/мл вводили в подвижную фазу (ацетонитрил “ос. ч.”) со скоростью потока 0.4 мл/мин, распыляли и ионизировали. Экспериментальные пики сравнивали с расчетными, в том числе и по изотопному распределению. Для расчетов использовали программу Molecular Weight Calculator by Matthew Monroe.
Синтез (Bu4N)[Mo5OI13] ⋅ THF (I). Смесь порошка Mo (1.403 г, 0.014 моль), I2 (4 г, 0.016 моль), LiI (0.767 г, 0.0057 моль) в сухом боксе помещали в стеклянную ампулу, которую откачивали и запаивали, помещали в муфельную печь, нагревали 4 ч до 400°С и выдерживали данную температуру 48 ч. После остывания ампулы до комнатной температуры, плав извлекали, растирали с порошком Bu4NI (6 г, 0.016 моль) и полученную смесь нагервали при 150°С в стеклянной ампуле 24 ч. Остывший плав отмывали этиловым спиртом от избытка Bu4NI и растворяли в ацетоне. Остаток экстрагировали тетрагидрофураном, объединяли с ацетоновым экстрактом и упаривали. Твердый продукт снова промывали этиловым спиртом и диэтиловым эфиром на пористом стеклянном фильтре. Масса промытого и высушенного (током воздуха на стеклянном фильтре) порошка (шоколадного цвета) составила 3.330 г. Продукт содержал смесь (Bu4N)2- [Mo6I14] и (Bu4N)[Mo5OI13], для разделения которых октаэдричексий кластер экстрагировали 100 мл CH3CN. При этом в раствор переходил (Bu4N)2- [Mo6I14] (1.00 г, 0.35 ммоль), а в нерастворимом осадке оставался (Bu4N)[Mo5OI13] (2.33 г, 0.97 ммоль), который хорошо растворим в тетрагидрофуране. Полное разделение достигали двойной перекристаллизацией из тетрагидрофурана при медленном насыщении раствора парами гексана с образованием сольвата I. Выход чистой фазы 1.86 г (27%).
ИК-спектр (область 4000–400 см–1; KBr; ν, см–1): 2954 с, 2926 с, 2867 ср, 1716 сл, 1520 сл, 1467 с, 1378 ср, 1328 сл, 1281 сл, 1256 сл, 1165 ср, 1131 сл, 1060 ср, 1029 сл, 991 сл, 883 ср, 800 сл, 735 ср, 525 сл, 445 с. ИК-спектр (область 600–100 см–1; KBr; ν, см–1): 442.2 сл, 200.5 ср, 152.4 сл, 121.5 ср. КР-спектр (ν, см–1): 14.9 с, 23.2 ср, 45.8 ср, 55.1 ср, 99.2 сл, 107.6 ср, 115.5 ср, 122.7 ср, 129.3 ср, 154.9 с, 208.4 сл. Электроспрей-масс-спектр (MeCN, m/z): средн. 2145.3 ([Mo5OI13]–: выч. 2145.3). Энергодисперсионный рентгеновский анализ: атомное отношение Mo : I = = 5.0 : 13.0.
Монокристаллы, пригодные для РСА, получали диффузией паров гексана в раствор (Bu4N) [Mo5OI13] в тетрагидрофуране. При насыщении парами гексана раствора смеси кластеров в тетрагидрофуране получили монокристаллы сокристаллизата (Bu4N){[Mo5OI13]0.9[Mo6I14]0.1} ⋅ THF (II).
РСА образцов I и II выполнен при 150 K на дифрактометре Bruker D8 Venture (ЦКП ИНХ СО РАН), оснащенном детектором CMOS PHOTON III и микрофокусным источником IµS 3.0 (MoKα, фокусирующие зеркала Монтеля). Редукция данных проведена с помощью пакета программам APEX3 [11]. Структуры расшифрованы и уточнены с помощью программ SHELXT [12] и SHELXL [13] и программной оболочки Olex2 [14]. Исследованный образец соединения I представлял собой сросток из двух основных кристаллических доменов. Из-за слишком большой разницы в интенсивности мало- и высокоугловых рефлексов не удалось корректно редуцировать данные с учетом обоих доменов. Редукция данных для основного домена и последующее введение матрицы двойникования не позволили полностью избежать наличия артефактной остаточной электронной плотности, что привело к относительно высокому конечному R-фактору. В структуре соединения II имеется разупорядочение фрагментов O и {MoI}– двух разных кластеров. Атомы Bu4N+ также должны занимать разные позиции в соответствии с разным стерическим влиянием кластеров, но разупорядочение Bu4N+ не было введено из-за малой заселенности альтернативных атомных позиций и отсутствия соответствующих выраженных пиков. Кристаллографические данные, детали экспериментов и уточнения структур I и II приведены в табл. 1.
Таблица 1.
Кристаллографические данные и детали эксперимента для соединений I и II
| Параметр | Значение | |
|---|---|---|
| I | II | |
| Брутто-формула | C16H36NOI13Mo5 | C20H44NO1.9I13.1Mo5.1 |
| М | 2387.86 | 2480.01 |
| Температура, K | 150(2) | 150(2) |
| Пр. гр. | P21/c | P21/n |
| a, Å | 19.092(2) | 11.0227(2) |
| b, Å | 18.730(2) | 15.4975(3) |
| c, Å | 24.364(2) | 28.1269(5) |
| β, град | 94.857(3) | 95.4930(10) |
| V, Å3 | 8681.2(15) | 4782.69(15) |
| Z | 8 | 4 |
| ρ(выч.), г cм–3 | 3.654 | 3.444 |
| µ, мм–1 | 10.667 | 9.775 |
| F(000) | 8368.0 | 4378.0 |
| Размер кристалла, мм | 0.09 × 0.09 × 0.05 | 0.1 × 0.08 × 0.06 |
| Излучение (λ, Å) | MoKα (0.71073) | |
| Диапазон сбора данных по θ, град | 2.606–66.394 | 4.548–61.038 |
| Диапазоны h, k, l | –29 ≤ h ≤ 29, –28 ≤ k ≤ 24, –37 ≤ l ≤ 36 |
–15 ≤ h ≤ 15, –22 ≤ k ≤ 22, –37 ≤ l ≤ 40 |
| Число измеренных рефлексов | 182 692 | 86 136 |
| Число независимых рефлексов (Rint, Rσ ) | 33 225 (0.0541, 0.0433) | 14 604 (0.0338, 0.0245) |
| Число ограничений/уточняемых параметров | 0/668 | 16/308 |
| R-фактор (I > 2σ(I)) | R1 = 0.0924, wR2 = 0.3291 |
R1 = 0.0337, wR2 = 0.0788 |
| R-фактор (все данные) | R1 = 0.1109, wR2 = 0.3663 |
R1 = 0.0420, wR2 = 0.0830 |
| Δρmin/Δρmax, e Å–3 | –5.92/8.01 | –2.14/2.16 |
Данные РСА депонированы в Кембриджском банке структурных (ССDC № 2063029 (I), 2063030 (II); http://www.ccdc.cam.ac.uk/conts/retrieving.html) и могут быть получены у авторов.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
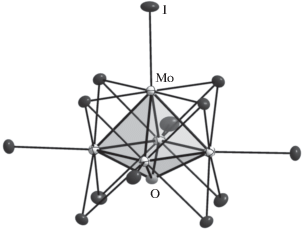
Синтез оксоиодидного кластерного комплекса (Bu4N)[Mo5OI13] ⋅ THF (I) осуществляется в две стадии. На первой стадии проводили нагрев смеси Mo, I, LiI при 400°С в течение 48 ч в соотношении, предполагающем получение кластерной соли пирамидального кластера Li2Mo5I13, образование которого предполагалось по аналогии с известными солями [W5I13]2– [6]. На второй стадии осуществляли замену катиона на тетрабутиламмоний нагреванием продукта реакции с Bu4NI при температуре плавления последнего (150°С) в течение 24 ч. Конечным продуктом оказался оксоиодидный кластер, выделенный из раствора тетрагидрофурана в виде сольвата I после двойной перекристаллизации. Кластерный анион I показан на рис. 2. Помимо этого, выделен (Bu4N)2Mo6I14, что предполагает образование Mo6I12 в качестве одного из продуктов высокотемпературной реакции (он становится единственным продуктом при проведении реакции выше 500°С). Можно предположить, что вторым продуктом является [Mo5I13]2–, который при экстракции на воздухе окисляется до [Mo5OI13]2–.
Рис. 2.
Строение кластерного аниона [Mo5OI13]– (эллипсоиды атомных смещений 50%-ной вероятности). Показан один из двух кристаллографически неэквивалентных анионов.
Температура 400°С была выбрана из предположения, что формирование пирамидального иодидного кластера Mo следует ожидать при относительно низких температурах, в интервале 300–500°С, тогда как выше 500°С будут получаться октаэдрические кластеры {Mo6I8}4+. Предварительные эксперименты показали, что при 300°С Mo реагирует с иодом в незначительной степени (в ампуле остается большое количество I2), хотя и в этих условиях удается выделить I в виде единичных монокристаллов. Температура 400°С представляется достаточно оптимальной, а с ее повышением доля (Bu4N)2[Mo6I14] будет только возрастать. В выбранных же условиях синтеза (Bu4N)[Mo5OI13] образуется в ~2 раза больше (по массе), чем (Bu4N)2[Mo6I14]. Образования иодидных кластеров молибдена меньшей нуклеарности при Т > 300°С не зафиксировано. С другой стороны, известно, если использовать другой источник молибдена (Mo(CO)6) и проводить реакцию с иодом при существенно более низкой температуре (110°С) в диглиме, конечным продуктом будет четырехъядерный кластер молибдена [Mo4OI12]2– [15]. Повышение нуклеарности в ряду [Mo4OI12]2–– [Mo5OI13]––[Mo6I14]2– идет параллельно с уменьшением степени окисления молибдена (+3.0; +2.8; +2.0 соответственно). Корреляция между степенью нуклеарности кластерных ядер (или количеством атомов металла в металлоостове кластерных ядер) и температурами синтеза наблюдается и в синтезах иодидных кластеров W и заключается в следующем: а) при более высоких температурах образуются кластеры с большей нуклеарностью, б) при повышении температуры может происходить превращение кластеров меньшей нуклерности в кластеры большей нуклеарности [5, 6].
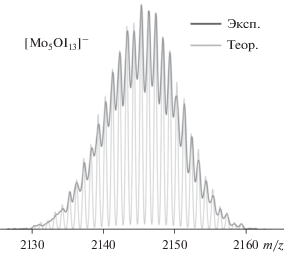
Кластер (Bu4N)2[Mo6I14] легко отделим из смеси благодаря существенной разнице в растворимости тетрабутиламмонийных кластерных солей в ацетонитриле. Кластер (Bu4N)[Mo5OI13] малорастворим в ацетонитриле, но хорошо растворим в тетрагидрофуране, и фаза I может быть получена в чистом виде после перекристаллизации из тетрагидрофурана, а степень чистоты фазы можно контролировать с помощью масс-спектрометрии и элементного анализа. При недостаточной очистке от (Bu4N)2[Mo6I14] в масс-спектре ацетонитрильного раствора продукта, помимо сигнала с центром при 2145.3 (m/z) (от кластерных анионов [Mo5OI13]–) (рис. 3), наблюдается сигнал с центром при 1176.5 (от анионов [Mo6I14]2–).
Рис. 3.
Найденное (обозначено темно-серым цветом) и рассчитанное (обозначено светло-серым цветом) изотопное распределение для частиц [Mo5OI13]– в ацетонитрильном растворе I.
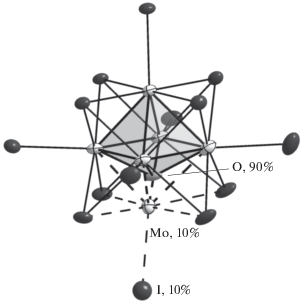
Оксоиодидный пирамидальный [Mo5OI13]– и октаэдрический анионный кластер [Mo6I14]2– могут образовывать сокристаллизат (Bu4N){[Mo5OI13]0.9- [Mo6I14]0.1} ⋅ THF (II), в структуре которого наблюдается разупорядочение атома кислорода и фрагмента {MoI}– (в основании квадратной пирамиды кластерного остова {Mo5}) (рис. 4).
Рис. 4.
Строение кластерного аниона в сокристаллизате II (эллипсоиды атомных смещений 50%-ной вероятности).
Согласно данным РСА, атомы молибдена в I (рис. 2) образуют квадратную пирамиду со средними расстояниями Mo–Mo 2.67 Å между базальными атомами и Mo–Mo 2.72 Å между базальным и апикальным атомами. К основанию пирамиды координирован атом кислорода, среднее расстояние Mo–O 2.10 Å. Кластерный анион [Mo5OI13]– можно представить как октаэдрический кластерный анион [Mo6I14]2– с отщепленной вершиной {MoI}– и координированным вместо нее атомом кислорода. При этом расстояния Mo–Mo в анионе [Mo5OI13]– идентичны расстояниям Mo–Mo в кластерных ядрах {Mo6I8}4+ (2.67–2.68 Å) [16, 17]. Это говорит о том, что замена фрагмента {MoI}– (d0, 6e) на изоэлектронный атом кислорода (s2p4, 6e) существенно не затрагивает связывающие кластерные скелетные орбитали, отвечающие за образование связей металл–металл. В оксоиодидном кластере (Bu4N)2[Mo4OI12] со структурой “бабочки” расстояния Mo–Mo не равнозначны и лежат в диапазоне от 2.65 Å (самая короткая связь в структуре “бабочки” {Mo4}) до 2.73 Å (остальные связи Mo–Mo), расстояния Mo–O тоже различны: от 2.07 и 2.15 Å [15]. Сопоставимые же (c найденными в I) расстояния металл–металл принадлежат и кластерам вольфрама (расстояние W–W в [PrN]2[W5I13] равно 2.64 Å [9], в [W6I14]2– оно несколько больше, 2.67 Å [6]). Более того, интересен факт изоструктурности (Bu4N)[Mo5OI13] ⋅ THF и (Bu4N)[W5(C)I13] · THF, при этом среднее расстояние W–W 2.69 Å в [W5(C)I13]– совпадает со средним расстоянием Mo–Mo в I (и также сопоставимо с расстоянием W–W в [W6I14]2–), а среднее расстояние W–C 2.16 Å несколько больше (на 0.06 Å) расстояния Mo–O в I.
Поскольку размеры и форма анионов [Mo6I14]2– и [Mo5OI13]– близки, не вызывает удивление образование сокристаллизата II, в котором все кристаллографические позиции атомов этих кластеров попарно совпадают за исключением атома O и фрагмента {MoI}–; заселенность позиций последних составляет 90 и10 соответственно.
Аналогия в строении октаэдрических клозо-кластеров [M6Х14]2– и производных от них нидо-аналогов [Mo5Х13]–/2–, [Mo5OI13]– и [W5CI13]– (M = Mo, W; X = Cl, Br, I) хорошо объясняется при рассмотрении их электронного строения в соответствии со схемой заполнения МО, предложенной Джонстоном и Мингосом в [4]. Эта схема подразумевает наличие 12 связующих орбиталей М–М (24 кластерных скелетных электрона, 84 валентных электрона). При удалении одной вершины образуется пирамидальный нидо-кластер [Mo5Х13]–, который располагает лишь девятью такими МО (18 скелетных электронов, 68 валентных электронов). При этом три МО бывшего октаэдрического кластера становятся разрыхляющими по отношению к связыванию М–М, хотя и возможно размещение одного электрона на самой низколежащей из них. Эти орбитали показаны на рис. 5. Очевидно, что обратная координация к квадратной грани металлофрагментов {MX}, изоэлектронных {MoI}– (например, {NbI}2–) [18], восстанавливает схему связывания с 12 связывающими МО. В случае неметалла (О или С), лишь две орбитали (набор е) подходят по симметрии для взаимодействия с р‑орбиталями (например, px и py), что дает дополнительно две связывающих орбитали, емкость которых составляет четыре электрона. Это не меняет число скелетных электронов (18), но суммарно дает 72 кластерных валентных электрона, если μ4-С и μ4-О рассматривать как доноры четырех электронов. В отличие от С и О, {MoI}– и {NbI}– (формально d6) используют d-орбитали, свойства симметрии которых допускают связующее с е-, так и с b1-орбиталью, используя все шесть электронов. Резюмируя, можно предположить, что нидо-кластеры [Mo5Х13]– будут образовывать устойчивые клозо-кластеры при присоединении металлофрагментов {MX} с электронной конфигурацией d6 или шапочных лигандов, способных донировать четыре электрона. В первом случае пятиядерный кластер будет выступать как своеобразный кластерный лиганд (нуклеофил), в то время как во втором – как полиядерный комплекс (электрофил), проявляя двойственную природу наподобие хорошо известных кластерных гидридов бора [19].
Авторы заявляют, что у них нет конфликта интересов.
Список литературы
Sokolov M.N., Naumov N.G., Samoylov P.P., Fedin V.P. // Comprehensive Inorganic Chemistry II, Oxford: Elsevier Ltd., 2013. V. 2. P. 271.
Welch E.G., Long J.R. // Progress Inorg. Chem. 2005. V. 54. P. 1.
Mikhaylov M.A., Sokolov M.N. // Eur. J. Inorg. Chem. 2019. P. 39.
Johnston R.L., Mingos D.M.P. // Inorg. Chem. 1986. V. 25. P. 1661.
Ströbele M., Meyer H.-J. // Dalton Trans. 2019. V. 48. P. 154.
Franolic J.D., Long J.R., Holm R.H. // J. Am. Chem. Soc. 1995. V. 117. P. 8139.
Jödden K., Schäfer H. // Z. Anorg. Allg. Chem. 1977. V. 430. P. 5.
Jödden K., von Schnering, H.G., Schäfer, H. // Angew. Chem. 1975. V. 87. P. 5945.
Ahmed E., Ahrens E., Heise M., Ruck M. // Z. Anorg. Allg. Chem. 2011. V. 637. P. 961.
Zietlow T.C., Gray H.B. // Inorg. Chem. 1986. V. 25. P. 631.
Bruker Apex3 Software Suite: Apex3, SADABS-2016/2 and SAINT (version 2018.7-2). Madison (WI, USA): Bruker AXS Inc., 2017.
Sheldrick G.M. // Acta Crystallogr. A. 2015. V. 71. P. 3.
Sheldrick G.M. // Acta Crystallogr. C. 2015. V. 71. P. 3.
Dolomanov O.V., Bourhis L.J., Gildea R.J. et al. // J. A-ppl. Crystallogr. 2009. V. 42. P. 339.
Mikhaylov M., Abramov P., Novozhilov I., Sokolov M. // Z. Anorg. Allg. Chem. 2018. V. 644. P. 438.
Bruckner P., Preetz W., Punjer M. // Z. Anorg. Allg. Chem. 1997. V. 623. P. 8.
Kirakci K., Cordier S., Roisnel T. et al. // Z. Kristallogr. New Cryst. Struct. 2005. V. 220. P. 116.
Artemkina S.B., Tarasenko M.S., Virovets A.V., Naumov N.G. // Russ. J. Coord. Chem. 2012. V. 38. № 4. P. 257.
Сиваев И.Б. // Журн. неорган. химии. 2020. Т. 65. № 12. С. 1643 (Sivaev I.B. // Russ. J. Inorg. Chem. 2020. V. 65. № 12. P. 1854). https://doi.org/10.1134/S0036023620120165
Дополнительные материалы отсутствуют.
Инструменты
Координационная химия