

Координационная химия, 2022, T. 48, № 10, стр. 632-644
Синтез, кристаллическое строение и свойства бис-катехолатных комплексов фенилкремния(IV)
Е. П. Крамарова 1, В. В. Негребецкий 1, А. Д. Володин 2, П. А. Буйкин 2, 3, А. В. Лалов 4, Р. А. Новиков 4, Т. М. Алиев 2, А. А. Корлюков 1, 2, А. В. Вологжанина 2, *
1 Российский национальный медицинский университет им. Н.И. Пирогова
Москва, Россия
2 Институт элементоорганических соединений им. А.Н. Несмеянова РАН
Москва, Россия
3 Институт общей и неорганической химии им. Н.С. Курнакова РАН
Москва, Россия
4 Институт органической химии им. Н.Д. Зелинского РАН
Москва, Россия
* E-mail: vologzhanina@mail.ru
Поступила в редакцию 10.02.2022
После доработки 10.03.2022
Принята к публикации 15.03.2022
- EDN: WHERUS
- DOI: 10.31857/S0132344X2209002X
Аннотация
Взаимодействием триметоксифенилсилана с двумя эквивалентами пирокатехина или его производных, содержащих акцепторные заместители при фенильном кольце, в присутствии дициклогексиламина синтезирован ряд бис-катехолатных анионных комплексов фенилкремния(IV) с анионами ((Cat1)– = катехолат, (Cat2)– = 3,4,5,6-тетрабромокатехолат, (Cat3)– = 4-цианокатехолат, (Cat4)– = = 4-нитрокатехолат, (Cat5)– = 3-фторокатехолат, (Cat6)– = 4,5-дибромокатехолат). Полученные комплексы охарактеризованы методами спектроскопии ИК и ЯМР 1H, 13C и 29Si, вольтамперометрии и ЭПР. Гетеролигандные анионные комплексы фенилкремния(IV) кристаллизуются из растворов в виде солей (Chex2NH2)[PhSi(Cat1)2] (I), (Chex2NH2)[PhSi(Cat2)2] · 0.5C6H14 (II), (Chex2NH2)[PhSi(Cat3)2] · · 2C6H6 (III), (Chex2NH2)[PhSi(Cat4)2] · H2O (IV), (Chex2NH2)[PhSi(Cat5)2] (V) и (Chex2NH2)[PhSi(Cat6)2] (VI) (Chex2${\text{NH}}_{2}^{ + }$ = дициклогексиламмоний), состав и строение которых подтверждены рентгенодифракционными данными (CCDC № 2150293–2150297 для I–V соответственно). Эффект заместителя при фенильном кольце на электронное строение аниона изучен с помощью квантовохимических расчетов методом PBE0/6-311G(d,p) с учетом неспецифической сольватации (модель PCM). Измерены потенциалы окисления II–VI, для II зарегистрировано образование радикалов при электрохимическом окислении и восстановлении.
Комплексы кремния с производными пирокатехина – одни из самых многочисленных типов соединений, где атом кремния имеет координационное число пять или шесть. Как правило, такие комплексы являются анионами и кристаллизуются в виде солей с органическими катионами или катионами щелочных металлов. Бис-катехолаты алкил- и арилкремния(IV) недавно привлекли внимание исследователей как источники алкильных- и арильных радикалов в фотокатализируемых реакцияхкросс-сочетания [1–8]. При этом в большинстве случаев в качестве прекурсора использовался бис-катехолат алкилкремния; свойства бискатехолатов арилкремния изучались лишь в [1, 8]. Данные об изучении эффекта заместителя на электрохимические и каталитические свойства соединений ограничены публикацией [8]. Авторам этой работы удалось установить, что для ряда замещенных бис-катехолатов фенилкремния, содержащих в фенильном кольце катехолат-аниона различные электронодонорные и электроноацепторные фрагменты, наилучшие результаты в отщеплении фенильного радикала продемонстрировал комплекс с 4-цианокатехолат-анионом.
В настоящей работе получен ряд дициклогесиламмониевых солей бис-катехолатов фенилкремния, изучена их кристаллическая структура и электрохимические свойства, с помощью квантовохимических расчетов оценены величины вертикального и адиабатического потенциалов ионизации, рассмотрены особенности электронного строения изолированных анионов и соответствующих окисленных форм (нейтральных дублетных радикальных семихинон-катехолатных комплексов).
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
В работе использовали коммерческие (Sigma Aldrich) производные пирокатехина, дициклогексиламин, С6Н6 и С6H14 (ч. д. а) без предварительной очистки. ИК-спектры записывали с использованием KBr на спектрометре Bruker Tensor-27. Спектры ЯМР (1H, 400 МГц; 13C, 75 МГц) регистрировали на приборе Bruker Avance II в CD3CN, DMSO-d6 или CDCl3 с использованием Me4Si в качестве внешнего стандарта. Температуры плавления определяли с использованием Stuart SMP10.
Синтез дициклогексиламмониевых солей (схема 1 ) проводили взаимодействием триметоксифенилсилана с пирокатехином (Cat1) и его производными (Catn; n = 2–6 для 3,4,5,6-тетрабромпирокатехина, 4-цианопирокатехина, 4‑нитропирокатехина, 3-фторпирокатехина, 4,5-дибромпирокатехина соответственно) в присутствии дициклогексиламина при температурах кипения соответствующих растворителей (ксилол или ацетонитрил).

Схема 1 .
Синтез бис(фенил-1,2-диолато-O,O')фенилсиликата(IV) дициклогексиламмония, (Chex2NH2)- [PhSi(Cat1)2] (I). Смесь 3.96 г (0.02 моль) триметоксифенилсилана, 4.4 г (0.04 моль) пирокатехина и 3.62 г (0.02 моль) дициклогексиламина в 10 мл ксилола кипятили 30 мин. На следующий день реакционную смесь выливали в 70 мл гексана. Образовавшиеся кристаллы отфильтровывали. Выход I 6.2 г (62%). Тпл = 272–275°С. Кристаллы, пригодные для РСА, выращивали из CH3CN.
ИК-спектр (ν, см–1): 1599 сл (Ph), 1483 c, 1460 сл (NH$_{2}^{ + }$), 1236 c (PhO). ЯМР 1Н (ДМСО-d6; δ, м.д.): 1.0–1.5 (м., 12Н), 1.50–2.00 (м., 8Н), 2.00 (м., 2Н), 6.50–7.54 (м., 12Н). ЯМР 13C (ДМСО-d6; δ, м.д.): 24.55, 25.53, 30.54, 52.61, 109.14, 110.26, 115.47, 116.12, 117.96, 119.71, 127.23, 128.28, 133.82, 135.23, 142.28, 145.76, 150.85. ЯМР 29Si (ДМСО-d6; δ, м.д.): –87.55.
Синтез бис(3,4,5,6-тетрабромофенил-1,2-диолато-O,O')фенилсиликата(IV) дициклогексиламмония н-гексан гемисольвата, (Chex2NH2)- [PhSi(Cat2)2] · 0.5C6H14 (II). Смесь 0.198 г (0.001 моль) триметоксифенилсилана, 0.83 г (0.002 моль) 3,4,5,6-тетрабромпирокатехина, 0.18 г (0.001 моль) дициклогексиламина в 5 мл ацетонитрила кипятили 2 ч. Через сутки смесь упаривали на роторном испарителе. Осадок растирали с гексаном до образования кристаллов. Выход II 0.59 г (52%). Тпл = 228–230°C. Кристаллы, пригодные для РСА, выращивали из диоксана.
ИК-спектр (ν, см–1): 1556 сл (Ar), 1447 с (NH$_{2}^{ + }$) 1272 сл, 1223 сл (ArO). ЯМР 1Н (CD3CN; δ, м.д.): 1.10–1.24 (м., 2H), 1.31 (м., 8H), 1.66 (д.м., J = 12.4 Гц, 2H), 1.74–1.88 (м., 4H), 1.94–2.07 (м., 4H), 3.19 (с., 2H), 6.38 (т., J = 37.6 Гц, 2H), 7.32–7.34 (м., 3H), 7.52–7.59 (м., 2H). ЯМР 13C (CD3CN; δ, м.д.): 24.95, 25.57, 30.04, 55.25, 107.26, 115.73, 128.72, 130.41, 135.54, 138.30, 149.43. ЯМР 29Si (CD3CN; δ, м.д.): –85.8.
HRMS (m/z) расч.: 952.3349, для C18H5O4SiBr8 найдено: 952.3316.
Синтез бис(4-цианофенил-1,2-диолато-O,O')фенилсиликата(IV) дициклогексиламмония бензол дисольвата, (Chex2NH2)[PhSi(Cat3)2] · 2C6H6 (III). Смесь 1.35 г (0.01 моль) 4-цианопирокатехина, 0.9 г (0.005 моль) дициклогекcиламина, 0.99 г (0.005 моль) триметоксифенилсилана в 15 мл ацетонитрила кипятили 2 ч. Через сутки смесь упаривали на роторном испарителе. Остаток растирали с петролейным эфиром. Выход III 2.45 г (89%). Тпл = = 252–254°C. Кристаллы, пригодные для РСА, выращивали из смеси C6H6–CN в соотношении 6 : 1.
ИК-спектр (ν, см–1): 2214 c (CN), 1601 ср (Ar), 1487 с $\left( {{\text{NH}}_{2}^{ + }} \right)$ 1260 с (ArO). ЯМР 1Н (CD3CN; δ, м.д.): 1.05–1.42 (м., 10H), 1.65 (д., J = 11.7 Гц, 2H), 1.73.–1.87 (м, 4H), 2.00. 2.12 (м., 4H), 3.16–3.31 (м., 2H), 6.82–6.91 (м., 2H), 7.03–7.14 (м., 4H), 7.19–7.31 (м., 3H), 7.51–7.58 (м, 2H). ЯМР 13C (CD3CN; δ, м.д.): 25.01, 25.61, 30.13, 55.15, 100.91, 101.33, 111.91, 112.02, 113.77, 113.91, 121.42, 125.94, 126.31, 128.36, 129.83, 135.29, 140.35, 151.01, 151.36, 156.00, 156.38. ЯМР 29Si (CD3CN; δ, м.д.): –85.23.
HRMS (m/z) расч.: 371.0494, для C20H11N2O4Si найдено: 371.0486.
Синтез бис(4-нитрофенил-1,2-диолато-O,O')фенилсиликата(IV) дициклогексиламмония моногидрата, (Chex2NH2)[PhSi(Cat4)2] · H2O (IV). Смесь 0.5 г (0.0025 моль) триметоксифенилсилана, 0.77 г (0.005 моль) 4-нитропирокатехина, 0.45 г (0.0025 моль) дициклогексиламина в 10 мл ксилола кипятили 40 мин. Через сутки смесь упаривали. Образовавшееся густое масло закристаллизировали путем растирания его в петролейном эфире. Выход IV 1 г (67%). Тпл = 12–215°C (разл.). Кристаллы, пригодные для РСА, выращивали испарением раствора в CH3CN.
ИК-спектр (ν, см–1): 1592 ср (Ar), 1485 с $\left( {{\text{NH}}_{2}^{ + }} \right)$ 1265 с (ArO). ЯМР 1Н (CD3CN; δ, м.д.): 1.06–1.45 (м., 10H), 1.64 (д., J = 12.4 Гц, 2H), 1.73–1.85 (м., 4H), 1.99–2.10 (м, 4H), 3.15–3.30 (м., 2H), 6.86 (д.д., J = 8.6, 4.4 Гц, 2H), 7.17–7.31 (м., 3H), 7.58 (д.д., J = 7.4, 2.1 Гц, 2H), 7.64 (д.д., J = 4.8, 2.6 Гц, 2H), 7.76 (т.д., J = 8.9, 2.7 Гц, 2H). ЯМР 13C (CD3CN; δ, м.д.): 25.02, 25.61, 30.04, 55.20, 105.39, 106.38, 106.67, 109.68, 110.26, 110.44, 117.43, 117.93, 118.58, 128.44, 129.31, 129.99, 135.35, 139.89, 140.47, 140.96, 150.37, 150.94, 158.36, 158.98. ЯМР 29Si (CD3CN; δ, м.д.): –83.52.
HRMS (m/z) расч.: 411.0290, для C18H11N2O8Si найдено: 411.0286.
Синтез бис(3-фторфенил-1,2-диолато-O,O')фенилсиликата(IV) дициклогексиламмония моногидрата (Chex2NH2)$\left[ {PhSiCat_{2}^{5}} \right]$ (V). Смесь 1 г (0.005 моль) триметоксифенилсилана, 1.28 г (0.01 моль) 3-фторпирокатехина, 0.9 г (0.005 моль) дициклогексиламина в 10 мл ацетонитрила кипятили 2 ч. Через сутки образовавшееся кристаллическое вещество отфильтровывали. Выход V 2.13 г (79%). Тпл = = 238–241°C. Кристаллы, пригодные для РСА, выращивали из CH3CN.
ИК-спектр (ν, см–1): 1625 ср, 1601 сл (Ar), 1494 c, 1472 c $\left( {{\text{NH}}_{2}^{ + }} \right)$, 1268 c ArO. ЯМР 1Н (ДМСО-d6; δ, м.д.): 1.18 (м., 10H), 1.59 (д., J = 12.6 Гц, 2H), 1.71 (д., J = 12.1 Гц, 4H), 1.91 (д., J = 10.8 Гц, 4H), 2.85–3.00 (м, 2H), 6.35–6.66 (м., 6H), 7.12–7.25 (м., 3H), 7.44–7.59 (м., 2H). ЯМР 13C (ДМСО-d6; δ, м.д.): 24.14, 25.12, 30.24, 52.28, 105.91 (д.д., J = 18.0, 11.2 Гц), 106.41 (д., J = 14.2 Гц), 106.55 (d., J = = 2.8 Гц), 111.45 (д., J = 2.4 Гц), 116.72 (д.д., J = = 13.2, 8.1 Гц), 118.16 (д., J = 9.5 Гц), 127.07, 128.31, 133.57 (д., J = 13.8 Гц), 134.67, 136.81, 140.43, 146.41, 147.95 (д., J = 5.6 Гц), 149.58, 150.71, 152.60 (д.д., J = = 9.2, 7.2 Гц), 153.85. ЯМР 29Si (ДМСО-d6; δ, м.д.): –84.94.
HRMS (m/z) расч.: 357.0400, для C18H11F2O4Si найдено: 357.0396.
Синтез бис(4,5-дибромофенил-1,2-диолато-O,O')фенилсиликата(IV) дициклогексиламмония, (Chex2NH2)[PhSi(Cat6)2] (VI). Смесь 0.5 г (0.0025 моль) триметоксифенилсилана, 1.38 г (0.005 моль) 4,5-дибромпирокатехина; 0.45 г (0.0025 моль) дициклогексиламина в 10 мл ацетонитрила кипятили 2 ч. Через сутки реакционную смесь упаривали. Остаток закристаллизовывали растиранием в петролейном эфире. Выход VI 1.86 г (92%). Тпл = 205–207°C (переосаждение диоксанового раствора соединения VI в петролейном эфире).
ИК-спектр (ν, см–1): 1595 с (Ar), 1477c, 1448 cp $\left( {{\text{NH}}_{2}^{ + }} \right),$ 1231 (ArO). ЯМР 1Н (CD3CN; δ, м.д.): 1.05–1.46 (м., 10H), 1.59–1.70 (м., 2H), 1.71–1.91 (м., 4H), 2.04 (д., J = 8.2 Гц, 4H), 3.12–3.31 (м., 2H), 7.06 (с., 3H), 7.18–7.29 (м., 3H), 7.46–7.56 (м., 2H). ЯМР 13C (CD3CN, δ, м.д.): 25.00, 25.59, 29.99, 55.07, 111.77, 115.58, 128.30, 135.15, 140.38, 146.37, 151.88. ЯМР 29Si (CD3CN; δ, м.д.): –84.53.
HRMS (m/z) расч.: 636.6970, для C30H33Br4NO4Si найдено: 636.6960.
РСА выполнен на дифрактометрах Bruker APEX DUO (графитовый монохроматор, λ(CuKα) = = 1.54178 Å (I), λ(MoKα) = 0.71073 Å (III)) при 120.0(2) К и Bruker D8 QUEST (графитовый монохроматор, λ(MoKα) = 0.71073 Å (II, IV, V)) при 100.0(2) К. Поглощение учтeно полуэмпирически согласно методике Р. Блессинга [9] в программе SADABS [10]. Для решения и уточнения структур использованы программные комплексы ShelX-2014 OLEX2 [11]. Структуры расшифрованы методом внутренней фазировки (программа ShelXT [12]) и уточнены методом наименьших квадратов (программа ShelXL [13]). Положения и тепловые параметры неводородных атомов уточнены в изотропном, а затем в анизотропном приближении полноматричным методом наименьших квадратов. Атомы фтора в двух независимых анионах 3-фторокатехолата разупорядочены по двум положениям с заселенностями 0.384 : 0.616 (уточнялась как независимая переменная). Все атомы водорода рассчитаны в соответствии с их идеализированной геометрией и уточнены с ограничениями, примененными к длинам связей CH и изотропным параметрам смещения (Uизо (H) = 1.2Uэкв (C) для CH2 и CH; Uизо (H) = 1.5Uэкв (C) для группы CH3). Кристаллографические данные и основные параметры уточнения для соединений I–V приведены в табл. 1.
Таблица 1.
Кристаллографические данные, параметры эксперимента и уточнения структур I–V
| Параметр | Значение | ||||
|---|---|---|---|---|---|
| I | II | III | IV | V | |
| Брутто-формула | C30H37NO4Si | C33H36NO4SiBr8 | C44H47N3O4Si | C30H37N3O9Si | C30H35F2NO4S |
| М | 503.69 | 1178.00 | 709.93 | 611.71 | 539.68 |
| Пр. группа | I41/a | $P\bar {1}$ | P21/c | P21/c | Pbca |
| a, Å | 19.4077(4) | 12.5736(14) | 10.5358(7) | 14.7233(3) | 10.6333(3) |
| b Å | 19.4077(4) | 13.6301(16) | 20.3097(17) | 11.4826(3) | 16.0600(5) |
| c Å | 28.3974(9) | 13.877(2) | 18.3939(14) | 17.6464(5) | 32.0397(10) |
| α, град | 90 | 60.703(4) | 90 | 90 | 90 |
| β, град | 90 | 67.288(6) | 102.318(2) | 99.6410(10) | 90 |
| γ, град | 90 | 77.553(4) | 90 | 90 | 90 |
| V, Å3 | 10 696.1(6) | 1912.2(5) | 3845.3(5) | 2941.20(13) | 5471.4(3) |
| ρ(выч.), г/см3 | 1.251 | 2.046 | 1.226 | 1.381 | 1.310 |
| μ, мм–1 | 1.059 | 8.457 | 0.108 | 0.140 | 0.136 |
| Область сканирования θ, град | 5.516–30.238 | 3.428–52.044 | 3.026–55.082 | 4.524–55.756 | 4.766–61.042 |
| Собранные отражения | 14 258 | 7487 | 16 836 | 26 510 | 34 239 |
| Независимые отражения | 4163 | 7487 | 16 836 | 7000 | 8363 |
| Rint | 0.0716 | 0.0426 | 0.0956 | ||
| GOOF | 1.012 | 1.036 | 1.056 | 1.055 | 1.040 |
| R1 (I > 2σ(I)) | 0.0439 | 0.0706 | 0.0890 | 0.0670 | 0.0579 |
| wR2 (по всем данным) | 0.1152 | 0.1921 | 0.1992 | 0.1838 | 0.1335 |
| Остаточные электронная плотность (min/max), e Å–3 | 0.20/–0.27 | 1.35/–1.25 | 0.38/–0.38 | 0.83/–0.44 | 0.36/–0.42 |
Дополнительная кристаллографическая информация для структур I–V представлена в Кембриджском банке структурных данных (CCDC № 2150293–2150297; http://www.ccdc.cam.ac.uk/structures).
Электрохимические исследования проводили на потенциостате-гальваностате Autolab PGSTAT128N с использованием программного пакета NOVA 2.0 и классической трехэлектродной ячейкой. В качестве рабочего электрода использовали стеклоуглеродный электрод, электрода сравнения – Ag/AgCl в насыщенном растворе KCl, противоэлектродом служила платиновая проволока. Дополнительным электролитом служил 0.1 М раствор TBAPF6 в ДМСО. Все измерения проводили при скорости развертки 100 мВ/с. Перед каждым экспериментом исследуемый раствор продувался газообразным азотом в течение 5 мин, а рабочий электрод тщательно полировали. Все измерения проводили при комнатной температуре. Измеренные потенциалы приведены относительно пары Fc/Fc+.
Спектры ЭПР регистрировали на приборе Bruker EMX 6/1, снабженным стандартным прямоугольным резонатором ER4102ST с центральной частотой 9.8 ГГц.
Квантовохимические исследования. Геометрию анионов I–VI и их окисленных форм в дублетном состоянии оптимизировали с помощью гибридного функционала PBE0 и базисного набора 6-311G(d,p) с использованием программы Q-Chem 5.4 [14, 15]. Расширение базисного набора дополнительными диффузными функциями привело к значительному увеличению затрат расчетных ресурсов и к проблемам со сходимостью из-за линейной зависимости, особенно в случае бромсодержащих анионов, однако серьезных различий в оптимизированной геометрии по сравнению с базисным набором 6-311G(d,p) не наблюдалось. Общая схема квантовохимических расчетов, описанная подробно в [16], включала оптимизацию геометрии с учетом влияния растворителя (PCM/ДМСО), расчет гармонических частот и термохимический анализ согласно статье [16]. При оптимизации использована эмпирическая дисперсионная поправка D3. Полученные величины полных энергий анионов и их окисленных форм использовали для определения расчетных значений первого потенциала окисления. Для анализа электроноакцепторного влияния использовали анализ средней локальной энергии ионизации, расчет которой выполняли с помощью программы Multiwfn [17], рисунки выполняли с использованием программы VMD [18].
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Взаимодействие пирокатехина (2 моль) или его коммерчески доступных производных с различными акцепторными заместителями при фенильном кольце с триметоксифенилсиланом(IV) (1 моль) в присутствии дициклогексиламина (1 моль) протекает при кипячении в ацетонитриле или ксилоле с получением солей (Chex2NH2)[PhSiIV(Catx)2]− (I–VI, Chex2${\text{NH}}_{2}^{ + }$ = бис-циклогексиламмоний, х = = 1–6, схема 1 ). Выход целевых продуктов изменяется от 52 до 89%, что хорошо соответствует ранее опубликованным данным для бис-катехолатфенилсиликатов(IV) [8]. Состав полученных комплексов был подтвержден данными спектроскопии ЯМР 1H, 13C и 29Si и масс-спектрометрии высокого разрешения. Согласно данным спектроскопии ЯМР 29Si, сигнал атома кремния смещен в область слабого поля в диапазон от –82 до –88 м.д., характерный для атомов пентакоординированного кремния.
Данные рентгеноструктурного анализа соединений I–V, полученных в виде монокристаллов, подтверждают, что комплексные анионы [PhSiIV(Catx)2]− содержат пентакоординированный атом кремния(IV), связанный с фенильной группой и двумя катехолат-анионами. В результате катионы и анионы в структурах I–V находятся в соотношении 1 : 1; кроме того, кристаллы II–IV содержат сольватные молекулы гексана, бензола или воды соответственно. Во всех случаях структурные единицы занимают общее положение. Независимая часть ячеек в структуре изученных соединений показана на рис. 1.
Рис. 1.
Независимая часть ячеек в структуре I–V в представлении атомов тепловыми эллипсоидами (р = 50%). В соединениях III и V изображена одна из двух разупорядоченных частей аниона и сольватной молекулы бензола.
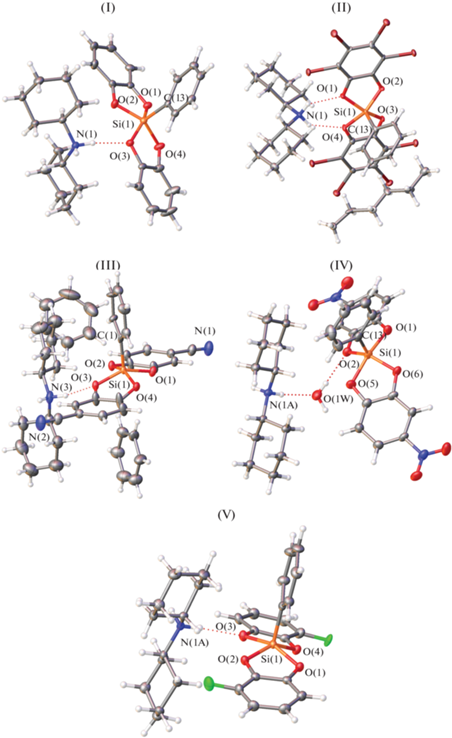
Как и в ранее структурно-охарактеризованных комплексах бис-катехолатфенилкремния(IV) [1, 8, 19–21], катехолат-анионы являются бидентатно-хелатными лигандами по отношению к центральному атому, связанными с ним двумя атомами кислорода, а фенильная группа – концевым лигандом. Таким образом, атом кремния(IV) образует координационный полиэдр состава SiIVCO4, в вершинах которого расположены атомы кислорода двух разных катехолат-анионов (рис. 1). Чтобы установить форму координационного полиэдра пятикоординированного кремния спироциклического строения, как правило, рассчитывается координата Берри, где координационый полиэдр отдельной структуры принимается за точку на координате перехода между квадратной пирамидой (КП) и тригональной бипирамидой (ТБП) [22]. Величина координаты Берри, которую можно оценить через телесные углы граней координационного полиэдра, показывает вклад ТБП в %. Расчетные значения координаты Берри для I–V составили: 84.5, 92.0, 84.5, 49.6 и 76.9%, т.е. лишь для соединения IV координационный полиэдр SiIVCO4 является промежуточным между ТБП и КП, и для комплекса V можно отметить значимый вклад КП в форму координационного полиэдра, а в остальных соединениях его можно считать искаженной ТБП. Cвязи Si–O в целом варьируют от 1.70(1) до 1.820(5) Å и короче связей Si–С (рис. 2). За аксиальные вершины были выбраны атомы кислорода, валентный угол OSiO которых ближе к 180°, поскольку длины связей лишь в структурах IV, V и (Me4N)[PhSiIV(Cat1)2] [19] позволяют разделить их на более длинные аксиальные и более короткие экваториальные. В остальных комплексах длины экваториальных и аксиальных связей в пределах 3σ могут совпадать друг с другом. Угол OaксSiOaкс изменяется от 149.3(1)° (III) до 167.7(1)° ((Me4N)[PhSiIV(Cat1)2]), причем отклонение от линейности обусловлено отталкиванием от фенильного кольца. Так, угол СSiOaкс составляет не менее 99.1(1)°, а угол СSiOэкв равен 105.0(1)°–115.3(1)°. Сравнение эквивалентных длин координационных связей показывает, что геометрия координационного узла атома кремния в значительной степени зависит от природы заместителей в фенильных группах катехолатного лиганда и наличия прочных водородных связей, в то время как координационная связь Si–С остается неизменной в пределах 3σ. Это совпадает с литературными данными о том, что комплексы гиперкоординированного кремния относятся к соединениям, на геометрию которых весьма заметно влияют межмолекулярные взаимодействия в кристалле или в растворе [23]. Даже в случае незамещенного катехолатного лиганда в отсутствие прочных водородных связей длины аксиальных и экваториальных связей могут совпадать как в комплексе K[PhSiIV(Cat1)2] · 2Me2CO [1] и резко отличаться как в случае (Me4N)[PhSiIV(Cat1)2] [19]. Аналогично близки друг к другу экваториальные и аксиальные связи в (Et4N)[PhSiIV(Cat7)2] (Cat7 = 3,4,5,6-тетрахлорокатехолат) [21], в котором также отсутствуют прочные водородные связи. Выраженное варьирование координационных связей Si–O наблюдается в комплексах с электронодонорными заместителями в фенильном кольце структур IV (Cat4 = 4-нитрокатехолат) и (Et3NH)[PhSiIV(Cat8)2] (Cat8 = = 3,5-ди-трет-бутилкатехолат) [20]. В соединении III, помимо прочных водородных связей, можно предположить образование прочного агостического контакта C–H…Si с участием сольватной молекулы бензола, поскольку для обеих разупорядоченных фрагментов на продолжении связи CPh–Si находится атом водорода сольватной молекулы (угол CPh–Si…H равен 158.0°–167.6°, r(Si…C) составляет 3.871(7)–3.899(12) Å), который достраивает координацию центрального атома до 5 + 1 (рис. 3). В результате координационный полиэдр атома кремния в данном соединении является, скорее, квадратной пирамидой, а длины связей Si–O примерно равны.
Рис. 2.
Длины координационных связей Si–C и Si–O с участием аксиальных и экваториальных атомов кислорода в структуре I–V и ранее опубликованных комплексах (Me4N)[PhSiIV(Cat1)2] {MAPPSI} [19], K[PhSiIV(Cat1)2] · 2Me2CO {YUVNEA} [1], (Et3NH)[PhSiIV(Cat1)2] {DALGAY} [20], (Et3NH)[PhSiIV(Cat8)2] {DALFUR} [20], (Et4N)[PhSiIV(Cat7)2] {CEPREU} [21], (Kcrow)(Kcrow(Me2CO))[PhSiIV(Cat5)2]2 {IXEBIZ} [8]. Cat6 = 3,4,5,6-тетрахлорокатехолат, Cat7 = 3,5-ди-трет-бутилкатехолат, crow = 18-краун-6. Буквами указан код соединения в КБСД. Отмечена экспериментальная ошибка определения связей.

Рис. 3.
Агостическое взаимодействие в структуре III. Контакт Si…H показан пунктиром. Изображена одна позиция из двух разупорядоченных.

Визуализация искажений координационного узла и сравнение конформаций анионного комплекса в целом представлено на рис. 4, на котором друг на друга наложены атомы координационного полиэдра атома кремния(IV). Положение фенильного кольца отображает факт его свободного вращения относительно одинарной связи Si–С и отсутствие либо низкую энергию связей CPh–H…OCat. Вместе с тем, несмотря на жесткую планарную конформацию отдельных катехолат-анионов, их взаимное расположение при атоме кремния также заметно варьирует в зависимости от заместителей при фенильном кольце и кристаллического окружения. Конформации [PhSiIV(CatR)2]− в структурах I, (Me4N)[PhSiIV(Cat1)2] [19], K[PhSiIV(Cat1)2] · · 2Me2-CO [1] и (Et3NH)[PhSiIV(Cat1)2] [20] также отличаются друг от друга (см. рис. 4).
Рис. 4.
Конформации анионов [PhSiIV(CatR)2]– в структурах I (красный), II (фуксия), III (синий), IV (голубой), V (сиреневый), (Me4N)[PhSiIV(Cat1)2] (серый) [19], K[PhSiIV(Cat1)2] · 2Me2CO (изумрудный) [1], (Et3NH)[PhSiIV${\text{Cat}}_{2}^{1}$] (оранжевый) [20], (Et3NH)-[PhSiIV${\text{Cat}}_{2}^{5}$] (фиолетовый) [20], (Et4N)[PhSiIV${\text{Cat}}_{2}^{6}$] (зеленый) [21], (Kcrow)(Kcrow(Me2CO))[PhSiIV${\text{Cat}}_{2}^{7}$]2 (розовый) [8]. Атомы водорода не изображены. Друг на друга наложены атомы координационного полиэдра SiIVCO4.

Влияние специфических взаимодействий можно проследить, сравнивая между собой эквивалентные связи, участвующие и не участвующие в прочных контактах. Так, в кристаллах ранее синтезированных и структурно охарактеризованных ацетамид- и капролактамсодержащих катехолатов кремния [24, 25] длины связей Si–O с участием катехолатных анионов, вовлеченные в водородные связи классического типа (O–H···O и N–H···O), как правило, на 0.1 Å длиннее, чем связи, не вовлеченные в контакты подобного типа. В комплексах I–V наличие донорных атомов водорода при катионе и ряда акцепторных атомов при анионах приводит к тому, что во всех соединениях обнаружены водородные связи, параметры которых приведены в табл. 2. В случае II катион образует две водородных связи с атомами кислорода двух разных катехолат-анионов в структуре одного комплексного иона с образованием островной водородно-связанной группировки (рис. 5). Соединения I, III и V относятся к соединениям, в структуре которых образуются водородно-связанные бесконечные цепи (рис. 6). В первом случае один из протонов связан с атомом О(3) одного аниона, а второй образует бифуркатную связь с атомами кислорода О(1) и О(4) другого аниона. Во втором случае катион образует две водородные связи и является мостиковым между атомами кислорода O(3) (или О(3А)) одного аниона и атомом азота N(1) цианогруппы другого. В структуре V атомы H(N) взаимодействуют с атомами кислорода аксиальных связей Si–Oaкс. Наличие молекулы воды в структуре IV приводит к тому, что, хотя катион участвует лишь в двух водородных связях, в целом водородно-связанный фрагмент является слоистым (рис. 5). При этом молекула воды участвует в трех водородных связях как донор двух протонов и акцептор водородной связи. Комплексный анион в структуре IV является акцептором трех водородных связей с участием одного катехолат-аниона и атомов кислорода двух нитрогрупп. В водородно-связанных слоях (рис. 5) можно выделить четырехчленные циклы с участием двух молекул воды и двух анионов и двенадцатичленные с участием четырех молекул воды, четырех катионов и четырех анионов. Если упростить сетку до 3,3-координированной (упростив мостиковые катионы), то ее топология относится к типу fes согласно номенклатуре Reticular Chemistry Structure Resource [26]. Участие атома кислорода катехолатной группы в образовании водородных связей приводит к удлинению связи Si–O с атомом кислорода – акцептором водородной связи. Как можно видеть из рис. 2, разница между длинами аксиальных или экваториальных связей составляет от 0.02 до 0.08 Å (в среднем 0.03 Å), тогда как разница между длинами координационных связей с атомами кислорода, не участвующими (или одновременно участвующими) в водородных связях в среднем составляет 0.01 Å.
Таблица 2.
Геометрические параметры водородных связей в структурах I–V*
| D–H…A | Расстояние, Å | Угол DHA, град | ||
|---|---|---|---|---|
| D–H | H…A | D…A | ||
| I | ||||
| N(1)–H(1A)…O(3) | 0.94 | 1.96 | 2.885(2) | 169 |
| N(1)–H(1B)…O(1)i | 0.94 | 2.20 | 3.024(2) | 154 |
| N(1)–H(1B)…O(4)i | 0.94 | 2.30 | 3.070(3) | 144 |
| II | ||||
| N(1)–H(1A)…O(1)ii | 0.91 | 2.19 | 2.930(11) | 138 |
| N(1)–H(1B)…O(4)ii | 0.91 | 2.18 | 2.924(14) | 138 |
| III | ||||
| N(3)–H(3B)…O(3) | 0.91 | 2.14 | 3.012(10) | 160 |
| N(3)–H(3B)…O(3A) | 0.91 | 1.98 | 3.012(10) | 168 |
| N(3)–H(3C)…N(1)iii | 0.91 | 2.13 | 3.027(4) | 170 |
| IV | ||||
| O(1W)–H(1WA)…O(2) | 0.85 | 2.07 | 2.912(3) | 171 |
| O(1W)–H(1WB)…O(7)iv | 0.91 | 2.15 | 2.964(3) | 157 |
| N(1)–H(1AB)…O(1W) | 0.91 | 1.98 | 2.804(3) | 157 |
| N(1)–H(1AA)…O(3)v | 3.013(3) | 159 | ||
| V | ||||
| N(1A)–H(1B)…O(3) | 2.870(2) | 166 | ||
| N(1A)–H(1A)…O(1)vi | 0.91 | 2.00 | 2.903(2) | 175 |
Рис. 5.
Водородно-связанные архитектуры в структурах II, IV. Атомы водорода, не участвующие в водородных связях, не показаны.

Рис. 6.
Водородно-связанные цепи в структурах I, III, V. Атомы водорода, не участвующие в водородных связях, не показаны.

Одним из актуальных применений комплексов пентакоординированого кремния является инициирование реакций полимеризации за счет генерирования радикальных частиц. Склонность к этому должна определяться природой заместителей на атоме кремния, которые могут в различной степени влиять на стабилизацию окисленных форм I–VI. Соединения II–VI содержат различное число акцепторных групп, которые могут проявлять не только индуктивный, но и мезомерный эффект. Для того чтобы исследовать влияние заместителей на электронное строение, были проведены квантовохимические расчеты неокисленных свободных анионов I–VI, а также их окисленных форм I*–VI*.
Оптимизация геометрии анионов и их окисленных форм, выполненная с помощью метода PBE0/6-311G(d,p), показала, что длины аксиальных связей Si–O отличаются от экваториальных на 0.03–0.06 Å, а координационный полиэдр сохраняет форму тригональной бипирамиды. В целом разница между аксиальными и экваториальными связями Si–O несколько возрастает при увеличении количества акцепторных заместителей. Тем не менее влияние заместителей на геометрию координационного узла атома кремния выражено относительно слабо (табл. S1–S6 ). В окисленных формах различие между аксиальными и экваториальными связями увеличивается, при этом оно зависит и от лиганда. Связи Si–O(1) и Si–O(2) в I*–VI* отличаются друг от друга намного больше, чем в случае Si–O(3) и Si–O(4). В результате удаления электрона аксиальная связь Si–O(1) оказалась на 0.02–0.03 Å короче, чем экваториальная Si–O(2). Наоборот, аксиальная связь Si–O(3) на 0.11–0.12 Å длиннее, чем экваториальная. В результате окисления наблюдается также и укорочение связи Si–C (на 0.02–0.03 Å).
Анализ граничных орбиталей анионных комплексов I–VI показал, что вертикальная ионизация затрагивает в основном область катехолатных лигандов. Действительно, наибольшие значения заселенности ВЗМО и НВМО наблюдаются на атомах углерода фенильных заместителей и атомов кислорода катехолатных лигандов (рис. 7). В I*–VI* максимумы спиновой плотности (рис. 8) локализованы на атомах кислорода и углерода (положение 3) катехолатного лиганда.
Рис. 7.
ВЗМО (a) и НВМО (б) аниона комплекса II. Положительные значения показаны синим цветом, положительные – красным, значение изоконтура равно 0.03.


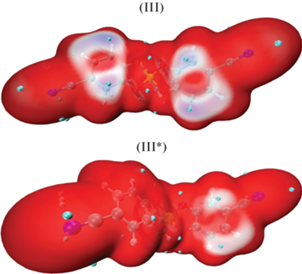
Распределение длин связей Si–O в I*–VI* свидетельствует о том, что электроноакцепторная способность одного из катехолатных лигандов заметно снижена. Для этого лиганда также снижается способность к взаимодействию с электрофильными агентами и свободными радикалами. Для иллюстрации данной способности П. Политцером была предложена функция среднего локального потенциала окисления (ALIE) [27]. На рис. 9 представлено наложение распределения функции ALIE на изоповерхность электронной плотности (ρ = 0.005 а.е.) для аниона III и его окисленной формы. Области минимальных значений функции ALIE отвечают предпочтительным положениям электрофильной атаки. Минимумы ALIE в III находятся в области фенильной группы, а также в области атомов углерода (положения 2 и 5) катехолатных лигандов. В случае III* уменьшение площади областей с минимальным значением функции ALIE по сравнению с III отражает ослабление реакционной способности при взаимодействии с электрофилами и свободными радикалами.
Рис. 9.
Распределение функции ALIE на изоповерхности функции электронной плотности (ρ = 0.005 а.е.) для III и III*. Наименьшие значения функции ALIE обозначены синим цветом, наибольшие – красным, минимумы ALIE – зелеными шарами. Интервал значений 0.31–0.38 а.е.
Величина потенциала окисления определяется вертикальным и адиабатическими потенциалами ионизации, которые, согласно квантовохимическим расчетам, явно зависят от природы заместителя. Наибольшие величины вертикального потенциала ионизации наблюдаются для VI с дибромокатехолатным лигандом, наименьшая – для IV с 4-нитрокатехолатным лигандом. Для адиабатического потенциала ионизации зависимость иная, наибольшие величины получены для анионного комплекса II с тетрабромкатехолатным лигандом, при этом величина для IV лишь ненамного меньше. Наименьшая величина получена для комплекса I с незамещенным катехолатным лигандом. По-видимому, большое число акцепторных заместителей является фактором стабилизации окисленных форм из-за их индуктивного эффекта. Нитрогруппа в положении 4 также способна эффективно стабилизировать нейтральный радикальный семихинон-катехолатный комплекс вследствие не только индуктивного, но и отрицательного мезомерного эффекта. Из величин адиабатических потенциалов можно вычислить и расчетные потенциалы окисления. Согласно литературным данным [16], для этого необходима величина свободной энергии, затрачиваемой на окисление и сольватацию анионных комплексов и их окисленных форм. Учет величины свободной энергии сольватации приводит к сильному завышению расчетной величины потенциала ионизации относительно экспериментального (табл. S7–S9 ). Причина этого, возможно, заключается в образовании специфических взаимодействий в растворе, что не может быть полностью учтено в квантовохимических расчетах. Другим источником погрешности может быть величина абсолютного электрохимического потенциала электрода относительно пары Fc/Fc+ в ДМСО. Нами использована расчетная величина (5.04 В [28]), так как экспериментальная величина в литературе не обнаружена. Поэтому расчетные величины потенциалов окисления могут быть взяты как относительные характеристики, отражающие способность к окислению анионных комплексов I–VI. Наибольшая величина потенциала окисления зарегистрирована в случае анионных комплексов II и IV, причем для последнего величина несколько выше. Хотя квантовохимические расчеты и предсказывают обратный порядок изменения величин E0 для II и IV, однако они все равно являются соединениями с наибольшими значениями потенциалов окисления (табл. S9 ).
Для комплексов II–V были измерены потенциалы окисления в ДМСО с помощью метода вольтамперометрии (рис. S1 ). Вольтамперные кривые для этих соединений (кроме II) демонстрируют наличие одного потенциала окисления в 0.80–0.99 В. Само окисление является необратимым. Наибольшие первые потенциалы окисления зарегистрированы для тетрабром- и 4-нитрокатехолатных лигандов (0.90 и 0.99 В). Для остальных измеренных соединений величины E0 фактически равны (для III, V и VI они составляют, соответственно 0.83, 0.80 и 0.81 В).
Образование катион- и анион-радикалов в случае II удалось зарегистрировать с помощью метода ЭПР, соответственно, при электрохимическом окислении и восстановлении в ацетонитриле (рис. S2 ). Были зарегистрированы уширенные спектры катион- и анион-радикалов (0.132 и 0.139 мТ соответственно), что в сочетании с невысокой интенсивностью и малым временем жизни не позволяет уверенно определить константы СТВ на атоме кремния, однако измеренные значения g-факторов соответствующих катион- и анион-радикалов равны 2.0060 и 2.0050, что соответствует наличию одного неспаренного электрона. Основываясь на полученных данных, можно заключить, что строение молекулярных фрагментов в случае окисления и восстановления может быть различным.
Авторы заявляют об отсутствии конфликта интересов.
Список литературы
Corcé V., Chamoreau L.-M., Derat E. et al. // Angew. Chem. Int. Ed. 2015. V. 54. № 39. P. 11414. https://doi.org/10.1002/anie.201504963
Raynor K.D., May G.D., Bandarage U.K. et al. // J. Org. Chem. 2018. V. 83. № 3. P. 1551. https://doi.org/10/gcts7f
Lin K., Wiles R.J., Kelly C.B. et al. // ACS Catal. 2017. V. 7. № 8. P. 5129. https://doi.org/10/gbrr5s
Levernier E., Corcé V., Rakotoarison L.-M. et al. // Org. Chem. Front. 2019. V. 6. № 9. P. 1378. https://doi.org/10/gpchtm
Lévêque C., Chenneberg L., Corcé V. et al. // Org. Chem. Front. 2016. V. 3. № 4. P. 462. https://doi.org/10/gjz9bm
Patel N.R., Kelly C.B., Siegenfeld A.P. et al. // ACS Catal. 2017. V. 7. № 3. P. 1766. https://doi.org/10/gpcj27
Cartier A., Levernier E., Corcé V. et al. // Angew. Chem. 2019. V. 131. № 6. P. 1803. https://doi.org/10/gpcj3m
Levernier E., Jaouadi K., Zhang H.-R. et al. // Chem. Eur. J. 2021. V. 27. № 34. P. 8782. https://doi.org/10/gpcj4s
Blessing R.H. // Acta Crystallogr. 1995. V. A51. № 1. P. 33. https://doi.org/10.1107/S0108767394005726
APEX3 Suite for Crystallographic Software – Single Crystal X-ray Diffraction. Bruker AXS, 2014.
Dolomanov O.V., Bourhis L.J., Gildea R.J. et al. // J. Ap-pl. Crystallogr. 2009. V. 42. № 2. P. 339. https://doi.org/10.1107/S0021889808042726
Sheldrick G.M. // Acta Crystallogr. A. 2015. V. 71. № 1. P. 3. https://doi.org/10.1107/S2053273314026370
Sheldrick G.M. // Acta Crystallogr. C. 2015. V. 71. № 1. P. 3. https://doi.org/10.1107/S2053229614024218
Krylov A.I., Gill P.M.W. // WIREs Comput. Mol. Sci. 2013. V. 3. № 3. P. 317. https://doi.org/10/f4wfdf
Epifanovsky E., Gilbert A.T.B., Feng X. et al. // J. Chem. Phys. 2021. V. 155. № 8. P. 084801. https://doi.org/10/gmxp67
Neugebauer H., Bohle F., Bursch M. et al. // J. Phys. Chem. A 2020. V. 124. № 35. P. 7166. https://doi.org/10/gnv5m6
Lu T., Chen F. // J. Comput. Chem. 2012. V. 33. № 5. P. 580. https://doi.org/10.1002/jcc.22885
Humphrey W., Dalke A., Schulten K. // J. Mol. Graph. 1996. V. 14. № 1. P. 33. https://doi.org/10/b3tgfk
Boer F.P., Flynn J.J., Turley J.W. // J. Am. Chem. Soc. 1968. V. 90. № 25. P. 6973. https://doi.org/10/dx7qfc
Holmes R.R., Day R.O., Chandrasekhar V. et al. // Inorg. Chem. 1985. V. 24. № 13. P. 2009. https://doi.org/10/dpfxj2
Holmes R.R., Day R.O., Harland J.J. et al. // Organometallics. 1984. V. 3. № 3. P. 341. https://doi.org/10/b9kkvp
Holmes R.R., Deiters J.A. // J. Am. Chem. Soc. 1977. V. 99. № 10. P. 3318. https://doi.org/10.1021/ja00452a021
Корлюков А.А. // Успехи химии. 2015. V. 84. № 4. P. 422.
Kramarova E.P., Volodin A.D., Negrebetsky V.V. et al. // Molecules. 2021. V. 26. № 12. P. 3548. https://doi.org/10.3390/molecules26123548
Корлюков А.А., Шипов А.Г., Крамарова Е.П. et al. // Изв. АН. Сер. хим. 2008. № 10. P. 2055.
O’Keeffe M., Peskov M.A., Ramsden S.J. et al. // Acc. Chem. Res. 2008. V. 41. № 12. P. 1782. https://doi.org/10/fwg963
Politzer P., Murray J.S. Chapter 8. The Average Local Ionization Energy: Concepts and Applications / Ed. Toro-Labbé A. // Theoretical and Computational Chemistry. 2007. V. 19. P. 119. https://doi.org/10.1016/S1380-7323(07)80009-4
Namazian M., Lin C.Y., Coote M.L. // J. Chem. Theory Comput. 2010. V. 6. № 9. P. 2721. https://doi.org/10/bqb274
Дополнительные материалы
- скачать ESM.docx
- Приложение 1.
Инструменты
Координационная химия