

Координационная химия, 2022, T. 48, № 6, стр. 334-342
Спиновое состояние комплекса кобальта(II) с N,N'-дизамещенным 2,6-бис(пиразол-3-ил)пиридином
Е. А. Хакина 1, Г. Л. Денисов 1, И. А. Никовский 1, А. В. Полежаев 1, 2, Ю. В. Нелюбина 1, 2, *
1 Институт элементоорганических соединений им. А.Н. Несмеянова РАН
Москва, Россия
2 Московский государственный технический университет им. Баумана
Москва, Россия
* E-mail: unelya@ineos.ac.ru
Поступила в редакцию 12.11.2021
После доработки 29.11.2021
Принята к публикации 30.11.2021
- EDN: YBIYFB
- DOI: 10.31857/S0132344X22060020
Аннотация
При взаимодействии нового N,N'-дизамещенного 2,6-бис(пиразол-3-ил)пиридинового лиганда (L) с Co(ClO4)2 · 6H2O получен комплекс кобальта(II) [Co(L)2](ClO4)2 (I), охарактеризованный методами элементного анализа, масс-спектрометрии, спектроскопии ЯМР и рентгеновской дифракции (CIF file CCDC № 2121030). Согласно данным методов Эванса, спектроскопии ЯМР и анализа температурной зависимости спектров ЯМР, позволяющих определять спиновое состояние парамагнитных соединений в растворе, ион кобальта(II) в комплексе I находится в высокоспиновом состоянии и не претерпевает температурно-индуцированного спинового перехода в диапазоне температур 235–345 К.
Некоторые комплексы переходных металлов с 3d4–3d7 электронной конфигурацией [1] могут существовать в двух спиновых состояниях – низкоспиновом (НС) и высокоспиновом (ВС) – и переключаться между ними под действием внешних возмущений, таких как изменение температуры [2], давления [3], приложение магнитного поля [4] или другие физические воздействия [5]. Различия в магнитных, диэлектрических, оптических и других свойствах [6] таких соединений в разных спиновых состояниях лежат в основе их использования для создания устройств сверхплотного хранения информации [7, 8], молекулярных переключателей и других устройств [4]. Чаще всего способность к переключению между НС и ВС состояниями (так называемый спиновый переход) встречается у комплексов железа(II) и кобальта(II) в (псевдо)октаэдрическом окружении азотсодержащих гетероциклических лигандов [1]. Одним из наиболее изученных классов подобных лигандов являются 2,6-ди(пиразол-1-ил)пиридины [9], что связано с простотой их химической модификации и синтетической доступностью [10]. Эти особенности, например, позволили обнаружить четкую зависимость между природой заместителей в различных положениях 2,6-ди(пиразол-1-ил)пиридина и температурой спинового перехода в соответствующих комплексах железа(II) [11].
Отсутствие аналогичной зависимости для изомерных лигандов – 2,6-ди(пиразол-3-ил)пиридинов – обусловлено наличием в первом положении пиразолильного кольца NH-групп, способных образовывать водородные связи с молекулами растворителя и/или противоионами [12]. Появление таких водородных связей вблизи атомов азота, координированных к иону металла, непредсказуемым образом влияет на его спиновое состояние [13]. Однако до недавнего времени все попытки введения заместителей в это положение 2,6-ди(пиразол-3-ил)пиридинов приводили к комплексам металлов, находящимся исключительно в ВС-состоянии [13], что не позволяло осуществлять молекулярный дизайн соединений со спиновыми переходами на основе данного класса N-гетероциклических лигандов по аналогии с изомерными 2,6-ди(пиразол-1-ил)пиридинами.
Недавно мы предложили такой дизайн N-заместителя – орто-замещенной арильной группы, который не только не препятствует протеканию спинового перехода в комплексах железа(II) [14] и кобальта(II) [15], но и позволяет управлять его температурой введением заместителей в другие положения 2,6-ди(пиразол-3-ил)пиридинового лиганда [16, 17].
В настоящей работе мы синтезировали новый представитель данного ряда N,N'-дизамещенных лигандов – 2,6-бис(1-(2,6-дихлорофенил)-5-метокси-1H-пиразол-3-ил)пиридин (L) – и комплекс кобальта(II) на его основе [Co(L)2](ClO4)2 (I) (cхема 1). Спиновое состояние полученного комплекса изучено при помощи рентгеноструктурного анализа и спектроскопии ЯМР, традиционно используемой для поиска новых соединений с температурно-индуцированным спиновым переходом [13].
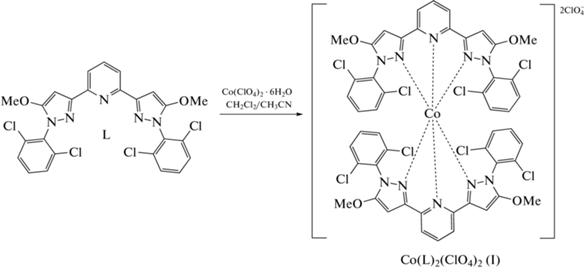
Схема 1 .
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Все операции, связанные с синтезом лиганда L и его комплекса, выполняли на воздухе с использованием коммерчески доступных органических растворителей, перегнанных в атмосфере аргона. Гексагидрат перхлората кобальта Co(ClO4)2 · 6H2O (Sigma-Aldrich) использовали без дополнительной очистки. 3,3'-(Пиридин-2,6-диил)бис(1-(2,6-дихлорфенил)-1H-пиразол-5-ол), используемый в качестве предшественника для синтеза лиганда L, получали по методике [14]. Анализ на углерод, азот и водород проводили на микроанализаторе Carlo Erba, модель 1106.
Синтез 2,6-бис(1-(2,6-дихлорофенил)-5-метокси-1H-пиразол-3-ил)пиридина (L). В колбе емкостью 50 мл растворяли 3,3'-(пиридин-2,6-диил)бис(1-(2,6-дихлорфенил)-1H-пиразол-5-ол) (0.5 г, 0.937 ммоль) в 20 мл ДМФА и к раствору добавляли карбонат цезия (0.763 г, 2.343 ммоль) и диметилсульфат Me2SO4 (186 мкл, 1.969 ммоль). Полученную суспензию перемешивали при 70°C в течение 8 ч. Затем реакционную смесь охлаждали до комнатной температуры и выливали в 70 мл дистиллированной воды. Образовавшийся осадок отделяли фильтрованием, промывали водой и сушили в высоком вакууме. Полученный продукт использовали без дополнительной очистки. Выход 448 мг (85%).
ЯМР 1H (ДМСО-d6; 400 MГц; δ, м.д.): 3.97 (c., 6H, OMe), 6.59 (с., 2H, Pz–CH), 7.59 (т., 2H, 3JHH= = 8.0 Гц, п-Ph), 7.70 (д., 4H, 3JHH= 8.0 Гц, м-Ph), 7.83 (с., 3H, п-Py + м-Py). ЯМР 13C (ДМСО-d6; 101 MГц; δ, м.д.): 59.97 (c., Me), 84.13 (c., 4-Pz), 118.80 (c., 3-Py), 129.49 (c., 4-Ph), 132.74 (c., 3-Ph), 133.25 (c., 2-Ph), 134.74 (c., 1-Ph), 137.89 (c., 4-Py), 151.47 (c., 3-Pz), 152.40 (c., 5-Pz), 157.32 (c., 2-Py). HR-MS (ESI+). m/z: [C25H17Cl4N5O2]+, рассчитано 582.0029; найдено 582.0014.
Синтез [Co(L)2](ClO4)2 (I). К раствору 0.0112 г (0.02 ммоль) лиганда L в смеси 0.3 мл хлористого метилена и 0.1 мл ацетонитрила добавляли раствор Co(ClO4)2 · 6H2O (0.0037 г, 0.01 ммоль) в 0.1 мл ацетонитрила. Полученную смесь перемешивали при комнатной температуре в течение 1 ч, затем хлористый метилен отгоняли с помощью ротационного испарителя. Остаток после отгонки выдерживали в течение 12 ч при температуре –18°C. Образовавшийся кристаллический осадок отделяли от жидкой фазы декантированием и высушивали на воздухе. Выход 11 мг (80%).
Масс-спектр (ESI), m/z: [Co(L)2]2+, рассчитано 590.5, найдено 590.8; [Co(L)2(ClO4)]+, рассчитано 1279.9, найдено 1279.9. ЯМР 1H (ацетонитрил-d3; 300 МГц; 292 K; δ, м.д.): –3.02 (уш.с., 2H, п-Py), 1.47 (уш.с., 8H, м-Ph), 2.06 (уш.с., 4H, п-Ph), 13.88 (уш.с., 12H, Me), 19.21 (уш.с., 4H, Pz), 56.66 (уш.с., 4H, м-Py).
РCA монокристаллов комплекса I, полученных методом медленного испарения растворителя (смеси ацетонитрила и хлористого метилена в соотношении 2 : 1 по объему) на воздухе, проведен на дифрактометре Bruker APEX2 DUO CCD (MoKα-излучение, графитовый монохроматор, ω-сканирование). Структура расшифрована с использованием программы ShelXT [18] и уточнена в полноматричном МНК с помощью программы Olex2 [19] в анизотропном приближении по $F_{{hkl}}^{2}.$ Положения атомов водорода рассчитаны геометрически и уточнены в изотропном приближении по модели наездника. Разупорядоченные молекулы растворителя (ацетонитрила) описаны в виде диффузного вклада в общее рассеяние с помощью опции Solvent Mask программы Olex2 [19]. Основные кристаллографические данные структуры I представлены в табл. 1.
Таблица 1.
Кристаллографические данные, параметры эксперимента и уточнения структуры I
| Параметр | Значение |
|---|---|
| Брутто формула | C50H34N10O12Cl10Co |
| М | 1380.30 |
| T, K | 120 |
| Сингония | Моноклинная |
| Пр. группа | P21/c |
| Z | 4 |
| a, Å | 22.2478(14) |
| b, Å | 24.8150(16) |
| c, Å | 22.6356(16) |
| α, град | 90 |
| β, град | 149.147(2) |
| γ, град | 90 |
| V, Å3 | 6409(8) |
| ρ(выч.), г см‒3 | 1.431 |
| μ, см−1 | 7.47 |
| F(000) | 2788 |
| 2θmax, град | 56 |
| Число измеренных отражений | 73 821 |
| Число независимых отражений | 15 457 |
| Число отражений с I > 3σ(I) | 11 423 |
| Количество уточняемых параметров | 772 |
| R1 | 0.0598 |
| wR2 | 0.1836 |
| GOОF | 1.036 |
| Остаточная электронная плотность (max/min), e Å‒3 | 1.336/‒1.664 |
Координаты атомов и другие параметры структуры I депонированы в Кембриджском банке структурных данных (CCDC № 2121030; http://www.ccdc.cam.ac.uk/).
Масс-спектрометрический анализ комплекса I выполняли с использованием жидкостного хромато-масс-спектрометра модели LCMS-2020 (Шимадзу, Япония) с ионизацией электрораспылением и квадрупольным детектором (регистрация положительных и отрицательных ионов с m/z в диапазоне 50–2000). Температуры линии десольватирования и нагревательного блока составляли 250 и 400°C соответственно. В качестве распылительного и осушающего газа использовали азот (99.5%), в качестве подвижной фазы – ацетонитрил (99.9+%, Chem-Lab) со скоростью потока 0.4 мл/мин. Объeм анализируемой пробы – 3 мкл (концентрация 0.2 мг/мл, растворитель – ацетонитрил).
Спектры ЯМР 1H и 13С регистрировали в ацетонитриле-d3 и ДМСО-d6 на спектрометрах Bruker Avance 300 и 400 с рабочими частотами для протонов 300.15 и 400 МГц соответственно. Значения химических сдвигов (δ, м.д.) в спектрах определяли относительно остаточного сигнала растворителя (1Н – 1.94 м.д. для ацетонитрила-d3, 1Н – 2.5 и 13С 39.52 м.д. для ДМСО-d6) или сигнала 1% примеси Me4Si (1H – 0.0 м.д.). Спектры ЯМР 1Н комплекса I регистрировали с использованием следующих параметров: диапазон спектра – 250 м.д., время регистрации – 0.2 с, длительность релаксационной задержки – 0.6 с, длительность импульса – 9.5 мкс, количество накоплений – 64. Полученные спады свободной индукции для повышения соотношения сигнал/шум обрабатывали при помощи экспоненциального взвешивания с коэффициентом до 3.
Температурную зависимость магнитной восприимчивости комплекса I в ацетонитриле-d3 оценивали с помощью метода Эванса [20] в интервале температур 235–345 К с использованием ампулы для спектроскопии ЯМР с коаксиальной вставкой. Внутреннюю (контрольную) ампулу заполняли ацетонитрилом-d3 с добавлением 1% Me4Si, а внешнюю ампулу – раствором парамагнитного комплекса (5.1 мг/см3) в ацетонитриле-d3 с той же концентрацией Me4Si. Молярную магнитную восприимчивость рассчитывали по разнице между химическим сдвигом Me4Si в чистом ацетонитриле-d3 и его сдвигом в растворе комплекса (Δδ в Гц) в ацетонитриле-d3 с использованием следующего уравнения:
где M – молекулярная масса комплекса, г/моль; ν0 – частота спектрометра, Гц; Sf – коэффициент формы магнита (4π/3); c – концентрация комплекса, г/см3; $\chi _{M}^{{{\text{dia}}}}$ – молярный диамагнитный вклад в парамагнитную восприимчивость, рассчитанный с использованием констант Паскаля [21]. Концентрацию с пересчитывали для каждой температуры в соответствии с изменением плотности растворителя ρ: cT = msρ/msol, где msρ – масса комплекса, msol – масса раствора.РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Для синтеза 2,6-бис(1-(2,6-дихлорофенил)-5-метокси-1H-пиразол-3-ил)пиридина (L) (схема 1 ) исходный 3,3'-(пиридин-2,6-диил)бис(1-(2,6-дихлорфенил)-1H-пиразол-5-ол), полученный по методике [14], вводили в реакцию метилирования диметилсульфатом в присутствии карбоната цезия (схема 2 ).

Схема 2 .
В связи с низкой растворимостью лиганда L в чистом ацетонитриле в качестве растворителя для проведения последующей реакции с гексагидратом перхлората кобальта(II) использовали смесь хлористого метилена и ацетонитрила в объемном соотношении 3 : 2. В результате получили гомолептический комплекс [Co(L)2](ClO4)2 (I) (cхема 1), охарактеризованный при помощи масс-спектрометрии (рис. 1), спектроскопии ЯМР (рис. 2) и РСА монокристалла (рис. 3). Последний, в частности, позволил определить спиновое состояние иона кобальта(II), который оказался высокоспиновым при температуре 120 К. При этом расстояния Co–N с атомами азота двух бис(пиразол-3-ил)пиридиновых лигандов (КЧ 6) (табл. 2) характерны для комплексов кобальта(II) в ВС-состоянии (2.0–2.2 Å [1]).

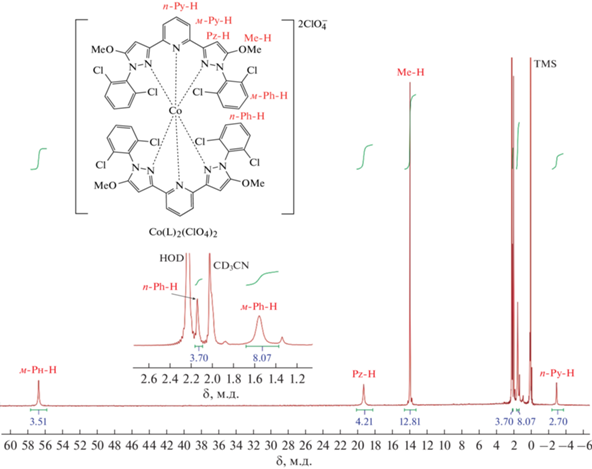
Рис. 3.
Общий вид комплекса I. Здесь и далее перхлорат-анионы и атомы водорода не показаны, остальные атомы представлены в виде эллипсоидов тепловых колебаний (p = 30%). Нумерация приведена только для иона металла и избранных гетероатомов.

Таблица 2.
Основные геометрические параметры для комплекса I по данным РСА при 120 К*
| Параметр | I |
|---|---|
| Co–N(Py), Å | 2.025(3)/2.030(3) |
| Co–N(Pz), Å | 2.107(3)–2.131(3) |
| θ, град | 89.39(3) |
| N(Py)CoN(Py), град | 178.56(11) |
| S(TP-6) | 11.244 |
| S(OC-6) | 3.325 |
* θ – двугранный угол между среднеквадратичными плоскостями 2,6-бис(пиразол-3-ил)пиридиновых лигандов, атомы N(Py) и N(Pz) соответствуют атомам азота пиридинового и пиразол-3-ильного фрагментов. S(TP-6) и S(OC-6) – отклонения формы полиэдра CoN6 от идеальной тригональной призмы (TP-6) и идеального октаэдра (OC-6) соответственно.
Согласно данным РСА, координационное окружение иона кобальта(II) в комплексе I близко к октаэдрическому (рис. 4). Так, значения угла θ между среднеквадратичными плоскостями двух лигандов и угла N(Py)CoN(Py), равные 90° и 180° в случае идеального октаэдра, составляют 89.39(3)° и 178.56(11)°. Более точно форму координационного полиэдра CoN6 характеризуют так называемые “меры симметрии” [22], описывающие ее отклонение от идеального октаэдра (OC-6). Чем эти значения меньше, тем лучше форма полиэдра описывается соответствующим многогранником. В комплексе I величина октаэдрической “меры симметрии” S(OC-6), оцененная на основе рентгенодифракционных данных при помощи программы Shape 2.1 [22], составляет 3.325 (табл. 2). Для сравнения “мера симметрии”, характеризующая отклонение от еще одного идеального полиэдра с шестью вершинами – тригональной призмы (TP-6), традиционно встречающейся у ВС-комплексов железа(II) с N-гетероциклическими лигандами [23] – принимает заметно более высокое значение 11.244 (табл. 2).
Рис. 4.
Проекция комплекса I в перпендикулярном направлении, иллюстрирующая близкую к октаэдрической форму координационного полиэдра иона кобальта(II). Штрих-пунктиром обозначены внутримолекулярные галогенные связи.

Незначительное отклонение формы координационного полиэдра иона кобальта(II) в комплексе I от идеального октаэдра, в первую очередь, вызвано “жесткостью” бис(пиразол-3-ил)пиридинового лиганда [24]. Например, аналогичные значения “мер симметрии” ранее наблюдались для ВС-комплексов кобальта(II) с другими бис(пиразол-3-ил)пиридинами [25, 26]. Образованию подобной жесткой структуры способствуют внутримолекулярные стекинг-взаимодействия между ароматическими фрагментами пиридина и N-арильных заместителей с углом между ними в 2.68(13)°–5.17(12)° и расстоянием между их центроидами в 3.641(2)–3.725(2) Å. Кроме того, атомы хлора в орто-положениях указанных заместителей попарно участвуют в образовании внутримолекулярных галогенных связей (Cl…Cl 3.1912(17)–3.2865(15) Å, CClCl 133.18(14)°–140.47(13)°) [27].
Атомы хлора Cl(2A) и Cl(4A) также образуют межмолекулярную галогенную связь (Cl…Cl 3.3460(13) Å, CClCl 153.27(13)°), объединяющую катионы [Co(L)2]2+ в бесконечные цепочки вдоль диагонали кристаллографической плоскости ac (pис. 5). Эти цепочки связаны между собой в плотный трехмерный каркас более слабыми взаимодействиями, включая галогенные связи Cl(1)…Cl(1A) и Cl(4)…Cl(3A) (Cl…Cl 3.5236(14), 4.756(2) Å, CClCl 159.71(12)°, 168.51(14)°) и контакты C–H…Cl (C…Cl 3.676(4) Å, CHCl 145.0(2)°) между соседними катионами [Co(L)2]2+, а также C–H…O контакты с окружающими их перхлорат-анионами (C…O 3.189(10)–3.432(3) Å, CHO 133.4(3)°–165.2(3)°).
Таким образом, ион кобальта(II) в комплексе I находится в ВС-состоянии в кристалле при 120 K, как однозначно следует из данных его РСА при этой температуре. Поскольку эффекты кристаллической упаковки в виде описанных выше межмолекулярных взаимодействий могут в некоторых случаях приводить к отсутствию спинового перехода [23, 28], спиновое состояние данного комплекса было исследовано в растворе при помощи традиционно используемого для этих целей метода Эванса спектроскопии ЯМР [13]. Для проведения такого эксперимента в ампулу для спектроскопии ЯМР, содержащую раствор парамагнитного комплекса и стандартного соединения (например, тетраметилсилана (ТМС)) в известной концентрации, помещают специальную коаксиальную вставку, содержащую раствор ТМС в том же растворителе. Разница в значениях химического сдвига ТМС в спектре ЯМР, зарегистрированном от этих двух растворов одновременно, позволяет рассчитать магнитную восприимчивость раствора исследуемого парамагнитного соединения. Тем самым можно однозначно установить спиновое состояние иона металла и его возможное изменение с температурой при регистрации спектров при разных температурах.
Рис. 5.
Фрагмент кристаллической упаковки для комплекса I, иллюстрирующий образование в кристалле бесконечных цепочек за счет галогенных связей (обозначены штрих-пунктиром).

Рис. 6.
Температурная зависимость магнитной восприимчивости комплекса I в растворе ацетонитрила-d3 по данным спектроскопии ЯМР (метода Эванса).
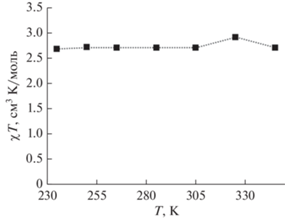
Рис. 7.
Спектры ЯМР 1Н, зарегистрированные при различных температурах для раствора комплекса I в ацетонитриле-d3.
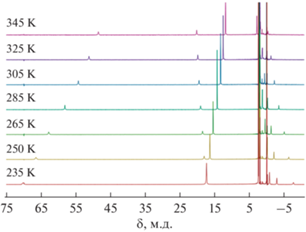

Для раствора комплекса I в ацетонитриле-d3 рассчитанное таким образом значение χТ (pис. 6) лишь незначительно отклоняется от величины 2.7 см3 моль–1 K, соответствующей иону кобальта(II) в ВС-состоянии (S = 3/2) во всем исследованном диапазоне температур 235–345 K, доступных для выбранного растворителя.
Данные о ВС-состоянии иона кобальта(II) в комплексе I, полученные с помощью метода Эванса, дополнительно подтверждаются линейным характером зависимости химических сдвигов сигналов комплекса в спектрах ЯМР 1Н (зарегистрированных при разных температурах) от обратной температуры (pис. 7, 8). Так, наблюдаемое значение химических сдвигов δн ядер в спектре ЯМР 1Н для соединений, которые могут существовать в двух спиновых состояниях, является средневзвешенным значением сдвигов этих же ядер для чистых НС- и ВС-состояний (ηнс и ηвс – заселенности состояний):
Для комплекса кобальта(II) с парамагнитным НС-состоянием наблюдаемый химический сдвиг (δн) каждого ядра можно представить в виде суммы диамагнитного (δдиа) и парамагнитного (δпар) вкладов [15]:
Диамагнитный вклад δдиа, который одинаков для НС- и ВС-состояний, приблизительно равен химическому сдвигу соответствующего ядра в свободном лиганде.
Для парамагнитных комплексов металлов, не претерпевающих температурно-индуцированный спиновый переход, парамагнитный химический сдвиг линейно зависит от обратной температуры (закон Кюри): $\delta _{{{\text{пар}}}}^{{{\text{вс}}}} = A + B{{T}^{{--1}}}.$ Напротив, спиновый переход приводит к заметным отклонениям химических сдвигов некоторых ядер от линейной зависимости [15]. Из полученных нами данных видно, что подобные отклонения не наблюдаются в случае комплекса I, что подтверждает отсутствие у него спинового перехода во всем исследованном диапазоне температур.
Таким образом, мы синтезировали и охарактеризовали новый комплекс кобальта(II) с N,N'-дизамещенным 2,6-бис(пиразол-3-ил)пиридином. Согласно результатам проведенного для него низкотемпературного рентгенодифракционного исследования (в первую очередь, длинам связей Со–N), ион кобальта(II) в указанном комплексе находится в ВС-состоянии даже при 120 К. Он также не претерпевает температурно-индуцируемого спинового перехода в растворе ацетонитрила в диапазоне температур 235–345 K, что подтверждается данными традиционно используемого для этой цели метода Эванса [13] и анализа температурной зависимости химических сдвигов в спектрах ЯМР [15]. Однако можно предположить, что замена иона кобальта(II) на ион железа(II), который в окружении 2,6-бис(пиразол-3-ил)пиридиновых лигандов с аналогичными N-арильными заместителями может менять свое спиновое состояние под действием температуры [14], позволит получить такой комплекс с температурно-индуцированным спиновым переходом.
Авторы заявляют, что у них нет конфликта интересов.
Список литературы
Spin-Crossover Materials: Properties and Applications / Ed. Halcrow M.A. Chichester (UK): John Wiley & Sons, Ltd., 2013.
Pavlov A.A., Aleshin D.Y., Nikovskiy I.A. et al. // Eur. J. Inorg. Chem. 2019. V. 2019. № 23. P. 2819.
Gütlich P., Ksenofontov V., Gaspar A.B. // Coord. Chem. Rev. 2005. V. 249. № 17. P. 1811.
Senthil Kumar K., Ruben M. // Coord. Chem. Rev. 2017. V. 346. P. 176.
Unruh D., Homenya P., Kumar M. et al. // Dalton Trans. 2016. V. 45. № 36. P. 14008.
Kahn O., Kröber J., Jay C. // Adv. Mater. 1992. V. 4. № 11. P. 718.
Ferrando-Soria J., Vallejo J., Castellano M. et al. // Coord. Chem. Rev. 2017. V. 339. P. 17.
Villalva J., Develioglu A., Montenegro-Pohlhammer N. et al. // Nat. Commun. 2021. V. 12. № 1. P. 1578.
Halcrow M.A. // Coord. Chem. Rev. 2005. V. 249. № 25. P. 2880.
Halcrow M.A. // Coord. Chem. Rev. 2009. V. 253. № 21. P. 2493.
Kershaw Cook L.J., Kulmaczewski R., Mohammed R. et al. // Angew. Chem. Int. Ed. 2016. V. 55. № 13. P. 4327.
Halcrow M.A. // Chem. Soc. Rev. 2011. V. 40. № 7. P. 4119.
Halcrow M.A. // Crystals. 2016. V. 6. № 5. P. 58.
Nikovskiy I., Polezhaev A.V., Novikov V.V. et al. // Chem. Eur. J. 2020. V. 26. P. 5629.
Pankratova Y., Aleshin D., Nikovskiy I. et al. // Inorg. Chem. 2020. V. 59. № 11. P. 7700.
Nikovskiy I.A., Polezhaev A.V., Novikov V.V. et al. // Crystals. 2021. V. 11. № 8. P. 922.
Melnikova E.K., Aleshin D.Y., Nikovskiy I.A. et al. // Crystals. 2020. V. 10. № 9. P. 793.
Sheldrick G.M. // Acta Crystallogr. A. 2008. V. 64. P. 112.
Dolomanov O.V., Bourhis L.J., Gildea R.J. et al. // J. A-ppl. Cryst. 2009. V. 42. P. 339.
Evans D.F. // J. Chem. Soc. 1959. P. 2003.
Bain G.A., Berry J.F. // J. Chem. Educ. 2008. V. 85. № 4. P. 532.
Alvarez S. // Chem. Rev. 2015. V. 115. P. 13447.
Kershaw Cook L., Mohammed R., Sherborne G. et al. // Coord. Chem. Rev. 2015. V. 289. P. 2.
Alvarez S. // J. Am. Chem. Soc. 2003. V. 125. № 22. P. 6795.
Pavlov A.A., Belov A.S., Savkina S.A. et al. // Russ. J. Coord. Chem. 2018. V. 44. № 8. P. 489. https://doi.org/10.1134/S1070328418080067
Pavlov A.A., Nikovskii I.A., Polezhaev A.V. et al. // Russ. J. Coord. Chem. 2019. V. 45. № 6. P. 402. https://doi.org/10.1134/S1070328419060046
Cavallo G., Metrangolo P., Milani R. et al. // Chem. Rev. 2016. V. 116. № 4. P. 2478.
Craig G.A., Costa J.S., Roubeau O. et al. // Chem. Eur. J. 2012. V. 18. № 37. P. 11703.
Дополнительные материалы отсутствуют.
Инструменты
Координационная химия