

Координационная химия, 2022, T. 48, № 7, стр. 404-413
Амиды олова на основе лигандов аценафтениминового ряда
В. А. Додонов 1, *, О. А. Кушнерова 1, Р. В. Румянцев 1, Е. А. Уливанова 1, А. Н. Лукоянов 1, Д. А. Разборов 1, И. Л. Федюшкин 1
1 Институт металлоорганической химии им. Г.А. Разуваева РАН
Нижний Новгород, Россия
* E-mail: dodonov@iomc.ras.ru
Поступила в редакцию 27.12.2021
После доработки 25.01.2022
Принята к публикации 31.01.2022
- EDN: OWISUQ
- DOI: 10.31857/S0132344X22070039
Аннотация
Реакция Dpp-Bian (Dpp-Bian = 1,2-бис[(2,6-диизопропилфенил)имино]аценафтен) с одним эквивалентом иода и избытком металлического олова приводит к парамагнитному комплексу [(Dpp-Bian)1–SnI] (I). Взаимодействие данного комплекса c одним эквивалентом н-бутиллития или трет-бутилата лития приводит к образованию [(Dpp-Bian)1–SnBu] (II) и [(Dpp-Bian)1–SnOtBu] (III) соответственно, быстро распадающиеся с образованием станнилена [(Dpp-Bian)2–Sn] (IV). Станилен IV проявляет восстановительные свойства по отношению к Dpp-Mian (Dpp-Mian = 2-моно[(2,6-диизопропилфенил)имино]аценафтен-1-он), образуя комплекс олова(IV) [{бис-(Dpp-Mian)}2Sn] (V). Восстановление дииминового лиганда с более акцепторными заместителями, CF3-Bian (CF3-Bian = 1,2-бис[(3,5-трифторметилфенил)имино]аценафтен), в отличие от Dpp-Bian, может протекать напрямую металлическим оловом и приводит к [${\text{(C}}{{{\text{F}}}_{{\text{3}}}}{\text{ - Bian}})_{2}^{{2--}}$(CF3-Bian)2–Sn] (VII). Новые соединения охарактеризованы методами ИК-, ЯМР-, ЭПР-спектроскопии, элементным анализом. Молекулярное строение комплексов I и VII установлено с помощью РСА (CIF files CCDC № 2130839 (I), 2130840 (VII)).
В последнее десятилетие в химии непереходных элементов произошел значительный прогресс [1–3]. В основном он связан с достижениями химии непереходных элементов в низковалентном состоянии [4, 5]. Обнаружена способность низковалентных производных главных подгрупп к активации малых молекул [6–8], окислительному присоединению/восстановительному элиминированию [9, 10], обратимым реакциям присоединения [11], что считалось до последнего времени невозможным для элементов главных подгрупп. Эти особенности реакционной способности комплексов низковалентных элементов главных подгрупп роднят их с комплексами переходных металлов, что открывает пути замены производных драгоценных металлов, используемых в промышленности, на более доступные производные элементов главных подгрупп.
Для производных 14 группы одно из низковалентных состояний – двухвалентное – является сравнительно стабильным. Низковалентные производные 14 группы обнаруживают необычную, нехарактерную для элементов главных подгрупп реакционную способность, которая скорее напоминает таковую у переходных металлов. Необычными производными, относящимися к группе низковалентных производных 14 группы, являются дистаннин ArSn≡SnAr [12], дигермин ArGe≡GeAr [13, 14], диплюмбин ArPb≡PbAr [15], (Ar = терфенил), стабильный диазадисиликациклобутановый бирадикалоид [16], а также низковалентные гидриды 14 группы. Все эти соединения способны активировать H2 [17–20], NH3 [21, 22], PH3, AsH3 [23, 24], S8 [25], олефины [26], алкины [27] и P4 [28] в мягких условиях. Низковалентные гидриды 14 группы, помимо способности к активации гетероалленов (CO2, CS2, RNCO) [29, 30], обнаруживают способность к катализу [31].
Один из подходов к регулированию реакционной способности низковалентных атомов элементов главных подгрупп заключается в использовании редокс-активных объемных лигандов, например 1,2-бис[(2,6-диизопропилфенил)имино]аценафтена (Dpp-Bian). Ранее были открыты реакции металлациклоприсоедиения изо(тио)цианатов к галлиевому [32, 33] и алюминиевому производному [34] Dpp-Bian. Проведены многочисленные реакции активации CO2 [35], SO2 [36], карбодиимидов [37] и других органических субстратов [38–41]. Важно отметить, что субстраты, находящиеся в активированном состоянии, могут быть вовлечены в дальнейшие превращения [35]. Еще одной особенностью лиганда Dpp-Bian является его способность хранить электроны и в нужный момент передавать их обратно на металл [42, 43]. Так, был реализован первый сольват-индуцированный перенос электрона [44] и лиганд-поддерживаемое двухэлектронное окислительное присоединение на непереходном металле [45]. Эти особенности позволили создать эффективные катализаторы гидроаминирования алкенов [46, 47] и полимеризации циклических эфиров [48, 49], превосходящие по характеристикам аналогичные комплексы переходных металлов.
Цель настоящей работы – получение низковалентных производных олова на основе Dpp-Bian и CF3-Bian.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Полученные соединения чувствительны к кислороду и влаге воздуха, поэтому все манипуляции по их синтезу, выделению и идентификации выполняли в вакууме с использованием техники Шленка. Растворители были осушены по известным методикам и отбирались конденсацией в вакууме непосредственно перед использованием [50].
ИК-спектры получали на спектрометре FSM-1201. Для их регистрации готовили суспензии соединений в вазелиновом масле. Спектры ЭПР получали на спектрометре Bruker ER 200 D-SRC, снабженном двойным резонатором ER 4105 DR (рабочая частота 9.5 ГГц) и термоконтроллером ER 4111 VT. В качестве стандарта при определении g-фактора использовали дифенилпикрилгидразил (ДФПГ, g = 2.0037). Соединения Dpp-Bian [51], CF3-Bian [52], [(Dpp-Bian)SnCl] [53] получали по опубликованным методикам. Остальные соединения получали из коммерческих источников и использовали без очистки. Спектры ЯМР регистрировали на Bruker DPX-200 (200 MГц), AvanceNEO300 (300 МГц) или Advance III (400 МГц) спектрометрах. Элементный анализ на C, H, N выполняли на приборе Elementar Variomicrocube. Компьютерное моделирование спектров ЭПР и расчет констант сверхтонкого взаимодействия проводили с применением WinEPR Simfonia и пакета EasySpin [54].
Синтез [(Dpp-Bian)SnI] (I). Суспензию Dpp-Bian 0.5 г (1.0 ммоль) в эфире (25 мл) приливали в ампулу с I2 (0.13 г, 0.5 ммоль) и избытком металлического олова (5 г). Смесь веществ перемешивали при комнатной температуре до растворения Dpp-Bian. При этом образовался раствор красно-коричневого цвета. Образовавшийся раствор комплекса I отфильтровывали и удаляли растворитель. Выход сухого продукта 0.634 г (85%).
ИК-спектр (ν, см–1): 3065 сл, 1671 сл, 1591 сл, 1531 ос, 1479 сл, 1447 с, 1426 сл, 1362 ср, 1319 с, 1255 ср, 1187 ср, 1144 сл, 1113 сл, 1084 сл, 1055 сл, 1042 сл, 934 сл, 926 сл, 861 ср, 835 сл, 821 с, 804 с, 787 ср, 771 с, 762 с, 752 ср, 697 сл, 670 сл, 544 сл. Спектр ЭПР (толуол, 293 K): g = 1.99898; A(119Sn) = 13.508, A(117Sn) = 12.911, A(127I) = 3.144, A(2 × 14N) = 0.573, A(2 × 1H) = 0.422, A(2 × 1H) = 0.222 мТл.
Синтез [(Dpp-Bian)Sn] (IV). Раствор комплекса [(Dpp-Bian)SnCl] в диэтиловом эфире (полученный из 0.5 г Dpp-Bian) отделяли от металлического олова, охлаждали до –78°C и добавляли при перемешивании 1.6 мл (1.05 ммоль) раствора nBuLi в гексане (0.66 моль/л), затем нагревали до комнатной температуры. Цвет раствора изменялся с красно-коричневого на фиолетовый, и выпадал бесцветный осадок. Полученную суспензию центрифугировали для отделения LiCl и побочных продуктов, таких как Dpp-Bian и металлическое олово. Концентрирование раствора приводит к образованию мелких темных кристаллов. Выход продукта IV 0.36 г (58%).
ИК-спектр (ν, см–1): 3061 ср, 3034 сл, 1917 сл, 1858 сл , 1797 сл, 1669 ср, 1611 с, 1597 с, 1439 ср, 1420 ср, 1391 с, 1364 ср, 1323 с, 1252 с, 1212 ср, 1186 ср, 1177 сл, 1108 ср, 1076 ср, 1057 ср, 1036 ср, 934 с, 878 сл, 833 сл, 816 с, 804 с, 795 ср, 780 сл, 764 ос, 670 ср, 642 сл, 619 ср, 594 сл, 547 ср. Спектр ПМР (400 MГц; C6D6; 297 K; δ, м.д.; J, Гц): 7.31 (с., 6H, аром.) 7.13 (д., 2H, CH нафт., J = 8.3), 6.85 (д.д., 2H, CH нафт., J = 8.3, J = 7.0), 6.38 (д., 2H, CH нафт., J = = 7.0), 3.50 (псевдосепт, 4H, 4 × CH(CH3)2, J = 6.8, J = 7.0), 1.27 (д., 12H, 4 × CH(CH3)(CH3), J = 6.8), 1.05 (д., 12Н, 4 × CH(CH3)(CH3), J = 7.0). Спектр ЯМР 119Sn (74.63 MГц; C6D6; 297 K; δ, м.д.): 360.0.
Реакция комплекса [(Dpp-Bian)SnCl] с nBuLi. В ампулу для проведения ЭПР-исследований насыпали 60 мг (0.092 ммоль) соединения [(Dpp-Bi-an)SnCl]. Образец охлаждали до температуры жидкого азота (195°С) и добавляли 1 мл Et2O, затем 0.42 мл 0.22 M раствора nBuLi в н-гексане. Размораживание ампулы проводили в резонаторе ЭПР-спектрометра. В результате фиксировали сигнал комплекса II, который появлялся при комнатной температуре через 5–10 мин после размораживания и постепенно (в течение суток) превращался в сигнал исходного [(Dpp-Bian)SnCl].
Спектр ЭПР (293 K, Et2O): g = 1.99893; A(119Sn) = = 8.160, A(117Sn) = 7.800, A(2 × 14N) = 0.547, A(2 × × 1H) = 0.151, A(2 × 1H) = 0.096, A(2 × 1H) = 0.028 мТл.
Реакция комплекса [(Dpp-Bian)SnCl] с tBuOLi. В ампулу для проведения ЭПР-исследований насыпали 50 мг (0.076 ммоль) соединения [(Dpp-Bian)SnCl] и 6 мг tBuOLi. Затем образец охлаждали до температуры жидкого азота (195°С) и добавляли 1 мл толуола. Размораживание ампулы проводили в резонаторе ЭПР-спектрометра. В результате фиксировали сигнал комплекса III, который появлялся при комнатной температуре через 15–20 мин после размораживания и постепенно исчезал в течение суток.
Спектр ЭПР (293 K, толуол): g = 1.99941; A(119Sn) = 10.500, A(117Sn) = 10.036, A(2 × 14N) = 0.503, A(2 × 1H) = 0.118, A(2 × 1H) = 0.083, A(2 × 1H) = = 0.017 мТл.
Синтез [(бис-Dpp-Mian)2Sn] (V). К раствору 0.148 г (0.23 ммоль) [(Dpp-Bian)Sn] в 2 мл толуола добавляли раствор 0.08 г (0.23 ммоль) Dpp-Mian в 2 мл толуола. Образовался желтый кристаллический осадок. Через 2 ч осадок отфильтровывали. Спектр ЯМР осадка совпадает со спектром ЯМР Dpp-Bian [55]. Летучие вещества удаляли в вакууме. Остаток растворяли в диэтиловом эфире (10 мл). Через сутки из полученного раствора при комнатной температуре образовался кристаллический осадок желтого цвета. Выход V 0.051 г (60%). По данным ЯМР, это соединение [(бис-Dpp-Mian)2Sn] [56] с примесью Dpp-Bian. Параметры кристаллической ячейки, согласно РСА, совпадают с данными комплекса [(бис-Dpp-Mian)2Sn] [56].
Синтез [(CF3-Bian)2Sn] (VI). Раствор CF3-Bian 0.6 г (1.0 ммоль) в ТГФ (25 мл) прилили в ампулу с избытком металлического олова (5 г). Образовавшуюся смесь перемешивали при комнатной температуре в течение 5 сут. При этом цвет раствора изменился с красно-оранжевого на ярко-синий. Образовавшийся раствор комплекса декантировали с избытка металлического олова и удалили растворитель. Выход сухого продукта 0.65 г (98%).
ИК-спектр (ν, см–1): 3056 сл, 1912 сл, 1758 ос, 1655 с, 1613 ср, 1588 с, 1507 о.с, 1429 ср, 1341 с, 1259 с, 1210 сл, 1190 сл, 1175 ср, 1150 ср, 1092 с, 1057 с, 999 ср, 962 сл, 924 с, 873 с, 833 ср, 804 с, 760 с, 744 ср, 723 ср, 699 ср, 658 сл, 619 с.
Спектр ПМР (200 MГц; ТГФ-d8; 297 K; δ, м.д.; J, Гц): 7.69 (с., 4H, 2 × o-C6H3) 7.41 (д., 2H, CH нафт., J = 8.1), 7.33 (с., 2H, 2 × м-C6H3) 7.17 (д.д., 2H, CH нафт., J = 8.1, J = 6.9), 7.01 (д., 2H, CH нафт., J = 6.9). Спектр ЯМР 119Sn (74.63 MГц; ТГФ-d8; 24°C; δ, м.д.; J, Гц): –103.59. Спектр ЯМР 19F (376.54 MГц; ТГФ-d8; 297K; δ, м.д.; J, Гц): –63.97.
Синтез [(CF3-Bian)3Sn] (VII). К раствору комплекса VI, полученному из 0.6 г (1.0 ммоль) CF3-Bi-an в ТГФ, добавляли половину эквивалента 0.3 г (0.5 ммоль) CF3-Bian. Затем ТГФ заменили на толуол. В результате образовался фиолетовый раствор комплекса [(CF3-Bian)3Sn], который при охлаждении обратимо становился синим. При концентрировании толуольного раствора комплекс кристаллизуется в виде оранжевых кристаллов. Выход кристаллического продукта 0.73 г (69%) в расчете на взятый в реакцию лиганд.
ИК-спектр (ν, см–1): 3066 сл, 3044 сл, 2726 сл, 1815 сл, 1676 сл, 1651 сл, 1605 ср, 1593 ср, 1530 с, 1438 ср, 1425 ср, 1408, 1373 с, 1300 сл, 1276 о.с, 1248 сл, 1175 с, 1148 с, 1138 о.с, 1105 ср, 1077 сл, 1042 сл, 1013 ср, 984 ср, 957 сл, 932 ср, 911 ср, 901 ср, 893 ср, 884 сл, 858 ср, 847 ср, 832 сл, 822 ср, 766 с, 727 с, 706 ср, 683 с.
РСА I и VII проведен на автоматическом дифрактометре Bruker AXS SMART APEX (графитовый монохроматор, МоКα-излучение, φ- и ω-сканирование, λ = 0.71073 Å). Экспериментальные наборы интенсивностей интегрированы с помощью программы SAINT [57]. Программа SADABS [58] использована для введения поправок на поглощение. Обе структуры расшифрованы методом “dual-space” с использованием программы SHELXT [59]. Все неводородные атомы в соединениях I и VII уточнены полноматричным МНК по $F_{{hkl}}^{2}$ в анизотропном приближении с помощью программного пакета SHELXTL [60]. Атомы водорода помещены в геометрически рассчитанные положения и уточнены в модели наездника (Uизо(H) = 1.5Uэкв(C) для СH3-групп и Uизо(H) = = 1.2Uэкв(C) для остальных групп). В кристалле VII на каждую молекулу комплекса приходится две разупорядоченные по двум положениям молекулы толуола. Атомы фтора двух CF3-групп в комплексе VII разупорядочены по трем положениям, а еще двух CF3-групп – по двум положениям. Для получения адекватных геометрических и тепловых характеристик при уточнении разупорядоченных фрагментов в VII применяли инструкции DFIX и ISOR. Основные кристаллографические характеристики и параметры рентгеноструктурного эксперимента для соединений I и VII приведены в табл. 1.
Таблица 1.
Кристаллографические данные, параметры эксперимента и уточнения структур I и VII
| Параметр | Значение | |
|---|---|---|
| I | VII | |
| Брутто-формула* | C36H40N2ISn | C98H52N6F36Sn |
| М | 746.29 | 2116.14 |
| Температура, K | 100(2) | 100(2) |
| Сингония | Моноклинная | Моноклинная |
| Пр. гр. | P21/c | С2/c |
| Параметры ячейки: | ||
| a, Å | 10.5124(5) | 28.901(3) |
| b, Å | 15.7287(7) | 13.4159(13) |
| c, Å | 20.0481(9) | 22.687(2) |
| α, град | 90 | 90 |
| β, град | 97.1150(10) | 101.881(4) |
| γ, град | 90 | 90 |
| V, Å3 | 3289.4(3) | 8607.8(15) |
| Z | 4 | 4 |
| ρ(выч.), мг/см3 | 1.507 | 1.633 |
| μ, мм–1 | 1.741 | 0.429 |
| Размер кристалла, мм | 0.96 × 0.67 × 0.40 | 0.52 × 0.35 × 0.17 |
| F(000) | 1492 | 4224 |
| θ, град | 1.952–28.999 | 1.680–29.999 |
| Число отражений собранных/независимых | 34 329/8725 | 46 099/12 302 |
| Rint | 0.0179 | 0.0416 |
| R1, wR2 (I > 2σ(I)) | 0.0220, 0.0582 |
0.0440, 0.1013 |
| R1, wR2 (по всем данным) | 0.0242, 0.0591 |
0.0605, 0.1075 |
| S | 1.042 | 1.070 |
| Остаточная электронная плотность (max/min), e/Å3 | 1.117/–0.776 | 1.165/–0.405 |
Дополнительные структурные результаты депонированы в Кембриджском банке кристаллографических данных (ССDС № 2130839 (I), 2130840 (VII); deposit@ccdc.cam.ac.uk; www: http:// www.ccdc.cam.ac.uk).
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
В 2006 году нашей группой было установлено, что Dpp-Bian может быть восстановлен металлическим алюминием до анион-радикала (в толуоле) или до дианиона (в Et2O) в присутствии галогенидов алюминия [61]. В настоящей работе была предпринята попытка восстановить Dpp-Bian металлическим оловом. Было установлено, что олово не восстанавливает Dpp-Bian в ароматических, алифатических растворителях, ТГФ, 1,2-диметоксиэтане или Et2O. Однако добавление половины эквивалента иода к суспензии Dpp-Bian в среде толуола, бензола или Et2O дает смесь, которая при действии на нее избытком олова приводит к образованию комплекса I с высоким выходом (схема 1 ).
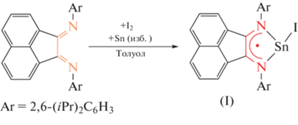
Схема 1 .
Соединение I парамагнитно, его спектр ЭПР и компьютерная модель представлены на рис. 1. Парамагнетизм обусловлен локализацией неспаренного электрона на сопряженной системе Dpp-Bian комплекса I. В связи с этим основной вклад в сверхтонкую структуру (СТС) вносят взаимодействия неспаренного электрона с атомом олова, атомом иода, двумя атомами азота дииминовой части и двумя парами протонов нафталиновой системы лиганда Dpp-Bian.
Рис. 1.
Спектр ЭПР комплекса I (толуол, 293 K): экспериментальный (а); симулированный (б) (g = = 1.99898; A(119Sn) = 13.508, A(117Sn) = 12.911, A(127I) = = 3.144, A(2 × 14N) = 0.573, A(2 × 1H) = 0.422, A(2 × 1H) = = 0.222 мТл).

Согласно данным рентгеноструктурного анализа, атом олова в комплексе I связан с бидентатно координированным Вian-лигандом и иодид-анионом (рис. 2). Длины связей в NCCN-фрагменте (табл. 2) указывают на анион-радикальную форму Вian-лиганда. Расстояния Sn–N в комплексе I (2.192(2) и 2.205(2) Å) несколько длиннее, чем в родственном I-производном олова(II) с менее стерически загруженным NCCN-лигандом (2.145(6)–2.154(5) Å) [62]. Пятичленный металлоцикл SnNCCN незначительно отклоняется от плоскости. Двугранный угол между плоскостями Sn(1)N(1)N(2) и N(1)C(1)C(2)N(2) составляет 5.24°. Связь Sn(1)–I(1) в комплексе I с плоскостью аценафтендииминового лиганда образует угол 103.98°. Такая геометрия характерна для родственных комплексов металлов 14-ой группы [62–64].
Рис. 2.
Молекулярное строение комплекса I. Тепловые эллипсоиды приведены с 30%-ной вероятностью. Атомы водорода не показаны для наглядности.

Таблица 2.
Избранные длины связей (Å) и углы (град) в соединениях I и VII
| Связь | I | VII |
|---|---|---|
| d, Å | ||
| Sn(1)–N(1) | 2.192(2) | 2.175(2) |
| Sn(1)–N(2) | 2.205(2) | 2.197(2) |
| Sn(1)–N(3) | 2.105(2) | |
| Sn(1)–I(1) | 2.823(1) | |
| N(1)–C(1) | 1.331(2) | 1.333(2) |
| N(2)–C(2) | 1.329(2) | 1.341(2) |
| N(3)–C(29) | 1.396(2) | |
| C(1)–C(2) | 1.439(2) | 1.445(2) |
| C(29)–C(29A)* | 1.379(3) | |
| Угол | ω, град | |
| N(1)Sn(1)N(2) | 76.65(5) | 76.95(6) |
| N(1)Sn(1)I(1) | 97.57(3) | |
| N(2)Sn(1)I(1) | 97.01(3) | |
| N(1)Sn(1)N(3) | 92.47(6) | |
| N(1)Sn(1)N(1A) | 163.58(8) | |
| N(1)Sn(1)N(2A) | 91.02(6) | |
| N(1)Sn(1)N(3A) | 99.98(6) | |
| N(2)Sn(1)N(3) | 96.08(6) | |
| N(1)Sn(1)N(1A) | 91.02(6) | |
| N(1)Sn(1)N(2A) | 86.54(8) | |
| N(1)Sn(1)N(3A) | 176.03(6) | |
| N(3)Sn(1)N(3A) | 81.46(8) | |
В 2005 г. нашей группой было показано, что комплекс магния [(Dpp-Bian)Mg-iPr(Et2O)] способен к восстановительному элиминированию изопропильного радикала [65]. При замене растворителя с диэтилового эфира на тетрагидрофуран изопропильный радикал отрывается от магния, при этом атом магния остается связанным с лигандом Dpp-Bian, который, в свою очередь, переходит от анион-радикала к дианиону.
Мы предположили схожий механизм образования комплекса IV при действии на комплексы [(Dpp-Bian)SnCl] [53] и I нуклеофилов. Для подтверждения нашего предположения необходимо было зафиксировать спектральным методом образование интермедиатов с анион-радикальным лигандом Dpp-Bian. Для этого мы провели реакцию комплекса [(Dpp-Bian)SnCl] с nBuLi и LiOtBu в резонаторе ЭПР-спектрометра. В результате нам удалось зафиксировать соответствующие сигналы парамагнитных соединений II и III (рис. 3 и 4).
Рис. 3.
Спектр ЭПР комплекса II (Et2O, 293 K): экспериментальный (а); спектр симулированный (б) (g = = 1.99893; A(119Sn) = 8.160, A(117Sn) = 7.800, A(2 × × 14N) = 0.547, A(2 × 1H) = 0.151, A(2 × 1H) = 0.096, A(2 × 1H) = 0.028 мТл).

Рис. 4.
Спектр ЭПР комплекса III (толуол, 293 K): экспериментальный (а); симулированный (б) (g = = 1.99941; A(119Sn) = 10.500, A(117Sn) = 10.036, A(2 × × 14N) = 0.503, A(2 × 1H) = 0.118, A(2 × 1H) = 0.083, A(2 × 1H) = 0.017 мТл).

Сигнал комплекса II нам удалось зафиксировать и в результате взаимодействия мононатриевой соли [(Dpp-Bian)Na] [66] с реакционной смесью, состоящей из одного эквивалента SnCl2(di-oxane) и одного эквивалента nBuLi (схема 2 ).

Схема 2 .
ЯМР-исследования этих реакций показывают, что станнилен IV образуется сразу при смешении реагентов и дает уширенный сигнал образовавшегося соединения, который со временем становится более разрешенным. Хромато-масс-спектрометрический анализ продуктов реакции соединения с nBuLi показал, что в смеси содержатся бутан и октан. На основании этого мы полагаем, что образование станнилена IV при взаимодействии комплекса [(Dpp-Bian)SnCl] с нуклеофилами протекает через стадию образования продукта обмена галогена на соответствующий кислотный остаток с последующим элиминированием, что приводит к восстановлению анион-радикала Dpp-Bian до дианиона (схема 3 ).

Схема 3 .
Станнилен IV проявляет восстановительные свойства по отношению к другому имино-аценафтену, Dpp-Mian (Dpp-Mian = 2-моно[(2,6-диизопропилфенил)имино]аценафтен-1-он). Нами было показано, что реакция комплекса IV с Dpp-Mian в толуоле приводит к образованию соединения, спектральные и дифракционные характеристики которого совпадают с данными для комплекса V [56], в котором атом олова связан с двумя лигандами бис-Dpp-Mian, образованными путем сдваивания двух Dpp-Mian по карбонильному атому углерода. При этом степень окисления олова становится равна +4. Вторым продуктом реакции является свободный Dpp-Bian (схема 4 ).

Схема 4 .
По данным циклической вольтамперометрии, первое восстановление CF3-Bian происходит при потенциале –1.35 В, а второе при –1.90 В, что соответственно на 0.72 и 0.8 В больше, чем для Dpp-Bian (–2.07 и –2.73 В) в аналогичных условиях (растворитель – тетрагидрофуран, рабочий электрод – стеклоуглерод (CУ), электрод сравнения – Ag/AgNO3, 0.01 M (CH3CN), вспомогательный электрод – Pt, фоновая соль – NBu4BF4, 0.2 M, скорость развертки 100 мВ/с). Следовательно, CF3-Bian восстанавливается легче, чем Dpp-Bian, а значит для лиганда CF3-Bian можно ожидать восстановления металлами, которые не восстанавливают напрямую Dpp-Bian.
Действительно, в отличие от Dpp-Bian, лиганд CF3-Bian восстанавливается металлическим оловом в среде координирующих растворителей, таких как диметоксиэтан или тетрагидрофуран. Реакция протекает при комнатной температуре и дает последовательно два продукта: на первой стадии образуется трис-лигандный комплекс VII, а затем бис-лигандный VI (схема 5 ). Трис-лигандный комплекс VII может быть получен в результате взаимодействия бис-лигандного комплекса VI с одним эквивалентом CF3-Bian. По данным ИК-спектроскопии, в спектре комплекса VII наблюдаются интенсивные полосы при 1605, 1593 см–1, которые относятся к валентным колебаниям полуторной и одинарной связям CN соответственно.

Схема 5 .
В отличие от комплекса I, атом олова в комплексе VII окружен тремя лигандами Bian (рис. 5). В независимой части элементарной ячейки находится только половина молекулы (ось симметрии второго порядка проходит через атом Sn(1) и середину связи C(29)–C(29A)). Таким образом, координационное число атома олова в комплексе VII равно шести, а координационный полиэдр представляет собой искаженный октаэдр. Распределение длин связей в двух лигандах (N(1)C(1)C(2)N(2) и N(1A)C(1A)C(2A)N(2A)) соответствует анион-радикальной форме Вian-лиганда, и длины связей находятся в хорошем согласии с комплексом I (табл. 2). В свою очередь, в N(3)C(29)C(29A)N(3A)-фрагменте наблюдается удлинение связей C–N с одновременным укорочением связи C–C, что свидетельствует о дианионной форме данного лиганда [67]. Степень окисления олова(IV) хорошо согласуется с укорочением длин связей Sn(1)–N(1) и Sn(1)–N(2) в соединении VII по сравнению с комплексом I. Пятичленные металлоциклы в комплексе VII практически плоские. Максимальное отклонение атомов от соответствующих плоскостей для всех трех лигандов не превышает 0.019 Å. Двугранные углы между плоскостями Sn(1)N(1)N(2) и N(1)C(1)C(2)N(2), Sn(1)N(3)N(3A) и N(3)C(29)C(29A)N(3A) составляют соответственно 0.37° и 1.10°. Плоскости аценафтендииминовых фрагментов располагаются друг относительно друга под углом 81.42° (лиганды N(1)N(2) и N(1A)N(2A)) и 86.53° (лиганды N(1)N(2) и N(3)N(3A)).
Рис. 5.
Молекулярное строение комплекса VII. Тепловые эллипсоиды приведены с 30%-ной вероятностью. Атомы водорода не показаны, CF3-группы представлены одним атомом углерода для наглядности.

Авторы заявляют, что у них нет конфликта интересов.
Список литературы
Jones C., Stasch A. The Chemistry of the Group 13 Metals in the +1 Oxidation State / Eds. Aldridge S., Downs A.J. John Wiley & Sons, Ltd., 2011. P. 285.
Arnold J. // Dalton Trans. 2008. № 33. P. 4334.
Melen R.L. // Science. 2019. V. 363. № 6426. P. 479.
Weetman C., Inoue S. // ChemCatChem. 2018. V. 10. № 19. P. 4213.
Power P.P. // Nature. 2010. V. 463. P. 171.
Stoy A., Böhnke J., Jiménez-Halla J.O.C. et al. // A-ngew. Chem. Int. Ed. 2018. V. 57. № 20. P. 5947.
Arrowsmith M., Böhnke J., Braunschweig H. et al. // A-ngew. Chem. Int. Ed. 2017. V. 56. № 45. P. 14287.
Légaré M.-A., Bélanger-Chabot G., Dewhurst R.D. et al. // Science. 2018. V. 359. № 6378. P. 896.
Kavara A., Boron T.T., Ahsan Z.S. et al. // Organometallics. 2010. V. 29. № 21. P. 5033.
Padělková Z., Švec P., Pejchal V. et al. // Dalton Trans. 2013. V. 42. № 21. P. 7660.
Rodriguez R., Contie Y., Mao Y. et al. // Angew. Chem. Int. Ed. 2015. V. 54. № 50. P. 15276.
Perla L.G., Kulenkampff J.M., Fettinger J.C. et al. // Organometallics. 2018. V. 37. № 21. P. 4048.
Peng Y., Fischer R.C., Merrill W.A. et al. // Chem. Sci. 2010. V. 1. № 4. P. 461.
Stender M., Phillips A.D., Wright R.J. et al. // Angew. Chem. Int. Ed. 2002. V. 41. № 10. P. 1785.
Pu L., Twamley B., Power P.P. // J. Am. Chem. Soc. 2000. V. 122. № 14. P. 3524.
Takeuchi K., Ichinohe M., Sekiguchi A. // J. Am. Chem. Soc. 2011. V. 133. № 32. P. 12478.
Schäfer A., Reißmann M., Schäfer A. et al. // Chem. Eur. J. 2014. V. 20. № 30. P. 9381.
Peng Y., Brynda M., Ellis B.D. et al. // Chem. Commun. 2008. № 45. P. 6042.
Hadlington T.J., Jones C. // Chem. Commun. 2014. V. 50. № 18. P. 2321.
Wang S., Sherbow T.J., Berben L.A. et al. // J. Am. Chem. Soc. 2018. V. 140. № 2. P. 590.
Jana A., Schulzke C., Roesky H.W. // J. Am. Chem. Soc. 2009. V. 131. № 13. P. 4600.
Brown Z.D., Power P.P. // Inorg. Chem. 2013. V. 52. № 11. P. 6248.
Hansen K., Szilvási T., Blom B. et al. // Chem. Eur. J. 2014. V. 20. № 7. P. 1947.
Präsang C., Stoelzel M., Inoue S, et al. // Angew. Chem. Int. Ed. 2010. V. 49. № 51. P. 10002.
Bouška M., Dostál L., Růžička A. et al. // Chem. Eur. J. 2011. V. 17. № 2. P. 450.
Han J.S., Sasamori T., Mizuhata Y. et al. // J. Am. Chem. Soc. 2010. V. 132. № 8. P. 2546.
Freitag S., Krebs K.M., Henning J. et al. // Organometallics. 2013. V. 32. № 22. P. 6785.
Sarkar D., Weetman C., Munz D. et al. // Angew. Chem. Int. Ed. 2021. V. 60. № 7. P. 3519.
Weiß S., Widemann M., Eichele K. et al. // Dalton Trans. 2021. V. 50. № 14. P. 4952.
Mandal S.K., Roesky H.W. // Acc. Chem. Res. 2012. V. 45. № 2. P. 298.
Hadlington T.J., Driess M., Jones C. // Chem. Soc. Rev. 2018. V. 47. № 11. P. 4176.
Dodonov V.A., Chen W., Zhao Y. et al. // Chem. Eur. J. 2019. V. 25. № 35. P. 8259.
Zhang W., Dodonov V.A., Chen W. et al. // Chem. Eur. J. 2018. V. 24. № 56. P. 14994.
Dodonov V.A., Chen W., Liu L. et al. // Inorg. Chem. 2021. V. 60. № 19. P. 14602.
Dodonov V.A., Kushnerova O.A., Baranov E.V. et al. // Dalton Trans. 2021. V. 50. № 25. P. 8899.
Fedushkin I.L., Skatova A.A., Dodonov V.A. et al. // I-norg. Chem. 2016. V. 55. № 17. P. 9047.
Dodonov V.A., Xiao L., Kushnerova O.A. et al. // Chem. Commun. 2020. V. 56. № 54. P. 7475.
Dodonov V.A., Skatova A.A., Fedushkin I.L. // Russ. J. Coord. Chem. 2019. V. 45. № 4. P. 301.
Sokolov V.G., Koptseva T.S., Dodonov V.A. et al. // Russ. Chem. Bull. 2018. V. 67. № 12. P. 2164.
Makarov V.M., Koptseva T.S., Sokolov V.G. et al. // Russ. J. Coord. Chem. 2020. V. 46. № 4. P. 215.
Chen W., Dodonov V.A., Sokolov V.G. et al. // Organometallics. 2021. V. 40. № 4. P. 490.
Dodonov V.A., Skatova A.A., Cherkasov A.V. et al. // Russ. Chem. Bull. 2016. V. 65. № 5. P. 1171.
Fedushkin I.L., Maslova O.V., Morozov A.G. et al. // A-ngew. Chem. Int. Ed. 2012. V. 51. № 42. P. 10584.
Fedushkin I.L., Skatova A.A., Dodonov V.A. et al. // I-norg. Chem. 2014. V. 53. № 10. P. 5159.
Fedushkin I.L., Dodonov V.A., Skatova A.A. et al. // Chem. Eur. J. 2018. V. 24. № 8. P. 1877.
Fedushkin I.L., Nikipelov A.S., Lyssenko K.A. // J. Am. Chem. Soc. 2010. V. 132. № 23. P. 7874.
Fedushkin I.L., Nikipelov A.S., Morozov A.G. et al. // Chem. Eur. J. 2012. V. 18. № 1. P. 255.
Morozov A.G., Martemyanova T.V., Dodonov V.A. et al. // Eur. J. Inorg. Chem. 2019. V. 2019. № 39–40. P. 4198.
Dodonov V.A., Morozov A.G., Rumyantsev R.V. et al. // Inorg. Chem. 2019. V. 58. № 24. P. 16559.
Armarego W.L.F. Purification of Laboratory Chemicals (Eighth Edition). Butterworth-Heinemann, 2017. Ch. 3. P. 95.
Paulovicova A., El-Ayaan U., Shibayama K. et al. // Eur. J. Inorg. Chem. 2001. № 10. P. 2641.
Gasperini M., Ragaini F., Cenini S. // Organometallics. 2002. V. 21. № 14. P. 2950.
Додонов В.А., Кушнерова О.А., Разборов Д.А. et al. // Изв. АН. Сер. хим. 2022. T. 2. C. 322.
Stoll S., Schweiger A. // J. Magn. Reson. 2006. V. 178. № 1. P. 42.
El-Ayaan U., Paulovicova A., Fukuda Y. // J. Mol. Struct. 2003. V. 645. № 2. P. 205.
Lukoyanov A.N., Ulivanova E.A., Razborov D.A. et al. // Chem. Eur. J. 2019. V. 25. № 15. P. 3858.
SAINT. Data Reduction and Correction Program. V. 8.38A. Madison (WI, USA): Bruker AXS, 2017.
Krause L., Herbst-Irmer R., Sheldrick G.M. et al. // J. Appl. Crystallogr. 2015. V. 48. № 1. P. 3.
Sheldrick G. // Acta Crystallogr. A. 2015. V. 71. № 1. P. 3.
Sheldrick G. // Acta Crystallogr. C. 2015. V. 71. № 1. P. 3.
Fedushkin I.L., Lukoyanov A.N., Fukin G.K. et al. // Russ. Chem. Bull. 2006. V. 55. № 7. P. 1177.
Ayers A.E., Dias H.V.R. // Inorg. Chem. 2002. V. 41. № 12. P. 3259.
Fedushkin I.L., Khvoinova N.M., Baurin A.Y. et al. // Inorg. Chem. 2004. V. 43. № 24. P. 7807.
Chia S.-P., Li Y., Ganguly R. et al. // Eur. J. Inorg. Chem. 2014. V. 2014. № 3. P. 526.
Fedushkin I.L., Skatova A.A., Hummert M. et al. // Eur. J. Inorg. Chem. 2005. V. 2005. № 8. P. 1601.
Fedushkin I.L., Skatova A.A., Chudakova V.A. et al. // Angew. Chem. Int. Ed. 2003. V. 42. № 28. P. 3294.
Sokolov V.G., Koptseva T.S., Rumyantcev R.V. et al. // Organometallics. 2020. V. 39. № 1. P. 66.
Дополнительные материалы отсутствуют.
Инструменты
Координационная химия