

Координационная химия, 2021, T. 47, № 8, стр. 505-515
Комплексы [Fe2(μ-S2ER2)(CO)6] (E = Si, Sn) – реагенты для получения гетерометаллических кластеров: синтез, строение и реакции с галогенсодержащими комплексами металлов
М. А. Огиенко 1, Н. А. Пушкаревский 1, Д. А. Баширов 1, Н. В. Куратьева 1, А. В. Вировец 1, С. Н. Конченко 1, *
1 Институт неорганической химии им. А.В. Николаева СО РАН
Новосибирск, Россия
* E-mail: konch@niic.nsc.ru
Поступила в редакцию 26.02.2021
После доработки 10.03.2021
Принята к публикации 12.03.2021
Аннотация
Синтезированы биядерные комплексы [Fe2(µ-S2ER2)(CO)6] (ER2 = SiMe2 (Iа), SiEt2 (Iб), SnEt2 (Iв)) – перспективные предшественники гетерометаллических кластеров. Изучены их реакции с галогенидными комплексами переходных металлов: [Cp"RhCl2]2 (Cp" = η5-C5H3tBu2), [(Dppe)NiCl2] (Dppe = = Ph2PCH2CH2PPh2), [(Ph3P)AuCl], [Mn(CO)5Cl], в результате которых получены гетерометаллические кластеры [Fe2Rh(µ3-S)2(CO)6Cp"] (II), [Fe2Ni(µ3-S)2(CO)6(Dppe)] (III), [Fe2(CO)6(µ-SSnClEt2)(µ-SAu(PPh3))] (IV), [Fe2(CO)6(µ4,η2-S2SnEt2){Mn(CO)4Cl}2] (V), [Fe2Mn(CO)9Mn(CO)5(µ3-S)(µ4-S)] (VI). Установлено, что кластер V превращается в VI при фотохимической активации. Строение соединений I–VI установлено РСА (CIF files CCDC № 751214 (Iв), 751215 (III · 0.5C7H8), 2062206 (V), 2062207 (Iб), 2062208 (Iа), 2062209 (IV · 0.5CH3C5H9).
Карбонилхалькогенидные кластеры железа известны уже несколько десятилетий и являются классическими объектами, на которых в значительной степени были изучены основные реакции кластеров и систематизированы их превращения, используемые для направленного синтеза более сложных гомо- и гетерометаллических производных [1, 2]. Существенный вклад в развитие этой области внесли работы А.А. Пасынского и его коллег [3–7]. Особое место в ряду халькогенкарбонилов железа занимает известный с 50-х годов прошлого столетия легкодоступный [Fe2(µ-S2)(CO)6] [8, 9]. Интерес к нему и его производным обусловлен тем, что, претерпевая разрыв связей S–S или/и Fe–Fe, данный комплекс является удобным источником фрагментов {FeS} и {Fe2S2} для построения более крупных кластеров, рассматриваемых как абиологические аналоги активных центров ряда ферментов [10–12]. Вследствие этого химия [Fe2(µ-S2)(CO)6] оказалась достаточно полно изучена, но интерес к ней стал постепенно угасать. Новый толчок развитию химии [Fe2(µ-S2)(CO)6] и его аналогов придала недавно обнаруженная активность соединений с фрагментом {Fe2S2} в процессах фотохимического восстановления протона до диводорода, причем в этих процессах оказались активны не только молекулярные соединения, но и наночастицы, полученные сорбцией [Fe2(µ-S2)- (CO)6] на поверхности полупроводников, например InP [13], CdTe [14] или ZnS [15, 16]. Эти результаты возродили интерес к [Fe2(µ-S2)(CO)6] [17–21] и побудили нас на поиск решения некоторых проблемных моментов его использования для синтеза гетерометаллических кластеров, в которых сохраняется фрагмент {Fe2S2}.
Одна из таких проблем присутствует в синтетических маршрутах, основанных на использовании аниона [Fe2(µ-S)2(CO)6]2–, получаемого in situ восстановлением [Fe2(µ-S2)(CO)6] [9]. Казалось бы, этот дианион подобен органическим дитиолатам, является хелатирующим лигандом с нуклеофильными центрами – мостиковыми атомами серы, которыми он и должен присоединяться к наиболее положительному атому или атомам органических и неорганических электрофилов (схема 1 ).
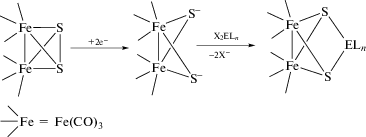
Схема 1 .
Действительно, эта схема применима и с точностью до возможного разрыва связи Fe–Fe позволяет получать кластеры, в которых присутствует фрагмент {Fe2S2}, что проверено на многих примерах [9, 22, 23]. Однако проблема состоит в том, что [Fe2(µ-S)2(CO)6]2– в растворе неустойчив и претерпевает дальнейшие превращения, вероятно, связанные с взаимодействием между анионной и нейтральной формами [Fe2(µ-S2)(CO)6] [24, 25]. Это лишает экспериментатора возможности “маневрировать” в методиках синтеза: использовать другие восстановители и растворители или более высокую температуру проведения синтеза, не опасаясь, что в результате в растворе будет получен набор анионных форм неизвестного состава. В попытке обойти эту проблему исследователи для синтеза гетерометаллических кластеров применяют методики, основанные на восстановлении [Fe2(µ-S2)(CO)6] в точно заданных условиях и последующем использовании без выделения восстановленной формы [Fe2(µ-S)2(CO)6]2–. Однако это не дает достаточной точности в дозировке последней, поскольку ее выход точно не известен. В значительной степени это ограничивает круг объектов, которые могут быть вовлечены в синтез в рамках данной схемы.
Решением этой проблемы могло бы стать использование не неустойчивого анионного [Fe2(µ-S)2- (CO)6]2–, а его стабильных нейтральных производных [Fe2(µ-S2ER2)(CO)6] (E = Si, Sn), которые в инертной атмосфере при комнатной температуре устойчивы, хорошо растворяются во всех апротонных растворителях и, как можно предположить, должны быть реакционноспособными по отношению к галогенидам, карбоксилатам или алкоголятам переходных металлов. Последнее предположение основано на том, что подобные подходы с использованием халькоген-силилированных и в меньшей степени станнилированных реагентов широко применяются в химии [26–29]. Базируются эти подходы на том, что группировки {R3E}+ и {R2E}2+ (E = Si, Sn) имеют высокое сродство к галогенид-иону или кислородсодержащему кислотному остатку (X− = Cl−, Br−, RCOO−, RO− и др.), поэтому образование соединений R3EX и R2EX2 обеспечивает “движущую силу” реакции. В то же время R3EX и R2EX2 относительно инертны, и обычно их легко удается отделить от целевого продукта синтеза. Иными словами, R3E и R2E (E = Si, Sn) можно рассматривать как “хорошие уходящие группы” в реакциях с галогенидами, карбоксилатами и алкоксидами переходных металлов. В результате этих взаимодействий должно происходить образование связи халькоген–металл, а в дальнейшем формирование кластерного остова.
Соединения типа [Fe2(µ-S2ER2)(CO)6] (E = Si, Sn) были получены ранее [9], но их реакционная способность и строение не были изучены. В настоящей работе разработаны улучшенные методики синтеза соединений [Fe2(µ-S2ER2)(CO)6] (ER2 = SiMe2 (Iа), SiEt2 (Iб), SnEt2 (Iв)) и с помощью монокристального РСА установлена их структура. Также для подтверждения нашего предположения о возможности использования Iа–Iв для синтеза гетерометаллических кластеров изучен ряд их реакций с галогенсодержащими комплексами переходных металлов: [Cp"RhCl2]2 (Cp" = η5-C5H3tBu2), [(Dppe)NiCl2] (Dppe = Ph2PCH2CH2PPh2), [(Ph3P)AuCl] и [Mn(CO)5Cl], в результате которых формируются комплексы: [Fe2Rh(µ3-S)2(CO)6Cp′′] (II), [Fe2Ni(µ3-S)2(CO)6(Dppe)] (III), [Fe2(CO)6(µ-SSnClEt2)(µ-SАu(PPh3))] (IV), [Fe2(CO)6(µ4,η2-S2SnEt2){Mn(CO)4Cl}2] (V), [Fe2Mn(CO)9Mn(CO)5- (µ3-S)(µ4-S)] (VI).
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Все операции по синтезу и выделению продуктов выполняли в атмосфере аргона в стандартной аппаратуре Шленка. Для удаления воды и растворенного кислорода растворители кипятили с использованием соответствующих осушителей и перегоняли в атмосфере аргона [30]. Синтез соединений [Fe2(µ-S2ER2)(CO)6] (ER2 = SiMe2 (Iа), SiEt2 (Iб), SnEt2 (Iв)) проводили с использованием подхода, описанного ранее [9]. Монокристаллы для РСА отбирали из выделенных кристаллических продуктов. Комплексы [(Dppe)NiCl2] [31], [Mn(CO)5Cl] [32] и [(Ph3P)AuCl] [33] получали по известным методикам. Синтез [Cp"RhCl2]2 (Cp" = = η5-C5H3tBu2) проводили аналогично методике [34].
Элементный анализ выполняли в лаборатории № 416 ИНХ СО РАН. При выполнении элементного анализа допускался кратковременный контакт (несколько секунд) образца с воздухом.
ИК-спектры (νCO, см–1) записывали на спектрометре Specord IR-75 для растворов соединений I, II, IV, V в н-гексане, для растворов кластеров III, VI в CH2Cl2 при комнатной температуре.
Спектры ЯМР 1H, 13C, 31P регистрировали при комнатной температуре на спектрометре Bruker AC 250 на частотах 1Н – 250.133 МГц, 13C – 62.896 МГц, 31P – 101.256 МГц. В качестве внутреннего стандарта использовали сигналы соответствующего растворителя: δН = 7.16 м.д. для C6D6, δС = 77.0 м.д. для СDCl3. Для спектров ЯМР 31P в качестве стандарта использовали 85%-ный водный раствор H3PO4. Константы спин-спинового взаимодействия приведены в герцах.
Синтез [Fe2(CO)6(µ3-S)2SiMe2] (Iа). К охлажденному до 0°C раствору [Fe2(CO)6(μ-S2)] (0.45 г, 1.31 ммоль) в 15 мл ТГФ, добавляли 0.2 мл сплава Na-K (мольное отношение Na : K = 2 : 3). Смесь перемешивали при охлаждении в ледяной бане в течение 4 ч. Полученный красный раствор отделяили от непрореагировавшего сплава Na-K и аморфного осадка, который промывали 15 мл ТГФ. К объединенному раствору добавляли раствор Me2SiCl2 (0.30 мл, 2.46 ммоль) в 10 мл ТГФ. Реакционную смесь перемешивали при комнатной температуре в течение 3 ч, затем упаривали досуха. Твердый остаток высушивали в вакууме и экстрагировали двумя порциями гексана (30 и 15 мл). Полученный темно-красный раствор упаривали в 4 раза. При выдерживании этого раствора при ‒16°C выпадают кристаллы соединения Iа. Выход 0.33 г (50%).
ИК-спектр (νCO, см–1): 2078 с, 2046 ср, 2038 с, 2008 с, 1998 с, 1987 ср, 1957 сл.
Синтез [Fe2(CO)6(µ3-S)2SnEt2] (Iв). К раствору [Fe2(CO)6(μ-S2)] (1.50 г, 4.37 ммоль) в 35 мл ТГФ, охлажденному до 0°C, добавляли 0.2 мл сплава Na-K (мольное отношение Na : K = 2 : 3). Смесь перемешивали при охлаждении в ледяной бане в течение 4 ч. Полученный красный раствор отделяли от непрореагировавшего сплава Na-K и аморфного осадка, который промывали 15 мл ТГФ. К объединенному фильтрату добавляли раствор [Et2SnCl2] (1.00 г, 4.03 ммоль) в 40 мл ТГФ. Реакционную смесь перемешивали при комнатной температуре в течение 3 ч, затем упаривали досуха. Твердый остаток промывали гексаном (15 мл) для удаления примесей исходного [Fe2(CO)6(μ-S2)], затем высушивали в вакууме. Полученный таким образом темный порошок экстрагировали двумя порциями СН2Сl2 (80 и 15 мл). Темно-красный раствор упаривали в 4 раза. При выдерживании сконцентрированного раствора при –16°C выпадают кристаллы соединения Iв. Выход 1.14 г (50%).
ИК-спектр (νCO, см–1): 2083 ср, 2070 с, 2030 с, 2005 с, 2001с, 1990 пл, 1986 с, 1976 ср.
Синтез [Fe2(CO)6(µ3-S)2SiEt2] (Iб). Попытки получить Iб по стандартной методике, описанной выше для Iа и Iв приводят к кристаллической фазе [Fe2(CO)6(µ3‑S)2SiEt2]⋅[Fe2(CO)6(µ-S2)]. Варьированием условий синтеза решить проблему образования такого сокристаллизата не удалось. Поэтому для синтеза Iб была использована другая методика: восстановление [Fe2(CO)6(µ-S2)] проводили Li[BHEt3] при низкой температуре.
К охлажденному до –80°C раствору [Fe2(CO)6- (μ-S2)] (0.50 г, 1.45 ммоль) в 35 мл ТГФ добавляли 3 мл 1 М раствора Li[BHEt3] в ТГФ. Смесь перемешивали при этой температуре в течение 20 мин. К полученному красному раствору прикапывали раствор Et2SiCl2 (0.45 мл, 3.01 ммоль) в 5 мл ТГФ. Реакционную смесь нагревали до комнатной температуры и перемешивали в течение 3 ч, затем упаривали досуха. Твердый остаток высушивали и экстрагировали двумя порциями гексана (40 и 5 мл). Полученный темно-красный раствор упаривали в 4 раза. При выдерживании этого раствора при –16°C выпадают кристаллы соединения Iб. Выход 0.49 г (78%).
ИК-спектр (νCO, см–1): 2078 с, 2046 ср, 2038 с, 2008 с, 1998 с, 1987 ср, 1957 сл.
Синтез [Fe2Rh(µ3-S)2(CO)6Cp′′] (II) и [Fe2Ni(µ3-S)2- (CO)6(Dppe)] (III). Реакции Iа–Iв с комплексами [Cp′′RhCl2]2 и [(Dppe)NiCl2 проводили однотипно с использованием следующих загрузок реагентов: для синтеза II – 0.049 г (0.094 ммоль) Iв и 0.034 г (0.048 ммоль) [Cp′′RhCl2]2; для синтеза III – 0.025 г (0.062 ммоль) Iа и 0.033 г (0.062 ммоль) [(Dppe)NiCl2]; 0.022 г (0.051 ммоль) Iб и 0.027 г (0.051 ммоль) [(Dppe)NiCl2]; 0.029 г (0.056 ммоль) Iв и 0.029 г (0.055 ммоль) [(Dppe)NiCl2].
Общий метод. К смеси твердых реагентов приливали 10 мл толуола. Реакционную смесь перемешивали при комнатной температуре 12 ч. Полученный темно-красный раствор фильтровали, небольшое количество неидентифицированного осадка промывали 15 мл толуола, объединенный фильтрат упаривали в 2 раза и выдерживали при ‒16°C, что приводило к кристаллизации, соответственно, фаз II или III · 0.5C7H8. Выход 70–80%.
Продукты реакций идентифицировали с помощью ИК-спектров [9, 23] (νCO, см–1): 2064 c., 2038 c., 1997 c., 1987 c (II); 2046 с., 2004 с., 1967 с., 1957 пл (III).
Синтез [Fe2(CO)6(µ-SSnClEt2)(µ-SAu(PPh3))] (IV). К смеси твердых реагентов Iв (0.062 г, 0.12 ммоль) и [(Ph3P)AuCl] (0.54 г, 0.24 ммоль) добавляли 10 мл толуола. Смесь перемешивали при комнатной температуре в течение 3 сут, что приводило к образованию коричневого раствора и темного осадка, который отфильтровывали и промывали 15 мл толуола. В объединенных фильтратах присутствует набор из нескольких продуктов (контроль по ТСХ). Раствор упаривали досуха, остаток экстрагировали 20 мл петролейного эфира. Выдерживание полученного оранжевого раствора при –16°C привело к образованию кристаллов фазы IV ⋅ 5CH3C5H9. Выход 0.013 г (10%).
ИК-спектры (νCO, см–1): 2073 с, 2064 с, 2045 ср, 2033 с, 2027 с, 2004 с, 1988 с, 1978 с. ЯМР 1H (C6D6; δ, м.д.): 6.89 (c., 15H, PPh3), 1.64 (кв., 2H, CH2, JSnH = = 58), 1.27 (т., 3H, CH3). ЯМР 31P C6D6; δ, м.д.): 36.1 (с., 1Р, PPh3).
Синтез [Fe2(CO)6(µ4,η2-S2SnEt2){Mn(CO)4Cl}2] (V) и [Fe2Mn(CO)6(µ3-S)(µ4-S){Mn(CO)5}] (VI). К смеси твердых реагентов Iв (0.062 г, 0.12 ммоль) и [Mn(CO)5Cl] (0.054 г, 0.24 ммоль) добавляли 10 мл толуола. Реакционную смесь перемешивали при комнатной температуре в течение 2 нед., что приводило к образованию коричневого раствора и темного осадка, который отфильтровывали и промывали 15 мл толуола. Объединенный фильтрат упаривали досуха. Твердый остаток промывали несколько раз гексаном до бесцветных вытяжек, а нерастворимую в гексане часть растворяли в 30 мл толуола. Из гексанового экстракта после концентрирования до 20 мл и выдерживания при –16°C выпадали кристаллы соединения V. Выход 0.050 г (45%). Из толуольного раствора при температуре –16°C кристаллизуется соединение VI. Выход 0.037 г (45%).
ИК-спектр V (νCO, см–1): 2106 пл, 2096 с, 2073 с, 2049 с, 2042 ср, 2024 с, 2014 ср, 2007 с, 1997 ср, 1978 с. ЯМР 1H (C6D6; δ, м.д.): 1.87 (кв., 2H, CH2, JSnH = = 64.5), 1.36 (т., 3Н, СН3). ЯМР 13C (CDCl3; δ, м.д.): 214.3–206.8 (м, 1С, СО), 29.5 (с., 1С, СН2), 9.0 (с., 1С, СН3).
Соединение VI идентифицировано по ИК-спектру [35] (νCO, см–1): 2137 сл, 2072 с, 2058 с, 2030 с, 2014 с, 2004 пл, 1998 ср, 1994 пл, 1938 сл, 1917 сл.
Синтез соединения VI. К твердой смеси Iа (0.075 г, 0.19 ммоль) и [Mn(CO)5Cl] (0.089 г, 0.37 ммоль) или Iб (0.065 г, 0.15 ммоль) и [Mn(CO)5Cl] (0.70 г, 0.30 ммоль) добавляли 10 мл толуола. Реакционную смесь перемешивали при комнатной температуре в течение 2 нед., что приводило к образованию коричневого раствора и темного осадка, который отфильтровывали и промывали 15 мл толуола. Объединенный фильтрат упаривали досуха. Твердый остаток промывали 80 мл гексана. Нерастворимый в гексане твердый остаток высушивали в вакууме и растворяли в 30 мл толуола. Выдерживание этого раствора при температуре –16°C приводило к образованию кристаллов соединения VI. Выход 0.038 г (30%) в случае Iа, 0.026 г (25%) в случае Iб.
РСА комплексов проведен по стандартной методике на автоматическом четырехкружном дифрактометре Bruker-Nonius X8Apex, оснащенном двухкоординатным CCD детектором, при температуре 150 К с использованием молибденового излучения (λ = 0.71073 Å) и графитового монохроматора. Интенсивности отражений измерены методом φ-сканирования узких (0.5°) фреймов. Поглощение учтено полуэмпирически по программе SADABS [36]. Структуры расшифрованы прямым методом и уточнены полноматричным МНК в анизотропном для неводородных атомов приближении по комплексу программ SHELXTL [37]. Атомы водорода уточнены в приближении жесткого тела. Детали экспериментов и параметры уточнения структур приведены в табл. 1, основные межатомные расстояния и валентные углы – в табл. 2.
Таблица 1.
Кристаллографические данные и параметры эксперимента для соединений Iа – IIIв, III ⋅ 0.5C7H8, IV · 0.5CH3C5H9, V
| Параметр | Значение | |||||
|---|---|---|---|---|---|---|
| Iа | Iб | Iв | III · 0.5C7H8 | IV · 0.5CH3C5H9 | V | |
| M | 402.04 | 430.09 | 520.69 | 847.05 | 1057.45 | 925.55 |
| Сингония | Триклинная | Триклинная | Моноклинная | Моноклинная | Триклинная | Моноклинная |
| Пр, гр, | P1 | P1 | P21/n | P21/n | P1 | P21/c |
| a, Å | 9.1326(3) | 8.0113(2) | 10.3807(3) | 23.4098(8) | 11.4668(4) | 14.1917(2) |
| b, Å | 9.2073(4) | 9.8447(2) | 9.2793(3) | 12.4314(3) | 13.2126(4) | 11.0030(2) |
| c, Å | 9.3959(4) | 11.7599(2) | 17.8586(7) | 25.7385(9) | 13.5206(4) | 19.4710(4) |
| α, град | 88.903(1) | 102.838(1) | 90 | 90 | 77.8150(10) | 90 |
| β, град | 64.930(1) | 102.838(1) | 105.661(1) | 107.463(1) | 68.5180(10) | 90.1480(10) |
| γ, град | 85.186(1) | 104.465(1) | 90 | 90 | 72.5010(10) | 90 |
| V, Å3 | 713.00(5) | 798.68(3) | 1656.38(10) | 7145.1(4) | 1806.14(10) | 3040.41(9) |
| Z | 2 | 2 | 4 | 8 | 2 | 4 |
| ρ(выч.), г/см3 | 1.873 | 1.788 | 2.088 | 1.575 | 1.944 | 2.022 |
| F(000) | 400 | 432 | 1008 | 3448 | 1020 | 1792 |
| µ(излучение), мм–1 | 2.425 | 2.171 | 3.494 | 1.574 | 5.793 | 2.923 |
| Размер кристалла, мм | 0.24 × 0.15 × 0.14 | 0.32 × 0.31 × 0.28 | 0.08 × 0.07 × 0.04 | 0.40 × 0.12 × 0.08 | 0.27 × 0.11 × 0.02 | 0.30 × 0.17 × 0.14 |
| T, K | 100.0(2) | 100.0(2) | 100.0(2) | 150(2) | 100.0(2) | 100.0(2) |
| 2θmax, град | 62.86 | 63.80 | 56.54 | 55.16 | 62.56 | 62.74 |
| Интервалы индексов отражения | –13 ≤ h ≤ 9, –12 ≤ k ≤ 13, –13 ≤ l ≤ 12 |
–11 ≤ h ≤ 11, –13 ≤ k ≤ 13, –11 ≤ l ≤ 17 |
–9 ≤ h ≤ 13, –10 ≤ k ≤ 12, –23 ≤ l ≤ 23 |
–30 ≤ h ≤ 30, –14 ≤ k ≤ 15, –33 ≤ l ≤ 33 |
–16 ≤ h ≤ 13, –18 ≤ k ≤ 18, –19 ≤ l ≤ 13 |
–20 ≤ h ≤ 14, –14 ≤ k ≤ 14, –27 ≤ l ≤ 15 |
| Измерено отражений | 8185 | 9372 | 13 273 | 61 587 | 34 957 | 17 438 |
| Независимых отражений (Rint) | 3800 (0.0244) | 4379 (0.0195) | 4112 (0.0371) | 16209 (0.0447) | 9765 (0.0291) | 8088 (0.0198) |
| Отражений с I ≥ 2σ(I) | 3126 | 3993 | 3288 | 11038 | 8728 | 6916 |
| Уточняемых параметров | 174 | 192 | 192 | 875 | 444 | 372 |
| R-факторы по I ≥ 2σ(I) | R1 = 0.0373, wR = 0.0848 |
R1 = 0.0228, wR2= 0.0473 |
R1 = 0.0333, wR2 = 0.0789 |
R1 = 0.0365, wR2 = 0.0826 |
R1 = 0.0211, wR2 = 0.0471 |
R1 = 0.0228, wR2= 0.0437 |
| R-факторы по всем отражениям | R1 = 0.0495, wR2 = 0.0907 |
R1 = 0.0261, wR2 = 0.0486 |
R1 = 0.0478, wR2 = 0.0836 |
R1 = 0.0771, wR2 = 0.1015 |
R1 = 0.0278, wR2 = 0.0498 |
R1 = 0.0309, wR2 = 0.0464 |
| GOOF по F 2 | 1.029 | 1.039 | 1.034 | 1.018 | 1.043 | 1.029 |
| Остаточная электронная плотность (min/max, e Å–3) | –0.459/0.962 | –0.248/0.389 | –0.556/3.948 | –0.391/0.489 | –0.811/1.159 | –0.471/0.686 |
Таблица 2.
Основные длины связей и валентные углы (град) в соединениях Iа–Iв, III · 0.5C7H8, IV · · 0.5CH3C5H9, V
| Связь | Iа (E = Si) | Iб (E = Si) | Iв (E = Sn) | III · 0.5C7H8 (M = Ni)* | IV · 0.5CH3C5H9 (M = Au, E = Sn) | V (M = Mn, E = Sn) |
|---|---|---|---|---|---|---|
| d, Å | ||||||
| Fe(1)–Fe(2) | 2.4823(5) | 2.4863(3) | 2.4928(8) | 2.4944(6) | 2.4968(5) | 2.5303(4) |
| Fe(1)–S(1) | 2.3103(8) | 2.3140(4) | 2.3032(11) | 2.2834(9) | 2.2844(7) | 2.2651(5) |
| Fe(1)–S(2) | 2.3114(7) | 2.3074(4) | 2.2987(11) | 2.2872(9) | 2.2934(7) | 2.2681(5) |
| Fe(2)–S(1) | 2.3132(7) | 2.3123(4) | 2.3064(11) | 2.2896(9) | 2.2666(7) | 2.2663(5) |
| Fe(2)–S(2) | 2.3090(7) | 2.3054(4) | 2.3017(11) | 2.2857(8) | 2.2973(7) | 2.2635(5) |
| E(1)–S(1) | 2.1530(9) | 2.1507(5) | 2.4762(10) | 2.5494(5) | ||
| E(1)–S(2) | 2.1537(10) | 2.1501(5) | 2.4714(10) | 2.4648(6) | 2.5448(5) | |
| M–P | 2.2662(6) | 2.1757(8), 2.1836(8) | 2.2662(6) | |||
| M–S(1) | 2.3110(6) | 2.1888(8) | 2.3110(6) | 2.3456(5) | ||
| M–S(2) | 2.8718(6) | 2.1893(8) | 2.8718(6) | 2.3421(5) | ||
| Sn(1)–Cl(1) | 2.4270(7) | 3.0481(5) | ||||
| Sn(1)–Cl(2) | 2.8635(5) | |||||
| Mn(1)–Cl(1) | 2.3903(5) | |||||
| Mn(2)–Cl(2) | 2.3953(6) | |||||
| Угол | ω, град | |||||
| S(1)Fe(1)Fe(2) | 57.59(2) | 57.457(11) | 57.30(3) | |||
| S(1)Fe(1)S(2) | 80.56(3) | 80.777(14) | 81.59(3) | 76.91(3) | 79.45(2) | 75.648(18) |
| S(1)Fe(2)S(2) | 80.55(3) | 80.854(14) | 81.45(3) | |||
| Fe(1)S(1)Fe(2) | 64.94(2) | 65.018(11) | 65.49(3) | 66.11(3) | 66.54(2) | 67.888(16) |
| Fe(1)S(2)Fe(2) | 64.99(2) | 65.232(11) | 65.63(3) | 66.12(3) | 65.90(2) | 67.885(16) |
| E(1)S(1)Fe(1) | 86.25(3) | 86.530(17) | 91.99(3) | |||
| E(1)S(1)Fe(2) | 87.03(3) | 85.864(16) | 90.51(3) | |||
| E(1)S(2)Fe(1) | 86.20(3) | 86.711(16) | 92.22(3) | 100.38(2) | 97.752(18) | |
| E(1)S(2)Fe(2) | 87.12(3) | 86.047(17) | 90.71(3) | 102.31(3) | 97.683(18) | |
| S(1)E(1)S(2) | 87.87(4) | 88.255(19) | 74.85(3) | 65.975(19) | 66.146(14) | |
| S(1)MS(2) | 80.97(3) | |||||
| MS(1)Fe(1) | 92.55(3) | 116.03(3) | 137.97(2) | |||
| MS(1)Fe(2) | 90.16(3) | 120.08(3) | 132.18(2) | |||
| MS(2)Fe(2) | 90.25(3) | 136.27(2) | ||||
| MS(2)Fe(1) | 92.43(3) | 138.28(2) | ||||
| S(1)Sn(1)Cl(1) | 159.77(2) | 68.150(14) | ||||
| S(2)Sn(1)Cl(2) | 70.689(14) | |||||
| Cl(1)Sn(1)S(2) | 95.20(2) | 134.001(14) | ||||
| MS(1)Sn(1) | 144.61(3) | 112.110(19) | ||||
Кристаллографические параметры комплексов депонированы в Кембриджском банке структурных данных (CCDC № 751214 (Iв), 751215 (III · 0.5C7H8), 2062206 (V), 2062207 (Iб), 2062208 (Iа), 2062209 (IV · 0.5CH3C5H9)) и доступны на сайте www.ccdc.cam.ac.uk/data_reguest/cif.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Комплексы Iа–Iв были синтезированы путем последовательного восстановления [Fe2(µ-S2)- (CO)6] до [Fe2(µ-S)2(CO)6]2− в растворе ТГФ и добавлением к нему соответствующих Cl2ER2 (ER2 = = SiMe2, SiEt2, SnEt2). Этот маршрут соответствует приведенной выше схеме 1 . В случае соединений Iа и Iв оптимальной оказалась методика, основанная на применении в качестве восстановителя жидкого сплава Na-K. Этот вариант был нами ранее предложен для синтеза гетерометаллических кластеров [23]. По непонятным причинам применение ее для синтеза соединения Iб не привело к успеху: воспроизводимо по стандартной схеме выделения удается получить только сокристаллизат [Fe2(CO)6(µ3‑S)2SiEt2]⋅[Fe2(CO)6(µ-S2)]. В этом случае более эффективной оказалась методика, в которой восстановление проводится с помощью Li[BEt3H]. Методика была ранее описана в [9] и использована нами в несколько модифицированном варианте.
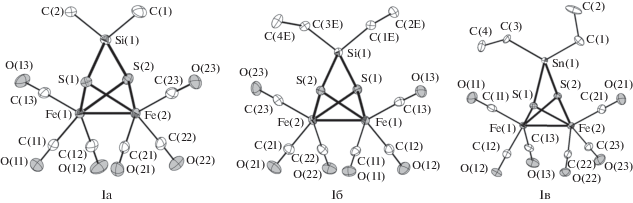
Соединения Iа–Iв были выделены в кристаллическом виде, и их строение установлено РСА (рис. 1, табл. 1 и 2). В молекулах Iа–Iв фрагмент {Fe2S2(CO)6} соединен с атомами Sn или Si через атомы S. Во всех трех случаях {Fe2S2(CO)6} имеет одинаковую геометрию. Замена кремния на олово мало сказывается на длине связей Fe–Fe и Fe–S. Небольшое закономерное укорочение Fe–Fe и удлинение Fe–S наблюдается, но различия не превышают нескольких тысячных ангстрема. Длины связей S–Si и S–Sn лежат в том же диапазоне, что и в других известных на сегодняшний день соединениях с мостиковыми станнотиолатными и силикотиолатными лигандами [38].
Рис. 1.
Строение молекул соединений Iа–Iв в кристалле. Показаны тепловые эллипсоиды 50%-ной вероятности. Атомы водорода не показаны.
Гетерометаллические кластеры II и III ранее были получены, согласно схеме 1 , последовательным восстановлением [Fe2(µ-S)2(CO)6] в толуоле и добавлением к нему, соответственно, комплексов [Cp′′RhCl2]2, [(Dppe)NiCl2] [23, 39]. Для проверки возможности получения тех же соединений в рамках предложенного нами подхода с использованием силильных и станнильных реагентов были проделаны реакции Iв с [Cp"RhCl2]2 и Iа–Iв с [(Dppe)NiCl2]. Как было установлено, действительно, в этих случаях происходит элиминирование Cl2ER2 и образование комплексов II и III (схема 2 ).
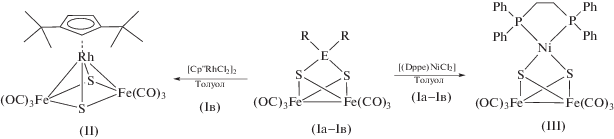
Схема 2 .
Выходы кристаллических продуктов в этих реакциях составляют 70–80%, что сравнимо с выходами в реакциях [Fe2(µ-S)2(CO)6]2− с [Cp"RhCl2]2 и [(Dppe)NiCl2]. Однако преимуществом использования Iа–Iв является то, что реагирующие формы – устойчивые нейтральные комплексы, существующие в индивидуальном виде и легко дозирующиеся для синтеза, в отличие от анионов [Fe2(µ-S)2- (CO)6]2−, которые получены in situ, неустойчивы в растворе и не могут быть выделены в твердом виде. Кроме того, при использовании [Fe2(µ-S)2(CO)6]2− ограничен выбор растворителей – до сих пор все реакции с его участием проводили в ТГФ. Реакции с силилированными и станнилированными кластерами протекают при комнатной температуре даже в неполярном растворителе, например толуоле. Таким образом, появляется некоторая степень свободы при планировании и выборе оптимальных условий проведение синтеза, а также упрощения процедуры выделения продукта. Например, при использовании толуола в качестве растворителя процедура сводится всего лишь к концентрированию и охлаждению реакционной смеси.
Строение кластера II было описано ранее [23], поэтому он был идентифицирован по ИК-спектру при сравнении такового с аутентичным образцом. Кластер III был структурно охарактеризован ранее в виде сольвата с эфиром: III ⋅ 0.5Et2O [39]. В нашем случае кристаллизация из толуола приводит к сольвату III ⋅ 0.5С7H8. В кристалле сольвата присутствуют две независимые молекулы [Fe2Ni(µ3-S)2(CO)6(Dppe)], строение которых отличается лишь незначительными отклонениями в углах и длинах связей, возникающими, вероятно, в результате специфики упаковки в кристалле. Включенные молекулы толуола упорядочены. В целом молекулы кластера III практически совпадают с таковыми в сольвате III ⋅ 0.5Et2O [39].
Реакции Iа–Iв с [Cp"RhCl2]2, [(Dppe)NiCl2] можно рассматривать как “тестовые”, так как в их результатах сомнений не было. Целью их изучения было прежде всего сравнение нового синтетического подхода с тем, который использовался ранее. Далее, в реакциях с другими галогенсодержащими комплексами переходных металлов мы преследовали цель получения новых, неизвестных ранее гетерометаллических кластеров. Так, образование железо-золотого кластера мы ожидали в реакции Iв с [(Ph3P)AuCl]. Действительно, из реакционной смеси выделено соединение IV (схема 3 ), однако выход его составляет всего 10%. Контроль за реакцией с помощью ТСХ и ЯМР 31P показывает, что на самом деле образуется набор продуктов, из которых выделить удается только один – соединение IV, остальные разлагаются в процессе выделения. При этом и соединение IV удалось выделить по воле случая. Попытки кристаллизации из чистого гексана не привели к успеху. Однако, когда вместо гексана был взят легкий петролейный эфир, образовались кристаллы, содержащие сольватные молекулы метилциклопентана CH3C5H9, содержавшегося в растворителе в небольшом количестве. По всей видимости, именно этот алкан наиболее подходит для сокристаллизации с “разветвленными” молекулами кластера IV.
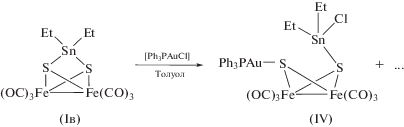
Схема 3 .
Строение сольвата IV · 0.5CH3C5H9 было определено с помощью монокристального РСА (рис. 2). В его молекуле присутствуют и фрагмент {Au(PPh3)}, соединенный с атомом серы, и фрагмент {SnEt2Cl}, присоединенный ко второму атому серы; т.е. в данном случае не происходит элиминирования {SnEt2}2+ в виде соответствующего хлорида. Продукт реакции может быть описан, скорее, как результат присоединения [(Ph3P)AuCl] по связи S–Sn. Попытки варьирования условий реакции (отношение реагентов, температура, растворитель), а также замена станнилированного комплекса Iв на силилированные аналоги Iа и Iб не позволили выделить и охарактеризовать какие-либо еще координационные соединения.
Рис. 2.
Строение кластера IV в кристалле. Показаны тепловые эллипсоиды 50%-ной вероятности. Атомы водорода не изображены, фенильные заместители показаны в упрощенном виде.
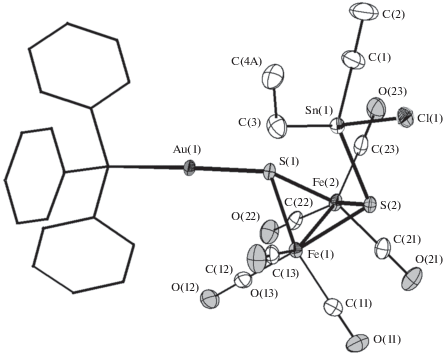
Соединение IV образует молекулярную кристаллическую структуру, в которой кластерный фрагмент соответствует формуле [Fe2(µ-SSnClEt2)- (µ-SAuPPh3)(CO)6]. Расстояние между атомами Sn(1)–S(1) заметно увеличивается до 2.8718(6) Å, а между Sn(1)–S(2) немного уменьшается до 2.4648(6) Å по сравнению с молекулой кластера Iв, где расстояния Sn–S равны 2.4757(9) и 2.4713(9) Å. Валентные углы FeSAu равны 116.03(3)° и 120.08(3)°. Они практически совпадают по значению с валентными углами FeSHg (116(1)°) в молекуле [Fe2(µ-CH3HgS)2(CO)6] [40].
Первая попытка проведения реакции Iв с [Mn(CO)5Cl] в отношении 1 : 2 в толуоле при комнатной температуре привела к двум продуктам V и VI с примерно одинаковым выходом (схема 4 ).
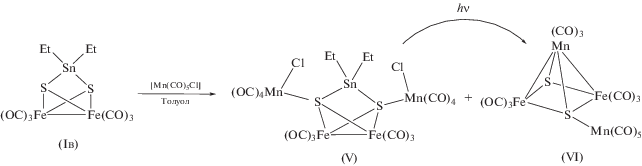
Схема 4 .
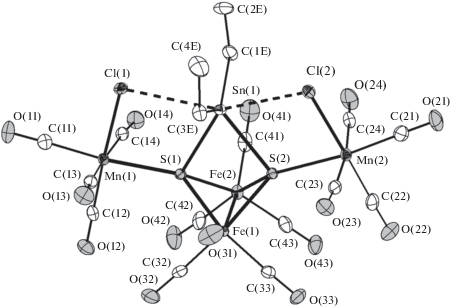
При проведении синтеза V и VI концентрирование и охлаждение реакционного раствора приводит к их совместной кристаллизации: V – большие светло-красные, VI – мелкие темно-зеленые кристаллы. Кластер VI описан ранее [35]. Он был идентифицирован по данным РСА и ИК-спектроскопии. Соединение V получено впервые. Разделить их удалось за счет разной растворимости в гексане. Структура кластера V установлена методом РСА (рис. 3). В молекуле V фрагменты {Mn(CO)4Cl} соединены с атомами S. Атомы Cl координированы к марганцу и олову по μ-типу. Расстояния Sn(1)–Cl(1) и Sn(1)–Cl(2) довольно велики (3.0481(5) и 2.8635(5) Å соответственно) и характерны для хлоридных мостиков в полиядерных гомо- и гетерометаллических оловосодержащих соединениях, например от 2.920(1) до 3.097(1) Å в [(η6-C6H6)Sn(AlCl4)2 ⋅ C6H6]n [41]. Увеличение координационного числа олова приводит к некоторому удлинению связей Sn–S (на ~0.07 Å) (табл. 2).
Рис. 3.
Строение кластера V в кристалле. Показаны тепловые эллипсоиды 50%-ной вероятности. Атомы водорода не показаны.
В растворе в спектре ЯМР 1H кластера V присутствуют сигналы атомов водорода этильных фрагментов: триплет от CH3 с химическим сдвигом 1.36 м.д. и квартет от CH2 с химическим сдвигом 1.87 м.д., также наблюдаются сателлиты расщепления протонов CH2-групп на ядрах 117Sn и 119Sn c константой спин-спинового взаимодействия JSnH = 64.5 Гц. В спектре ЯМР 13C кластера V наблюдаются мультиплет от атомов углерода СО-лигандов (хим. сдвиг 206.8–214.3 м.д.), СH2-групп (хим. сдвиг 29.8 м.д.) и CH3-групп (хим. сдвиг 9.0 м.д.).
Сравнивая кластеры V и VI, можно заметить, что их молекулы отличаются на фрагмент {Et2SnCl2}, который в V “не ушел” из молекулы. Логично предположить, что кластер V – промежуточный продукт и при отщеплении фрагмента Et2SnCl2 должен превратиться в соединение VI, но кинетически этот процесс заторможен. Для проверки этого предположения были проведены исследования по инициации такого отщепления. Оказалось, что при фотохимической инициации (облучение дневным светом раствора V в толуоле или бензоле) действительно происходит его полное превращение в кластер VI, однако термическая инициация (нагревании раствора V в хлористом метилене) данного превращения не вызывает.
В отличие от Iв взаимодействие кластеров Iа, Iб с комплексом [Mn(CO)5Cl] протекает с образованием VI в качестве основного продукта. В небольших количествах в реакционной смеси присутствуют также [Fe2(CO)6(µ-S2)] и [Fe3(CO)9- (µ3-S)2] (схема 5 ).
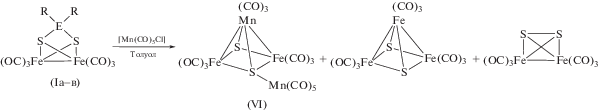
Схема 5 .
При этом образование Si-содержащего соединения, аналогичного по строению кластеру V, не наблюдалось. Если предположить, что реакция Iа, Iб с комплексом [Mn(CO)5Cl] идет по тому же маршруту, кинетическая заторможенность “второй стадии” отсутствует, и R2SiCl2 легко удаляется без дополнительной активации. Скорее всего, это связано с тем, что для кремния не характерны координационные числа >4 в отличие от Sn. Присутствие в смеси кластеров [Fe2S2(CO)6] и [Fe3S2(CO)9] можно объяснить частичным разложением промежуточных продуктов реакции с образованием более термодинамически стабильных комплексов.
Таким образом, в данной работе мы показали, что реакции силилированных и станнилированных комплексов [Fe2(µ-S2ER2)(CO)6] (E = Si, Sn) с галогенсодержащими комплексами d-металлов действительно приводят к гетрометаллическим кластерам и могут быть предложены как основа нового синтетического подхода к таковым. Результатом данных реакций являются кластеры, аналогичные полученным по классической схеме – восстановлением [Fe2(CO)6(µ-S2)] до дианиона и дальнейшей обработкой его электрофильными агентами. Преимуществом предложенного подхода является возможность точной дозировки [Fe2(µ-S2ER2)(CO)6] и их растворимость во всех органических растворителях. В то же время обнаружены некоторые особенности, которые следует учитывать при проведении синтетических экспериментов, например при использовании станнилированных реагентов следует учитывать заторможенность элиминирования R2SnCl2, для удаления которого из молекулы промежуточного продукта может быть необходимой фотохимическая инициация.
Авторы заявляют, что у них нет конфликта интересов.
Список литературы
Schriver D.F., Whitmire K.H. // Comprehensive Organometallic Chemistry / Ed. Stone F.G.A., Wilkinson G., Oxford: Pergamon Press, 1982. P. 243.
Shieh M., Lai Y.-W. // J. Chin Chem. Soc. 2002. V. 49. P. 851.
Пасынский А.А., Еременко И.Л. // Успехи химии. 1989. Т. 58. № 2. С. 303.
Пасынский А.А., Семенова Н.И., Торубаев Ю.В. и др. // Изв. АН. Сер. хим. 2003. С. 944.
Пасынский А.А., Шаповалов С.С., Тихонова О.A. и др. // Коорд. химия. 2015. Т. 41. № 11. С. 6699 (Pasynskii A.A., Shapovalov S.S., Tikhonova O.A. et al. // Russ. J. Coord. Chem. 2015. V. 41. № 11. P. 74). https://doi.org/10.1134/S1070328415110068
Torubaev Y.V., Pasynskii A.A., Pavlova A.V. et al. // J. Organomet. Chem. 2015. V. 777. P. 88.
Torubaev Y.V., Shapovalov S.S., Tikhonova O.G. et al. // Polyhedron. 2020. V. 177. P. 114298.
Hieber W., Beck J. // Z. Anorg. Allg. Chem. 1958. V. 296. S. 91.
Seyferth D., Henderson R.S., Song L. // Organometallics. 1982. V. 1. P. 125.
Tard C., Pickett C.J. // Chem. Rev. 2009. V. 109. P. 2245.
Song L.-C., Tang M.-Y., Mei S.-Z. et al. // Organometallics. 2007. V. 26. P. 1575.
Song L.-C., Wang H.-T., Ge J.-H. et al. // Organometallics. 2008. V. 27. P. 1409.
Nann T., Ibrahim S.K., Woi P.-M. et al. // Angew. Chem. Int. Ed. 2010. V. 49. P. 1574.
Wang F., Wang W.-G., Wang X.-J. et al. // Angew. Chem. Int. Ed. 2011. V. 50. P. 3193.
Wen F., Wang X., Huang L. et al. // ChemSusChem. 2012. V. 5. P. 849.
Wang F., Wang W.-G., Wang H.-Y. et al. // ACS Catal. 2012. V. 2. P. 407.
Simmons T.R., Berggren G., Bacchi M. et al. // Coord. Chem. Rev. 2014. V. 270–271. P. 127.
Хризанфорова В.В., Карасик А.А., Будникова Ю.Г. // Успехи химии. 2017. Т. 86. С. 298.
Hai L., Zhang T., Zhang X. et al. // Electrochem. Commun. 2017. V. 82. P. 66.
Shupp J.P., Rose A.R., Rose M.J. // Dalton Trans. 2017. V. 46. P. 9163.
Arsenyeva K.V., Ershova I.V., Chegerev M.G. et al. // J. Organomet. Chem. 2020. V. 927. P. 121524.
Seyferth D., Song L.-C., Henderson R.S. // J. Am. Chem. Soc. 1981. V. 103. P. 5103.
Пушкаревский Н.А., Огиенко М.А., Куратьева Н.В., Конченко С.Н. // Изв. АН. Сер. хим. 2008. № 1. С. 35.
Zhuang B., Chen J., He L. et al. // J. Organomet. Chem. 2003. V. 682. P. 59.
Konchenko S.N., Sanden T., Pushkarevsky N.A. et al. // Chem. Eur. J. 2010. V. 16. P. 14278.
Dehnen S., Eichhöfer A., Fenske D. // Eur. J. Inorg. Chem. 2002. P. 279.
Tran D.T.T., Kowalchuk C.M., Taylor N.J., Corrigan J.F. // Inorg. Chem. 2002. V. 41. P. 5693.
Komuro T., Matsuo T., Kawaguchi H., Tatsumi K. // Angew. Chem. 2003. V. 115. № 4. P. 481.
Fuhr O., Dehnen S., Fenske D. // Chem. Soc. Rev. 2013. 42. P. 1871.
Гордон А., Форд Р. Спутник химика. М.: Мир, 1976. 541 с.
Busby R., Hursthouse M.B., Jarrett P.S. et al. // Dalton Trans. 1993. P. 3767.
Finn M.G. Pentacarbonylchloromanganese. e-EROS Encyclopedia of Reagents for Organic Synthesis, John Wiley & Sons, Ltd., 2001. p. 1.
Sinha P., Wilson A.K., Omary M.A. // J. Am. Chem. Soc. 2005. V. 127. P. 12488.
White C., Yates A., Maitlis P.M., Heinekey D.M. // Inorg. Synth. 1992. V. 29. P. 228.
Seyferth D., Henderson R.S., Fackle J.P., Mazany A.M. // J. Organomet. Chem. 1981. V. 213. P. C21.
APEX2 (version 1.08), SAINT (version 7.03), and SADABS (version 2.11). Madison (WI, USA): Bruker Advanced X-ray Solutions, 2004.
Sheldrick G.M. // Acta Crystallogr. A. 2008. V. 64. № 1. P. 112.
Groom C.R., Bruno I.J., Lightfoot M.P., Ward S.C. // Acta Crystallogr. B. 2016. 72. P. 171.
Lozano A.A., Santana M.D., Garcia G. et al. // Z. Anorg. Allg. Chem. 2005. V. 631. P. 2062.
Seyferth D., Gallagher M.K. // Inorg. Chem. Acta. 1983. V. 73. P. 159.
Schmidbaur H., Probst T., Steigelmann O., Muller G. // Z. Naturforsch. B. 1989. V. 44. P. 1175.
Дополнительные материалы отсутствуют.
Инструменты
Координационная химия