

Координационная химия, 2022, T. 48, № 5, стр. 277-286
Гетерометаллические комплексы германия(IV) на основе N-фенилзамещенного о-амидофенолятного лиганда
А. В. Пискунов 1, *, К. В. Арсеньева 1, А. В. Климашевская 1, А. В. Черкасов 1
1 Институт металлоорганической химии им. Г.А. Разуваева РАН
Нижний Новгород, Россия
* E-mail: pial@iomc.ras.ru
Поступила в редакцию 27.10.2021
После доработки 03.11.2021
Принята к публикации 06.11.2021
- EDN: LUEAKH
- DOI: 10.31857/S0132344X22050073
Аннотация
Новые биметаллические комплексы PhAPGe[M(CO)nCp]2 (PhAP = 3,5-ди-трет-бутил-N-(фенил)-о-амидофенолятный дианион; M = Fe, n = 2 (II); M = W, n = 3 (III)) получены в ходе реакции внедрения O,N-гетероциклического гермилена PhAPGe (I) по связи металл–металл в димерах [Fe(CO)2Cp]2 и [W(CO)3Cp]2. Окисление соединений II и III трифлатом серебра(I) приводит к образованию парамагнитных о-иминосемихинолятов Ge(IV), зафиксированных методом спектроскопии ЭПР. Окислительное присоединение 3,6-ди-трет-бутил-о-бензохинона к гермилену I протекает с окислением низковалентного центра до четырехвалентного состояния и сопровождается симметризацией с образованием соответствующих бис-о-амидофенолятного и бис-катехолатного производных германия(IV). Дигермиленоксид Iа, полученный гидролизом исходного гермилена I, выступает окислителем в реакции с димером циклопентадиенилкарбонила никеля и формирует в соединении (CpNi)2[PhAPGeOGePhAmP]2 (PhAmP = 3,5-ди-трет-бутил-N-(фенил)-о-аминофенолятный анион) (IV) восьмичленный металлоцикл, содержащий четыре донорно-акцепторные связи Ge(II)–Ni(II). Молекулярные структуры соединений II–IV установлены методом РСА (СIF files CCDC № 2118153–2118155).
Координационные и металлоорганические соединения, содержащие в своем составе редокс-активные лиганды, являются одной из перспективных точек развития современной химии и находят применение в целом ряде областей исследования, таких как каталитические превращения малых молекул, молекулярная электроника и молекулярный магнетизм [1–4]. Интерес к этому типу лигандов обусловлен способностью к обратимому окислительно-восстановительному превращению, сохраняя при этом связь с металлом. Химия комплексов металлов, содержащих этот тип лигандов интенсивно развивается преимущественно на примере производных переходных металлов [5–8]. Некоторые из них обладают уникальными магнитными и электронными свойствами [9–13]. Парамагнитные анион-радикальные формы редокс-активных лигандов могут успешно применяться в качестве спин-меченых лигандов [14]. Спектры ЭПР таких соединений обладают высокой информативностью и могут дать различные сведения об их структурe и механизмe превращения в растворе [15–21].
В последние годы наметилось развитие нового направления, включающего элементы главных подгрупп в комплексы с редокс-активными лигандами. Данное сочетание позволяет вовлечь такие соединения в реакции окислительного присоединения и восстановительного элиминирования без изменения степени окисления комплексообразователя [1, 22–24], а также наблюдать слабые магнитные взаимодействия, не осложненные присутствием парамагнитного иона переходного металла [25]. Целый ряд исследовательских групп предпринимает попытки получения гетерометаллических производных на основе редокс-активных лигандных систем, c непосредственным формированием связи металл–металл. При этом атом непереходного металла преимущественно 13 [26–32] или 14 [33–38] групп в таких соединениях ковалентно связан с переходным металлом или лантанидом [29, 39–41]. Подавляющее число таких исследований выполнено на примере дииминовых редокс-активных лигандов. Известно лишь несколько работ, где атом непереходного металла при этом хелатирован диолатным [35, 42] или амидофенолятным [43, 44] лигандами. Необходимо отметить, что окислительно-восстановительные свойства соединений такого типа, демонстрирующие редокс-свойства органических лигандов, как правило, не изучались.
В настоящем исследовании мы синтезировали и охарактеризовали органобиметаллические производные со связями Ge–M (M = Fe, Ni, W), с участием редокс-активного 3,5-ди-трет-бутил-N-(фенил)-о-амидофенолята (I).
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Все операции по синтезу и исследованию химических превращений комплексов германия проводили в условиях отсутствия кислорода и влаги воздуха. Использованные в работе растворители очищали и обезвоживали согласно рекомендациям [45]. Применяли коммерческий реактив [CpFe(CO)2]2. o-Aмидофенолят германия (гермилен) I и оксид о-аминофенолята германия (Ia) синтезировали в соответствии с методиками, описанными в [43, 46]. Димеры циклопентадиенилакарбонилов никеля [47] и вольфрама [48] получали согласно известным процедурам.
Спектры ЯМР регистрировали на спектрометре Bruker Avance Neo 300 MГц. Спектры ЭПР фиксировали на спектрометре Bruker EMX. В качестве стандарта при определении g-фактора использовали 2,2-дифенил-1-пикрилгидразил (g = 2.0037). Для определения точных параметров спектр ЭПР симулировали с помощью программы WinEPR SimFonia (Bruker).
Синтез комплексов (PhAP)Ge[Fe(CO)2Cp]2 (II) и (PhAP)Ge[W(CO)3Cp]2 (III). К навеске [CpM(CO)n]2 (0.4 ммоль; M = Fe (n = 2), 0.14 г; M = W (n = 3), 0.27 г) добавляли бледно-желтый раствор гермилена I (0.4 ммоль, 0.15 г) в TГФ. Практически моментально произошло изменение цвета на интенсивно бoрдовый. Реакционную смесь оставляли на 12 ч при постоянном перемешивании при комнатной температуре. Комплексы II и III выделяли в виде диамагнитных кристаллических веществ темно-красного цвета из концентрированного раствора в гексане при комнатной температуре.
Комплекс II. Выход 0.22 г, 0.31 ммоль (78%). Спектр ЯМР 1Н (C6D6; 20°С; δ, м.д.; J, Гц): 7.6 (д., 2H, HPh, JH,H = 7.7); 7.52 (д., 1H, HAP, JH,H = 2.2); 7.1 (д., 1H, HAP, JH,H = 2.2); 7.33 (м., 2H, HPh ); 6.95 (м., 1H, HPh ); 4.2 (с., 10H, Сp); 1.8 (с., 9H, t-Bu); 1.38 (с., 9H, t-Bu). Спектр ЯМР 13C (C6D; 20°С; δ, м.д.; J, Гц): 214.3, 213.3 (C≡O); 152.4, 148.4, 139.6, 122.0, 121,1 (СPh); 139.2 (N–CPh); 133.5 (C–N); 129.4 (С–О); 83.3 (Сp); 34.9, 34.2 (Cчетв); 29.8, 31.8 (Сt-Bu).
Комплекс III. Выход 0.26 г, 0.25 ммоль (63%). Спектр ЯМР 1Н (C6D6; 20°С; δ, м.д.; J, Гц): 7.63 (д., 1H, HAP, JH,H = 2.2); 7.61 (д., 1H, HAP, JH,H = 2.2); 7.44 (с., 1H, HPh); 7.34 (м., 2H, HPh); 7.01 (м., 2H, HPh); 4.81 (с., 10H, Сp); 1.82 (с., 9H, t-Bu); 1.38 (с., 9H, (t‑Bu). Спектр ЯМР 13C (C6D6; 20°С; δ, м.д.; J, Гц): 220.4, 217.7, 217.5 (C≡O); 151.6, 147.7, 140.3, 123.2, 121,8 (СPh); 139.8 (N–CPh); 133.5 (C–N); 129.5 (С–О); 90.23 (Сp); 34.95, 34.48 (Cчетв); 31.8, 30.1 (Сt-Bu).
Синтез комплекса (CpNi)2[PhAPGeOGePhAmP]2 (IV). К раствору оксида дигермилена Iа (0.138 ммоль), полученного in situ, в толуоле (10 мл) добавляли раствор [CpNiCO]2 (0.042 г, 0.138 ммоль) в том же растворителе (10 мл). Реакционную смесь выдерживали в темноте при комнатной температуре в течение 2 сут. За это время цвет раствора изменился на коричневый. Комплекс IV выделяли в виде желтого кристаллического порошка после смены растворителя на гексан. Выход 0.22 г (0.07 ммоль) (52%).
Спектр ЯМР 1Н (C6D6; 20°С; δ, м.д.; J, Гц): 7.71 (д., 1H, HAP,JH,H = 2.2); 7.59 (д., 1H, HAP,JH,H = = 2.0); 7.35–6.66 (м., 12H, HPh, NH); 6.65 (д., 1 H, HAP,JH,H = 2.2), 6.44 (д., 1 H, HAP,JH,H = 2.0), 4.99 (с., 10Н, Ср), 1.47 (с., 9H, t-Bu), 1.34 (с., 9H, t-Bu), 1.19 (с., 9 H, t-Bu), 1.04 (с., 9 H, t-Bu).
Реакция комплекса I с 3,6-ди-трет-бутил-о-бензохиноном. К раствору гермилена I (0.2 г, 0.54 ммоль) в толуоле (10 мл) медленно приливали раствор хинона (0.119 г, 0.54 ммоль) в тетрагидрофуране (10 мл). Реакционная смесь быстро приобретала коричневый цвет. Растворитель удаляли при пониженном давлении и остаток растворяли в хлористом метилене. Добавление в полученную смесь гексана приводило к выпадению белого кристаллического осадка, который, по данным спектрокопии ЯМР 1Н и 13С, является бис-(3,6-ди-трет-бутилкатехолато)германий(IV)дитетрагидрофуранатом (V), описанным ранее [49]. Выход 0.095 г (0.18 ммоль) (34%).
РСА соединений II, III и IV проведен на дифрактометре Bruker D8 Quest (ω-сканирование, МоКα-излучение, λ = 0.71073 Å). Измерение и интегрирование экспериментальных наборов интенсивностей, учет поглощения и уточнение структур проведены с использованием программных пакетов APEX3 [50], SADABS [51] и SHELX [52]. Структуры решены с помощью алгоритма dual-space [53] и уточнены полноматричным МНК по $F_{{hkl}}^{2}$ в анизотропном приближении для неводородных атомов. Атом водорода H(1A) в IV найден из разностного синтеза Фурье электронной плотности. Все остальные водородные атомы в II, III и IV помещены в геометрически рассчитанные положения и уточнены изотропно с фиксированными тепловыми параметрами U(H)изо = 1.2U(C)экв (U(H)изо = 1.5U(C)экв для метильных фрагментов). Кристаллографические данные и параметры рентгеноструктурных экспериментов и уточнения структур приведены в табл. 1.
Таблица 1.
Кристаллографические данные и параметры уточнения структур II, III и IV
| Параметр | Значение | ||
|---|---|---|---|
| II | III | IV | |
| Брутто-формула | C34H35NO5Fe2Ge | C36H35NO7GeW2, 1/2C6H14 | C90H112N4Ni2Ge4O6, C6H14 |
| М | 721.92 | 1077.02 | 1839.78 |
| Сингония | Моноклинная | Моноклинная | Триклинная |
| Пр. группа | P21/n | P21/c | $P\bar {1}$ |
| T, K | 100 | 120 | 100 |
| a, Å | 13.7818(5) | 13.1443(5) | 10.0556(9) |
| b, Å | 12.4681(4) | 17.8359(8) | 12.9106(11) |
| c, Å | 18.7331(6) | 16.8068(7) | 19.0244(16) |
| α, град | 90 | 90 | 75.069(3) |
| β, град | 105.074(2) | 104.8300(10) | 75.495(3) |
| γ, град | 90 | 90 | 79.371(3) |
| V, Å3 | 3108.20(18) | 3808.9(3) | 2291.3(3) |
| Z | 4 | 4 | 1 |
| ρ(выч.), г/см3 | 1.543 | 1.878 | 1.333 |
| µ, мм–1 | 1.925 | 6.858 | 1.751 |
| Размер кристалла, мм | 0.34 × 0.21 × 0.05 | 0.19 × 0.11 × 0.04 | 0.26 × 0.09 × 0.03 |
| Область сканирования θ, град | 2.12–30.20 | 2.10–26.10 | 2.17–25.08 |
| Количество измеренных/ независимых отражений | 33 210/9204 | 45 580/7552 | 25 909/8064 |
| Rint | 0.0565 | 0.0676 | 0.0756 |
| Количество независимых отражений с I > 2σ(I) | 6944 | 5955 | 5652 |
| Число уточняемых параметров/ ограничений | 394 | 488 | 682 |
| R (I > 2σ(I)) | R1 = 0.0429, wR2 = 0.0901 |
R1 = 0.0401, wR2 = 0.0646 |
R1 = 0.0606, wR2 = 0.1008 |
| R (по всем данным) | R1 = 0.0709, wR2 = 0.0990 |
R1 = 0.0614, wR2 = 0.0697 |
R1 = 0.1030, wR2 = 0.1179 |
| S (F 2) | 1.016 | 1.020 | 1.056 |
| Макс. и мин. остаточной электронной плотности, e Å–3 | 0.99/–0.50 | 1.41/–0.91 | 0.84/–0.68 |
Структуры зарегистрированы в Кембриджском банке структурных данных (CCDC № 2118153 (II), 2118154 (III), 2118155 (IV); ccdc.cam.ac.uk/structures).
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Внедрение низковалентных производных элементов группы 14 по связи металл–металл это достаточно надежный и распространенный способ для синтеза гетерометалических производных со связью переходный металл–тетрилен [35, 54]. Мы обнаружили, что взаимодействие мономерного амидофенолята германия(II) I с циклопентадиенилкарбонилами переходных металлов [CpM(CO)n]2 (M = Fe, n = 2; M = W, n = 3) приводит к формированию новых биметаллических комплексов (II, III) с хорошими выходами согласно схеме 1 . Реакция полностью заканчивается за 24 ч при комнатной температуре в растворе толуола. Конечные соединения были выделены в качестве твердых кристаллических продуктов красно-коричневого цвета. Соединения II и III стабильно ведут себя на воздухе в кристаллическом состоянии в течение нескольких дней, однако их растворы медленно разлагаются в аэробной атмосфере. Необходимо отметить, что реакция гермилена I с участием соединения никеля [Cp-Ni(CO)]2 в аналогичных условиях не сопровождается видимым изменением цвета реакционной смеси. Нам не удалось выделить и охарактеризоватьиндивидуальные вещества из данной реакционной смеси.
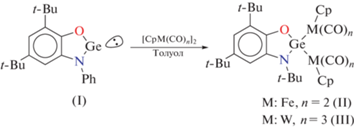
Схема 1 .
Образование диамагнитных амидофенолятных комплексов II и III подтверждается данными спектроскопии ЯМР 1Н и 13С. Спектры демонстрируют хорошee разрешениe при комнатной температуре, в них наряду с протонами о-амидофенолятного фрагмента наблюдается единственный дополнительный сигнал, отвечающий цикло-пентадиенильным заместителям. Спектр ЯМР 13С в диагностической области карбонильных атомов углерода показывает только два пика для II и три пика для III. Это указывает на эквивалентность двух фрагментов с переходными металлами в координационной сфере атома Ge в растворах комплексов II и III.
Мы провели рентгеноструктурные исследования монокристаллических образцов комплексов II и III. Согласно полученным данным, координационные полиэдры атомов германия в этих соединениях представляют собой искаженные тетраэдры. Молекулярные структуры II и III схожи между собой и представлены на рис. 1, 2 соответственно, а избранные длины связей и величины углов в этих комплексах приведены в табл. 2. В каждом из этих комплексов амидофенолятный фрагмент RApGe связан с двумя фрагментами CpM(CO)n (M = Fe, n = 2 для II; M = W, n = 3 для III) за счет связи Ge–M. Углы OGeN 87.09(8)° (II) и 86.4(2)° (III) типичны для подобных производных Ge(IV) [55] и лишь немногим больше аналогичного угла в исходном комплексе I (85.95(9)°) [43]. Длины связей C–O 1.367(2) (II), 1.371(7) (III) и C–N 1.414(3) (II), 1.395(8) (III) в амидофенолятных лигандах лежат в характерном диапазоне 1.35–1.42 Å и сопоставимы с аналогичными величинами в амидофенолятах четырехвалентных металлов. В целом геометрические параметры этого лиганда типичны для O,N-координированных амидофенолятных дианинов [55–58]. Угол WGeW 127.06(2)° в III несколько больше, чем FeGeFe 123.02(2)° в II и, по-видимому, обусловлен более высокой стерической загрузкой координационной сферы германия за счет более объемных металлоорганических фрагментов CpW(CO)3 по сравнению с CpFe(CO)2. Длины связей Ge–Fe в II равны 2.3782(4), 2.3867(4) Å. Расстояния Ge–W (2.6987(7), 2.7027(7) Å) в III лишь немногим отличаются от таковых в [CpW(CO)3]2GeCl2 [59].
Рис. 1.
Молекулярная структура комплекса PhAPGe[CpFe(CO)2]2 (II). Тепловые эллипсоиды избранных атомов приведены с 50%-ной вероятностью. Атомы водорода не изображены для ясности.
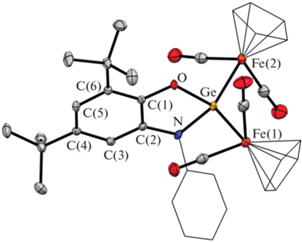
Рис. 2.
Молекулярная структура комплекса PhAPGe[CpW(CO)3]2 (III). Тепловые эллипсоиды избранных атомов приведены с 50%-ной вероятностью. Атомы водорода не изображены для ясности.
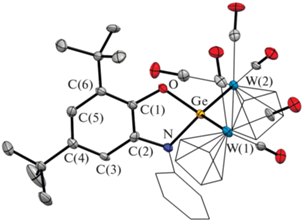
Таблица 2.
Избранные длины связей и углы для соединений II и III
| Связь | II | III |
|---|---|---|
| d, Å | ||
| Ge–N | 1.921(2) | 1.909(5) |
| Ge–O | 1.849(2) | 1.854(4) |
| C(1)–O | 1.367(3) | 1.371(7) |
| C(2)–N | 1.414(3) | 1.395(8) |
| C(1)–C(2) | 1.403(3) | 1.395(8) |
| C(2)–C(3) | 1.385(3) | 1.412(8) |
| C(3)–C(4) | 1.401(3) | 1.399(9) |
| C(4)–C(5) | 1.391(3) | 1.374(9) |
| C(5)–C(6) | 1.406(3) | 1.407(8) |
| C(6)–C(1) | 1.403(3) | 1.396(8) |
| Ge–W(1) | 2.6987(7) | |
| Ge–W(2) | 2.7027(7) | |
| Ge–Fe(1) | 2.3782(4) | |
| Ge–Fe(2) | 2.3867(4) | |
| Угол | ω, град | |
| OGeN | 87.09(8) | 86.4(2) |
| OGeM(1) | 106.63(5) | 112.1(2) |
| NGeM(1) | 113.96(6) | 111.5(2) |
| OGeM(2) | 107.85(5) | 100.8(2) |
| NGeM(2) | 111.75(6) | 111.5(2) |
| M(1)GeM(2) | 123.02(2) | 127.06(2) |
Мы показали [46], что гермилен I не вступает во взаимодействие c электронно ненасыщенным ванадоценом. В то же время данный металлоцен легко реагирует с дигермиленоксидом Ia, образующимся при осторожном гидролизе I. Взаимодействие Ia, синтезированного in situ по известной методике [46], и [CpNiCO]2 протекает медленно при комнатной температуре в мягких условиях (схема 2 ). В процессе реакции интенсивно красный цвет [CpNiCO]2 меняется на желто-коричневый. В результате из реакционной смеси удалось выделить германий-никелевый комплекс IV в виде желтого кристаллического вещества.
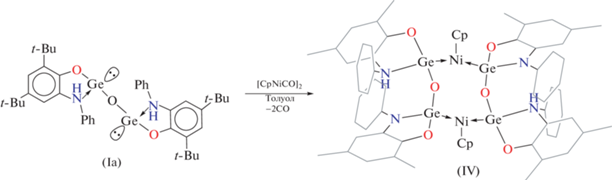
Схема 2 .
Молекулярная структура IV приведена на рис. 3 и представляет собой димерный гетеробиметаллический комплекс никеля(II) и германия(II), в котором каждый из атомов никеля связан с одним циклопентадиенильным лигандом и двумя дигермиленоксидными анионными лигандами. В данной реакции ион Ni+ (схема 2 ) восстанавливает один из аминофенолятных протонов дигермилена Ia, окисляясь при этом до иона Ni2+. Как следствие, один из аминофенолятных фрагментов переходит в амидофенолятное состояние, в то время как второй германиевый центр дигермиленоксида остается неизменным. Формальные КЧGe(II) и КЧNi(II) в IV равны 4 и 5 соответственно.
Рис. 3.
Молекулярная структура комплекса (CpNi)2[PhAPGeOGePhAmP]2 (IV). Тепловые эллипсоиды избранных атомов приведены с 50%-ной вероятностью. Атомы водорода не изображены для ясности.
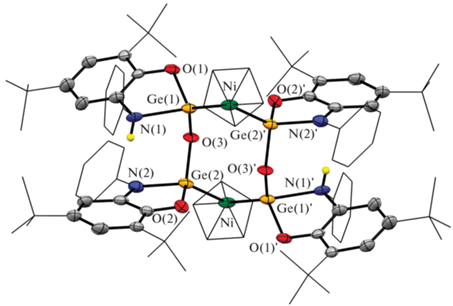
В образованном восьмичленном металлоцикле, имеющем твист-конформацию, оба атома низковалентного германия(II) связаны с катионными центрами никеля за счет донорно-акцепторного взаимодействия. Расстояния Ni–Ge в IV сопоставимы между собой: длина связи Ge(2) → Ni, формируемой анионным гермиленом с депротонированным органическим лигандом, равна 2.2129(9) Å и лишь немногим короче расстояния Ge(1)–Ni (2.2229(9) Å). Таким образом, распределение длин связей Ni–Ge в металлоцикле свидетельствует о делокализации электронной плотности по фрагменту GeNiGe. Важно отметить, что длины связей Ge–Ni в IV заметно короче величин, наблюдаемых для известных структур, содержащих фрагмент Ge–Ni–Cp (~2.3 Å) [60, 61]. При этом они существенно длиннее донорно-акцепторной связи (2.08 Å), образованной в ходе взаимодействия циклопентидиенилникелевого центра с N,N-гетероциклическим диамидогермиленом [62]. Наблюдаемое различие обусловлено более высоким значением КЧGe в IV по сравнению с комплексом никеля [62].
Между двумя германиевыми фрагментами в IV четко видна существенная разница в геометрии (сравнительный анализ длин связей в этих фрагментах приведен в табл. 3). В то время как в металлоцикле Ge(1)O(1)C(1)C(2)N(1) протонированного аминофенолятного фрагмента PhAmP наблюдается перегиб по линии O…N (двугранный угол между двумя плоскостями O(1)Ge(1)N(1) и O(1)C(1)C(2)N(1) равен 26.1(2)°), металлоцикл в амидофенолятном фрагменте практически плоский (6.6(2)°). Кроме того, протонированный атом азота N(1) имеет характерное тетраэдрическое окружение по сравнению с амидофенолятным N(2). Координационная связь Ge(1)–N(1) (2.124(5) Å) заметно длиннее ковалентной Ge(2)–N(2) (1.887(4) Å). При этом расстояния Ge–O практически равны между собой (1.834(3), 1.837(3) Å) и значительно превышают аналогичные расстояния в тетраэдрическом бис-амидофеноляте германия(IV) [55]. Все это подтверждает сохранение низкой степени окисления у атомов германия в IV.
Таблица 3.
Избранные длины связей в амидофенолятных (PhAP) и аминофенолятных (PhAmP) фрагментах IV
| Связь | PhAP | PhAmP |
|---|---|---|
| d, Å | ||
| Ge–N | 1.887(4) | 2.124(5) |
| Ge–O | 1.837(3) | 1.838(3) |
| C–O | 1.364(6) | 1.368(6) |
| C–N | 1.409(7) | 1.473(7) |
| Ge–Ni | 2.2289(9) | 2.2129(9) |
| Ge–Oоксид | 1.787(3) | 1.745(3) |
Каждый из атомов никеля в IV имеет искаженное плоско-тригональное координационное окружение; в вершинах располагаются атомы германия и центроид циклопентадиенильного лиганда. Отклонение Ni(II) от плоскости Ge(1)CpцентрGe(2) составляет 0.34 Å. Расстояние между атомами никеля в IV равно 6.036(2) Å, что исключает наличие валентных взаимодействий между ними. Длина связи Ni–Cpцентр равна 1.714(2) Å и значительно короче аналогичных расстояний в никелоцене (2.14–2.18 Å).
Гетерометаллический комплекс IV имеет хорошо разрешенный спектр ЯМР 1Н, где протоны циклопентадиенильных групп выходят синглетом при 4.99 м.д., спектр 13С получить не удалось в виду недостаточной растворимости комплекса IV.
В ходе исследования было установлено, что соединения II и III могут подвергаться одноэлектронному окислению трифлатом серебра по амидофенолятному редокс-активному лиганду с образованием парамагнитных о-иминосемихиноновых производных Ge(IV) (схема 3 ).
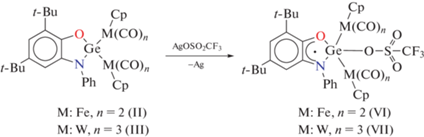
Схема 3 .
Взаимодействие сопровождается осаждением металлического серебра и изменением цвета реакционной смеси с темно-красного на интенсивно-фиолетовый. При этом в спектре ЭПР наблюдаются интенсивные сигналы, свидетельствующие об окислении амидофенолятного лиганда до парамагнитной формы (рис. 4). Сверхтонкая структура спектра (дублет (1 : 1) триплетов (1 : 1 : 1)) обусловлена сверхтонким взаимодействием неспаренного электрона с одним протоном и одним атомом азота о-иминосемихинонового лиганда. Однако интенсивно окрашенная реакционная смесь достаточно быстро теряет окраску и парамагнитные биметаллические производные разлагаются в растворе. Выделить их в индивидуальном состоянии не представилось возможным. При этом в случае соединения вольфрама VII даже на первых этапах реакции наблюдаются примесные сигналы в спектре ЭПР, что свидетельствует о его большей лабильности в сравнении с менее стерически-загруженным комплексом VI.
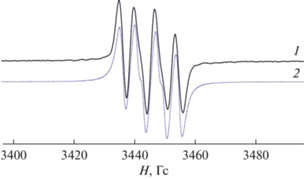
Рис. 4.
Экспериментальный (1) и симулированный (2) изотропный спектр ЭПР соединения VI в толуоле. Параметры спектра: ai(H) = 4.7 Гс, ai(14N) = 6.9 Гс, gi = 2.0032.
Производные германия(II) и олова(II) на основе редокс-активных лигандов способны вступать в окислительно-восстановительные реакции как с участием органического фрагмента, так и низковалентного центра тетрилена [57, 58]. Ожидалось, что окислительное присоединение 3,6-ди-трет-бутил-о-бензохинона к низковалентному металлоцентру гермилена I позволит получить смешанолигандное производное четырехвалентного германия, содержащее два типа редокс-активных дианионов – катехолатного и о-амидофенолятного. Реакция протекает в растворе тетрагидрофурана со скоростью смешения реагентов (cхема 4). Однако кристаллизацией из реакционной cмеси удалось выделить лишь симметричный бис-тетрагидрофуранат бис(3,6-ди-трет-бутил-катехолато)германия(IV) (V). Структурные и спектральные характеристики соединения V соответствуют опубликованным ранее [49]. Это свидетельствует о симметризации в растворе промежуточного несимметричного производного с образованием соединений VIII и V.
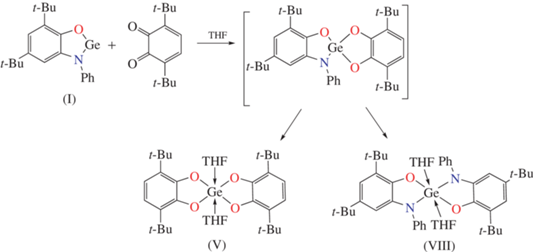
Схема 4 .
Таким образом, новые гетерометаллические комплексы со связью Ge–M (М = Fe, W, Ni) были получены в ходе реакции внедрения по связи М–М в димерах циклопентадиенилкарбонилов переходных металлов низковалентного производного германия(II), содержащего 3,5-ди-трет-бутил-N-фенил-о-амидофенолятный лиганд. Одноэлектронное окисление новых органобиметаллических соединений трифлатом серебра дает нестабильные парамагнитные о-иминосемихинолятные производные германия(IV). При двухэлектронном окислении гермилена о-хиноном образуется неустойчивый смешанолигандный интермедиат, который претерпевает симметризацию.
Авторы заявляют, что у них нет конфликта интересов.
Список литературы
Абакумов Г.А., Пискунов А.В., Черкасов В.К. и др. // Успехи химии. 2018. Т. 87. № 5. С. 393.
Luca O.R., Crabtree R.H. // Chem. Soc. Rev. 2012. V. 42. № 4. P. 1440.
Bas de Bruin, Gualco P., Paul N.D. Ligand Design in Metal Chemistry: Reactivity and Catalysis. John Wiley & Sons, Ltd., 2016. P. 448.
Schulz S. // Chem. Eur. J. 2010. № 16. V. 22. P. 6416.
Poddel’sky A.I., Cherkasov V.K., Abakumov G.A. // C-oord. Chem. Rev. 2009. V. 253. P. 291.
Kaim W. // Inorg. Chem. 2011. V. 50. P. 9752.
Старикова A.A., Минкин В.И. // Успехи химии. 2018. Т. 87. С. 1049.
Фоменко И.С., Гущин A.Л. // Успехи химии. 2020. Т. 89. С. 966.
Dei A., Gatteschi D., Sangregorio C., Sorace L. // Acc. Chem. Res. 2004. V. 37. P. 827.
Markevtsev I.N, Monakhov M.P., Platonov V.V. et al. // J. Magn. Magn. Mater. 2006. V. 300. P. e407.
Sato O. // J. Photochem. Photobiol. 2004. V. 5. P. 203.
Abakumov G.A., Cherkasov V.K., Nevodchikov V.I. et al. // Inorg. Chem. 2001. V. 40. P. 2434.
Pierpont C.G. // Coord. Chem. Rev. 2001. V. 216−217. P. 99.
Абакумов Г.A. // Журн. Всесоюз. хим. об-ва. им. Д.И. Менделеева. 1979. Т. 17. С. 156.
Kozhanov K.A., Bubnov M.P., Cherkasov V.K. et al. // Dalton Trans. 2004. P. 2957.
Пискунов А.В., Цыс К.В., Чегерев М.Г., Черкасов А.В. // Коорд. химия. 2019. Т. 45. № 9. С. 527 (Piskunov A.V., Tsys K.V., Cherkasov A.V., Chegerev M.G. // Russ. J. C-oord. Chem. 2019. V. 45. № 9. P. 626). https://doi.org/10.1134/S0132344X19090068
Meshcheryakova I.N., Arsenyeva K.V., Fukin G.K. et al. // Mendeleev Commun. 2020. V. 30. № 5. P. 592.
Bubnov M.P., Kozhanov K.A., Skorodumova N.A. et al. // J. Mol. Struct. 2019. V. 1180. P. 878.
Kozhanov K.A., Bubnov M.P., Teplova I.A. et al. // J. Mol. Struct. 2017. V. 1147. P. 541.
Kozhanov K.A., Bubnov M.P., Abakumov G.A., Cherkasov V.K. // J. Magn. Reson. 2012. V. 225. P. 62.
Bubnov, M.P., Teplova, I.A., Kozhanov, K.A. et al. // J. Magn. Reson. 2011. V. 209. № 2. P. 149.
Ершова И., Пискунов A. // Коорд. химия. 2020. Т. 46. С. 132 (Ershova I.V., Piskunov A.V. // Russ. J. Coord. Chem. 2020. V. 46. P. 154). https://doi.org/10.1134/S1070328420030021
Чегерев M., Пискунов A. // Коорд. химия. 2018. Т. 44. С. 109 (Chegerev M.G., Piskunov A.V. // Russ. J. Coord. Chem. 2018. V. 44. P. 258). https://doi.org/10.1134/S1070328418040036
Broere D.L., Plessius R., van der Vlugt J.I. // Chem. Soc. Rev. 2015. V. 44. P. 6886.
Ершова И.В., Пискунов A.В., Черкасов В.K. // Успехи химии. 2020. Т. 89. С. 1157.
Dange D., Choong S.L., Schenk C. et al. // Dalton Trans. 2012. V. 41. P. 9304.
Protchenko A.V., Saleh L.M.A., Vidovic D. et al. // Chem. Commun. 2010. V. 46. P. 8546.
Федюшкин И.Л., Соколов В.Г., Макаров В.М. и др. // Изв. АН. Сер. хим. 2016. № 6. С. 1495.
Fedushkin I.L., Lukoyanov A.N., Tishkina A.N. et al. // Organometallics. 2011. V. 30. P. 3628.
Fedushkin I.L., Sokolov V.G., Piskunov A.V. et al. // Chem. Commun. 2014. V. 50. P. 10108.
Jones C., Mills D.P., Rose R.P., Stasch A., Woodul W.D. // J. Organomet. Chem. 2010. V. 695. P. 2410.
Bonello O., Jones C., Stasch A., Woodul W.D. // Organometallics. 2010. V. 29. P. 4914.
Gendy C., Mansikkamaki A., Valjus J. et al. // Angew. Chem., Int. Ed. 2019. V. 58. P. 154.
Protchenko A.V., Dange D., Schwarz A.D. et al. // Chem. Commun. 2014. V. 50. P. 3841.
Piskunov A.V., Lado A.V. Ilyakina E.V. et al. // J. Organomet. Chem. 2008. V. 693. № 1. P. 128.
Klosener J., Wiesemann M., Niemann M. et al. // Chem. Eur. J. 2018. V. 24. P. 4412.
Veith M., Stahl L., Huch V. // Organometallics. 1993. V. 12. № 5. P. 1914.
Neto J.L., de Lima G.M., Porto A.O. et al. //J. Mol. Struct. 2006. V. 782. № 2–3. P. 110.
Jones C., Stasch A., Woodul W.D. // Chem. Commun. 2009. V. 1. P. 113.
Sanden T., Gamer M.T., Fagin A.A. et al. // Organometallics. 2012. V. 31. № 11. P. 4331.
Arnold P.L., Liddle S.T., McMaster J. et al. // J. Am. Chem. Soc. 2007. V. 129. № 17. P. 5360.
Hsueh-Ju Liu, Ziegler M.S., Don Tilley T. // Angew. Chem. Int. Ed. 2015. № 54. P. 6622.
Arsenyeva K.V., Ershova I.V., Chegerev M.G. et al. // J. Organomet. Chem. 2020. V. 927. 121524.
Chegerev M.G., Piskunov A.V., Tsys K.V. et al. // Eur. J. Inorg. Chem. 2019. P. 875.
Гордон А., Форд Р. Спутник химика. М.: Мир, 1976. P. 543.
Arsenyeva K.V., Chegerev M.G., Cherkasov A.V. et al. // Mendeleev Commun. 2021. V. 31. P. 330.
Руководство по неорганическому синтезу / Под ред. Брауэра Г. М.: Мир, 1986. Т. 6. С. 1994.
Piper T.S., Wilkinson G. // J. Inorg. Nucl. Chem. 1956. V. 3. P. 104.
Ладо A.В., Пискунов A.В., Жданович И.В. и др. // Коорд. химия. 2008. Т. 34. С. 251 (Lado A.V., Piskunov A.V., Zhdanovich I.V. et al. // Russ. J. Coord. Chem. 2008. Т. 34. № 4. С. 251). https://doi.org/10.1134/S1070328408040027
Smart. APEX3. Madison (WI, USA): Bruker AXS Inc., 2018.
Krause L., Herbst-Irmer R., Sheldrick G.M., Stalke D. // J. Appl. Cryst. 2015. V. 48. P. 3.
Sheldrick G.M. // Acta Crystallogr. C. 2015. V. 71. P. 3.
Sheldrick G.M. // Acta Crystallogr. A. 2015. V. 71. P. 3.
Holt M.S., Wilson W.L., Nelson J.H. // Chem. Rev. 1989. V. 89. P. 11.
Piskunov A.V., Aivaz’yan I.A., Poddel’sky A.I. et al. // Eur. J. Inorg. Chem. 2008. V. 9. P. 1435.
Chegerev M.G., Piskunov A.V., Starikova A.A. et al. // Eur. J. Inorg. Chem. 2018. P. 1087.
Chegerev M.G., Piskunov A.V., Maleeva A.V. et al. // Eur. J. Inorg. Chem. 2016. P. 3813.
Tsys K.V., Chegerev M.G., Pashanova K.I. et al. // Inorg. Chim. Acta. 2019. V. 490. P. 220.
Filippou A.C., Winter J.G., Kociok-Köhn G., Hinz I. // Dalton Trans. 1998. V. 12. P. 2029.
Titova S.N., Bychkov V.T., Domrachev G.A. et al. // J. Organomet. Chem. 1980. V. 18. № 2. P. 167.
Pankratov L.V., Nevodchikov V.I., Zakharov L.N. et al. // J. Organomet. Chem. 1992. V. 429. № 1. P. 13.
Veith M., Stahl L. // Angew. Chem. Int. Ed. 1993. V. 32. № 1. P. 106.
Дополнительные материалы отсутствуют.
Инструменты
Координационная химия